
Abstract
Autism spectrum disorders (ASD) are highly heritable and genetically complex conditions. Although highly penetrant mutations in multiple genes have been identified, they account for the etiology of <1/3 of cases. There is also strong evidence for environmental contribution to ASD, which can be mediated by still poorly explored epigenetic modifications. We searched for methylation changes on blood DNA of 53 male ASD patients and 757 healthy controls using a methylomic array (450K Illumina), correlated the variants with transcriptional alterations in blood RNAseq data, and performed a case–control association study of the relevant findings in a larger cohort (394 cases and 500 controls). We found 700 differentially methylated CpGs, most of them hypomethylated in the ASD group (83.9%), with cis-acting expression changes at 7.6% of locations. Relevant findings included: (1) hypomethylation caused by rare genetic variants (meSNVs) at six loci (ERMN, USP24, METTL21C, PDE10A, STX16 and DBT) significantly associated with ASD (q-value <0.05); and (2) clustered epimutations associated to transcriptional changes in single-ASD patients (n=4). All meSNVs and clustered epimutations were inherited from unaffected parents. Resequencing of the top candidate genes also revealed a significant load of deleterious mutations affecting ERMN in ASD compared with controls. Our data indicate that inherited methylation alterations detectable in blood DNA, due to either genetic or epigenetic defects, can affect gene expression and contribute to ASD susceptibility most likely in an additive manner, and implicate ERMN as a novel ASD gene.
Similar content being viewed by others

Introduction
Autism spectrum disorders (ASD; OMIM 209850) are a group of early-onset neurodevelopmental disorders diagnosed on the basis of two symptom areas: social communication impairments, and restricted repetitive behaviors, interests or activities (DSM-V). The currently estimated prevalence of ASD is 1/42 males and 1/189 females aged 8 years, after a documented prevalence increase during the past two decades.1 There is strong evidence for a genetic etiology of ASD with heritability ranging from 50 to 90%.2, 3 However, despite remarkable advances driven by genetics and genomics leading to the identification of single-gene mutations, copy-number changes and double hit models, the etiology of around 70% of ASD cases still remains unknown.4, 5 This relatively poor detection rate could be explained in part by the high phenotypic and genetic heterogeneity of ASD with multiple inheritance models, including monogenic, oligogenic and polygenic with gene–environment interactions.6, 7, 8 In addition, most genomic studies to date have focused on the analysis of sequence and copy-number changes in the coding part of the genome, therefore, missing putative epimutations and other changes located elsewhere in the genome.
Epigenetics comprises DNA methylation, long non-coding RNA and histone post-translational modifications, which act as regulatory mechanisms of gene expression. Epigenetic aberrations have been associated with a wide range of diseases including ASD.9, 10 Several single-gene disorders commonly associated with autism (that is, Fragile-X11 and Rett syndromes12 show multiple epigenetic deregulations, and recurrent genomic alterations in chromosomal regions subjected to imprinting (chr7q, 15q (ref. 13, 14)) commonly manifest as ASD. Other epigenetic defects secondary to mutations in genes such as UBE3A15 and OXCTR,16 as well as altered CpG methylation in genes such as RORA, BCL1 ref. 17) and EN-2,18 have also been implicated in autism. Therefore, methylation and imprinting alterations associated or not to genetic anomalies, may be responsible for additional cases of ASD. Although epigenetics is still a poorly explored field in ASD, the current genome-wide technologies permit to assess DNA methylome in the available tissues. To date, several small-size case–control studies have addressed methylomic abnormalities in ASD searching for differences in twin pairs discordant for the disease on cell line or blood DNA,17, 19 investigating DNA from different brain regions,20, 21 or looking for effects of advanced maternal age on buccal epithelium as a surrogate of ectodermal cells.22 Methylation analyses of promoter regions of genes in blood DNA failed to detect relevant changes in an initial study (analyzing 1505 regions in 70 patients),23 while reporting hypermethylation at ENO2 in 14.5% ASD cases (analyzing 16,000 regions in 131 patients).24
In this study we aimed to identify methylation aberrations in blood DNA of a cohort of ASD patients in a search for testable epigenetic biomarkers contributing to ASD etiology. Functional consequences were analyzed by RNAseq of blood RNA, integrating the analysis of epigenetic variants and their transcriptional consequences. Finally, we performed a case–control study of the resulting rare variants using an independent and larger ASD sample in order to increase the power and significance of the associations, as well as a screening for mutations affecting the coding sequence in the previously identified genes.
Materials and methods
Study population
We selected 53 unrelated idiopathic ASD male patients, aged from 2 to 14 years (Supplementary Table 1) and referred from several hospitals of the Spanish Health System. All individuals had a diagnosis of ASD based on clinical and psychological evaluations (DSM-IV and ADI-R test). Genetic and biochemical testing for comorbid conditions (Fragile-X and some metabolic disorders), and molecular karyotyping (aCGH or SNP array) were negative in all cases. The study was approved by the ethical committee of the centers involved (CEIC-Parc Salut Mar), and written informed consent was obtained from the parents or legal tutors.
Healthy controls consisted of 10 age-matched Spanish male children analyzed simultaneously. Extraction of DNA and RNA was performed from peripheral blood with the Blood DNA extraction kit (Qiagen, Hilden, Germany), and RNA was isolated from lymphocytes using a ficoll density-gradient (STEMCELL Technologies, Vancouver, BC, Canada), according to the manufacturer’s instructions.
Additional methylation data from 91 controls (males of a wide age range) were obtained from the Center for Research in Environmental Epidemiology (CREAL) and the Bellvitge Institute for Biomedical Research (IDIBELL),25, 26 as well as from 656 controls (males and females of European ancestry aged from 19 to 101 years) available through GEO (GSE40279). All 757 control samples were obtained from whole blood, except for 5 from cord blood.
In addition, DNA methylation data (blood, 415 ASD and 405 healthy siblings, males, 6 to 25 years old) obtained with the HumanMethylation27 platform (Illumina, San Diego, CA, USA) were downloaded from dbGAP database (accession, phs000619) and probes shared by both platforms were compared.
Methylomic analyses
The DNA was bisulfite converted and hybridized onto Illumina HumanMethylation450K BeadChips (Illumina) at the Spanish National Genotyping Centre (Barcelona, Spain), according to the manufacturer’s protocol. Samples were placed randomly through the chips and a technical replicate was included in each chip. Raw data were obtained with Genome Studio (Illumina; v1.8) and quality controls included technical replicates correlation, detection P-value filtering and a bisulfite conversion control. The obtained correlation Pearson’s mean coefficient values were over 0.903 and the Spearman’s coefficient (rho) over 0.962. Detection P-value was filtered removing probes with P-values over 0.01 and sample bisulfite conversion passed the intensity threshold of 4000 as indicated in manufacturer’s instructions.
We obtained the beta-values for each CpG, ranging from 0 (unmethylated) to 1 (completely methylated). Simple scaling normalization was applied using the HumMeth27QCReport27 and LUMI28 packages in R.
Differential methylation (DM) analyses were performed using Limma R Package,29 applying eBayes and linear BH-FDR adjustment; q-value was set at 0.01. Probes with low-quality detection signal in >5% of the samples, with cross-reactive hybridization,30 or harboring known single-nucleotide polymorphisms (SNPs; minor allele frequency (MAF)>0.005) within the CpG±5nt were removed (dbSNPv137.2 and CEU 1000-Genomes databases). We used two different approaches to perform DM analyses: we first compared the ASD group versus the control group, and then, based on the assumption that genetic alterations in ASD are highly heterogeneous, we compared each patient individually versus the control group. Following the individual approach, we curated the results discarding differentially methylated CpGs (DMCpGs) altered in any of the control samples, and those with values below the absolute methylation difference threshold (Delta Beta value⩾|1.8|, representing 18% of difference). We prioritized probes showing methylation-status changes as previously reported,31 being the hypomethylated status with beta-values <0.3, hypermethylated with beta values >0.7 and intermediate beta in the 0.3–0.7 range. After the first DM analysis, we performed a second DM analysis including new healthy control data from an epidemiology study (n=91, Spanish, males, wide age range), applying the same statistical analysis and filtering criteria, except for a less-strict fold change (under 1.5 or 15%). Finally, we compared the resulting DMCpGs with a publicly available control data set (GSE40279; n=656 Hispanic and CEU, males and females, wide age range) and discarded probes altered in this control set with a frequency >0.005, considered as relatively common epigenetic variants. We obtained DMCpGs and differentially methylated regions (DMRs). We defined DMRs as regions with more than one significant DMCpGs close to each other (using our strict filter of significant difference >18% at each DMCpG). We also used the COMB-P32 algorithm to detect more subtle methylation changes in selected windows of 1000 kb and weighted for 50-bp windows with a nominal P-value <0.01. Manhattan clustering and principal component analyses were performed using R.
We also performed subset-quantile within array normalization (SWAN) to correct type1/type2 errors using minfi tool,33 considering an absolute methylation difference of 7% as previous studies.20, 22 Then, the same pipeline as above was applied to define the final DMCpGs.
DMCpGs validation
We performed genetic and epigenetic validations of selected altered DMCpGs. Parental samples of ASD probands were also analyzed. The same DNA samples were subjected to PCR amplification and Sanger sequencing to analyze the sequence, and to pyrosequencing and BS-PCR direct sequencing34 to analyze the methylation levels quantitatively or qualitatively. Pyrosequencing analyses were performed using PyroMark Q96 (Qiagen, Hilden, Germany) following the manufacturer’s instructions and the results were analyzed with the software provided. Primer sequences for pyrosequencing are available upon request. Pyrosequencing was also used to replicate the analysis in additional samples. Methylation was also assessed qualitatively by BS-PCR direct sequencing.34 Primers were designed with Methyl Primer Express v.1. Software (Applied Biosystems, Carlsbad, CA, USA) and PCR conditions are available on request.
Analysis of cis-acting elements on DMRs
We searched for putative genetic variants in the flanking intervals of the DMRs. We designed primers at both sides of the DMRs (including ~1 kb in each side), and performed Sanger sequencing of the PCR amplified products on specific samples. We also used multiple-ligation probe-amplification (MLPA) with probes located next to the DMCpGs on these regions using standard protocols and analytical methods. Primers and probes were designed as explained above and are available upon request.
Large-scale genotyping
In order to define a putative association of the identified rare variants in ASD patients, we genotyped 32 rare SNVs (Supplementary Table 2) in an additional set of 394 ASD samples (Spanish and ECACC collection) and 500 Spanish controls by Sequenom (San Diego, CA, USA). We used 250 ng of DNA to perform the Sequenom assay, which was conducted at Universitat de Valencia (Valencia, Spain). To compute the statistical association we included all the data (450K, 27k and Sequenom) and compared allelic frequencies. Fisher’s exact test and correction by false discovery rate (FDR) were performed using R (fisher.test function and q-value35).
RNA sequencing
To identify the functional consequences of methylation alterations we performed an RNAseq study in 29 out of the 53 studied samples, from which we obtained good quality RNA from blood (RIN>7). The RNA library was prepared and sequenced (paired-end 100-bp reads) using Illumina HiSeq 2000 (Illumina) and sequence reads aligned (build hg37) using TopHat36 and Bowtie.37 Expression was quantified by htseqcount in Conditional Quantile Normalization counts.38 Data were corrected for batch effect by Combat algorithm (SVA package R).39 We assessed case–control differential expression of the genes flanking 50 kb upstream and downstream the DMCpG, distance used in similar studies.40, 41, 42 We selected altered genes by Z-score, selecting Z-scores over 2 (P-value <0.05) or 1.5 if two genes were altered in the same region, as recommended in expression analysis.43
Targeted NGS sequencing
The coding region of five selected genes (ERMN, STX16, SMG7-AS1, PIK3CD and ZCCHC9) was included in a targeted resequencing panel analyzing 279 additional idiopathic ASD cases and 105 controls. Samples were fragmented and processed with the TruSeq DNA Sample Preparation kit (Illumina). The libraries were captured by the NimbleGen SeqCap EZ custom Library (Hofman-La Roche, Basel, Switzerland). Quality control was assessed using FastQC tool,44 adapter sequences were trimmed using Trim Galore45 and mapping was performed using BWA.46 GATK47 was used for variant calling and Annovar48 for functional annotation. Rare mutations predicted deleterious (loss of function or missense predicted damaging with the CONDEL program49) were selected. We also analyzed exome data of ASD patients available from dbGAP (accession=phs000482.v1.p1 and phs000298.v1.p1; n=931) in order to define the number of deleterious mutations in selected genes. To define whether the burden of deleterious mutations is higher in ASD, we use exome data from controls in the EXAC database,50 and compared the mutational load in cases and controls (Fisher’s test FDR corrected).
Gene ontology analysis
We searched for enriched pathways in DAVIDGo database,51 and for over-represented functions using CPDB52 for all the genes associated to the DMCpGs, as well as for the subset of genes with significant association and located in the DMRs.
Results
Methylation alterations in blood DNA of ASD
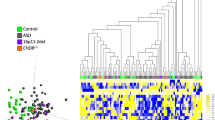
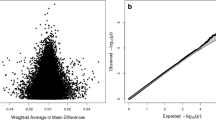
Clustering and principal component analyses showed a random distribution of the methylation data from patients and controls with no common patterns or systemic differences between groups independently of the normalization method applied (Supplementary Figures 1 and 2). A similar result was obtained in the differential methylation analysis comparing cases with controls as groups, with no significant differentially methylated CpG (DMCpGs) identified (q-value <0.01).
In order to detect rare methylation defects, we compared each individual ASD patient with the age- and gender-matched control group (n=10). After assessing differential methylation with the additional control sets with stringent filtering, we identified 700 DMCpGs present in ASD samples (4 to 33 per sample; Figure 1, and Supplementary Table 3). We observed a high concordance between the two normalization methods (93% of the DMCpGs identified). Only 34 additional DMCpGs were found with minfi (Supplementary Table 4). Therefore, the results were not affected by the normalization approaches. To define the consistency of the identified DMCpGs, we performed sequencing validation on the same DNA samples at 98 sites, including 85 isolated CpGs and 13 plausible DMRs. The validation rate, conceived as true genetic or epigenetic variants, was 85% (84/98), 78 sites corresponding to genetic alterations (meSNVs) and 5 to epigenetic modifications in the DMRs.

Pipeline followed for the differential methylation analyses with detailed filtering criteria. For the first two comparisons (DM1 and DM2), we established a P-value threshold <0.01, whereas in the third comparison (DM3) we searched for rare methylation alterations in a large control cohort (frequency <0.005). We obtained 700 DMCpGs, 156 intergenic probes and 544 of them encompassing 467 genes and 10 miRNA. We selected for further analyses the DMCpGs found in more than one case, including the dbGAP study (accession=phs000619), the differentially methylated regions, and the DMCpGs associated with significant cis-acting expression changes. Further studies with patients and controls were performed, for which some public accession data were added to the cohorts (data not added). Venn-diagram: correlation data between two normalization methods showing high concordance. ASD, autism spectrum disorder; C, controls; DM, differential methylation analysis; DMCpGs, differentially methylated CpGs; F, females; M, males.
Most of the 700 DMCpGs (587; 83.9%) in ASD cases showed a relative hypomethylation compared with controls, and mostly changed from a hypermethylated state to an intermediate state (Beta-values from 30 to 60%; Supplementary Figure 3). Regarding genomic location, 544 DMCpGs located within 467 genes and 10 miRNA, whereas 156 DMCpGs were intergenic probes (>1.5 kb from the gene Transcription Starting Sites; Figure 1). We searched for convergence between the detected genes and core ASD-associated genes compiled in SFARI and AutismKb databases (n=486 genes), and found 32 genes (6.5%; Supplementary Table 5).
Although most DMCpGs were case specific, we found 20 present in two or more ASD patients. Rare SNPs at the CG position (meSNPs) had been described at 9 (45%) of these sites (dbSNP137 and 1000G MAF<0.005), so we assumed the SNPs were the cause of the DMCpG. Validation and segregation analysis of the remaining DMCpGs (n=11) revealed that all were also rare single-nucleotide variants (meSNVs) inherited from healthy progenitors (Supplementary Table 2).
We also found five DMRs in four different patients (one patient had two DMRs). Methylation levels were validated, and we discarded underlying genetic changes by dosage analysis with MLPA probes in the region and by sequencing of the flanking intervals. These clustered epimutations were inherited in three cases, two from the father and one from the mother, with identical degree and extension in the parental samples (Table 1 and Supplementary Figures 4–7). A search for DMRs with less-stringent methylation differences using the COMB-P algorithm revealed six additional regions, but they were also found altered in several controls and then not considered as ASD specific.
Transcriptional consequences of DMCpGs
Out of the 476 DMCpGs altered in any of the 29 samples with blood transcriptomic data by RNAseq, we studied expression changes of nearby genes that were expressed in blood (n=630). We observed a strong correlation (Z-score>2) with expression changes of flanking genes in 7.6% of the loci harboring DMCpGs. Validation and segregation analysis of the 15 DMCpGs outside DMRs revealed that all contained rare inherited meSNVs (Supplementary Table 2, Supplementary Figure 8). Interestingly, a methylation alteration in the promoter of RNF166 led to a transcriptional alteration of three flanking genes (Figure 2).

Heterozygous meSNV associated with overexpression of several regional genes at 16q24.3. (a) Beta values across the region for the patient (green), the rest of the patients (red), the age-paired (blue) and the Spanish and GEO (GSE:40279; purple) controls sets. Diagram under the graphs shows the genes in the region, 450k array probes, the DHSs and chromatin domains in the region. (b) Sanger sequence for the DMCpG showed a paternally inherited meSNV (G>A). (c) Expression levels showed overexpression of RNF166 and MVD, and a tendency in CTU2 (d) correlation for methylation values and expression for the genes for all ASD samples. ASD, autism spectrum disorder; C, controls; DHSs, DNAseI hypersensitive sites.
Regarding the rare meSNVs present in more than one patient, a significant overexpression of the flanking gene(s) was observed in four cases and a downregulation in one. The meSNV causing hypomethylation within GALNT5 correlated with overexpression of the nearby ERMN gene (Table 1 and Figure 3) in one of the cases (ASD_5). Finally, RNAseq also revealed significant changes of the expression levels of flanking genes in 4/5 of the DMRs (Figure 4).

Example of a DMCpGs present in more than one case with associated effects on gene expression. (a) Hypomethylation in cg05777410 correlated with ERMN overexpression. Beta values for the region showing two individuals (ASD_1 and ASD_5) with DM at a single site in the region, cg05777410; the rest of ASD patients (red), the age-paired (blue) and the Spanish and GEO (GSE:40279; purple) control sets showed a similar pattern. The diagram under the graph shows the genomic context with the GALNT5 and ERMN genes, 450k array probes, DNSs and chromatin domains. (b) Sanger sequence of the region in the ASD_5 trio showing a maternally inherited meSNV (C>T in chr2:158162587) at cg05777410. (c) Quantitative RNAseq data for ERMN and GALNT5 transcripts showed a significant overexpression of the ERMN gene in ASD_5, and ASD_33 compared with the rest of individuals. (d) Location of the deleterious mutations at the coding region of Ermin, showing exclusively those detected in ASD patients. ASD, autism spectrum disorder; C, controls.

Diagram showing the region and expression for ZCCHC9 detected DMR. (a) Diagram for the region with 13 CpGs altered located in ZCCHC9 promoter (pointed with an arrow). The diagram shows the gene location, the 450k array probes, the DNAseI hypersensitive clusters, and chromatin domain regions. ASD_13 shows increased methylation levels. (b) Expression for all ASD patients for the region. ASD_13 shows a downregulation for the gene. ASD, autism spectrum disorder; C, controls; DMRs, differentially methylated regions.
Rare meSNVs associated to ASD
We first used methylomic (Illumina 27k, Illumina) data from blood DNA of 415 ASD cases and 405 controls as a replication cohort. Only 17 (2.4%) of the 700 DMCpGs were covered by this array. Relative hypomethylation at cg25781162 (ABCG5), caused by an inherited meSNP in one of our probands, was also detected in another ASD patient, and absent in controls (Table 1).
To determine whether a selected set of rare meSNVs (found in more than one ASD patient and/or associated with transcriptional effects) were statistically associated to ASD, we genotyped them by Sequenom in an additional 394 ASD and 500 Spanish controls. None of the variants were common in the Spanish control population (MAF<0.002), discarding population-specific variants. Significant association following multiple testing corrections (q-value<0.05) was found with five meSNVs (at the loci: GALNT5-ERMN, USP24, METTL21C, PDE10A and DBT; Table 1). Considering all the data for case–control analyses, six additional meSNVs identified in a single or two ASD patient(s) were absent in all the studied control samples (n=935–1713; Table 1).
We also used pyrosequencing in another cohort of ASD patients (n=75) to check whether three of the clustered DMRs were present in more cases, but all resulted negative. Therefore, most of these DMRs were patient specific and absent in controls (n=757), except for one present in 3 of the 757 controls. Taken together, these patient-specific meSNVs and DMRs are still good candidates that cannot be excluded from being associated with ASD risk, although replication in larger data sets would be required.
Deleterious mutations in ERMN associated to ASD
To identify other putative genetic mutations in some genes whose methylation and expression changes showed association with ASD disease, we performed targeted resequencing of the coding regions of 5 selected genes (ERMN, STX16, SMG7-AS1, PIK3CD and ZCCHC9). We completed the screening in additional 279 ASD patients and 105 Spanish controls, used to exclude population-specific variants. No loss-of-function mutations were identified in any of the genes in the ASD cohort, but deleterious missense rare variants were detected. Mutations in ERMN were identified in three unrelated ASD patients, one in two cases (c.830G>A/R277Q) and the second in one case (c.823 T>G/S275A) (Supplementary Table 6). We also reanalyzed 931 exomes of ASD patients (previously reported in O’Roak et al.53 and De Rubeis et al.54) available from dbGAP and the EXAC repository data to define the mutation load. We identified five additional ASD cases harboring the same R277Q mutation and four additional deleterious mutations in ASD patients (Figure 3 and Supplementary Table 6). In the EXAC repository, a total of 327 predicted pathogenic mutations could be found. By comparison, a significant increase in the mutation load of ERMN in ASD with respect to controls was observed (P-value=0.046), which is much more significant if we exclude three population-specific variants (P-value=7.97E−05). The finding of a higher mutation burden in ASD patients, along with a positive association with a single meSNV at this locus and the overexpression in carriers of the meSNV, strongly support that ERMN is implicated in ASD. No significant mutation burden was identified in any of the other screened genes in ASD patients.
General gene ontology analysis
Gene onthology analysis for the total amount of genes (n=467) revealed six significantly (P-value<0.05) enriched KEGG pathways (DavidGO; Supplementary Table 7): axon guidance (n=10 genes), PIP metabolism (n=5 genes), chemokine signaling (n=10 genes), focal adhesion (n=12 genes), adherence (n=6 genes) and tight junctions (n=8 genes). Regarding the biological function enrichment annotation chart (CPDB database), we found enrichment in functions such as nervous system development (n=73 genes), neuron development (n=30), synaptic transmission (n=28), cell death (n=59 genes) and immune response (n=18 genes). In addition, when analyzing all the significant associated regions and the DMRs we found only AMPK signaling pathway significantly enriched (CAMKK1 and PIK3CD).
Discussion
With the main goal of identifying methylation aberrations as testable epigenetic biomarkers for ASD, we have performed an epigenome-wide association study in blood DNA in a selected cohort of idiopathic ASD patients, and explored the functional consequences on blood transcriptome of a subset. Although ASD is mainly a disorder of neurodevelopment and there are genomic regulatory regions with brain-specific epigenetic states, there is evidence that blood cells can be used as proxies for epigenetic state at the many regions whose epigenetic state is common to multiple tissues.55 We used the 450k methylomic array containing selected probes for ~1.7% of the estimated 28 million CpG sites of the human genome. No systemic methylation alterations were identified in this ASD population, as previously shown in other studies of ASD using brain and blood DNA samples.19, 56 Despite the small size of our and the previously reported cohorts, the combined data discarded common DMRs in the regions analyzed shared by all ASD patients. In a similar manner to most genetic variants currently associated to ASD, and considering the low-recurrent nature of epigenetic events, low prevalent or individual-specific epimutations would not be detected with this type of group analyses.56 We then searched for individual-specific ASD-related variants using a large number of controls (n=757), both age matched and adults. We considered that including many controls with a wide age range would provide more power to select ASD-related and significant changes without biased comparisons. Although blood DNA methylation has been shown to vary with aging, the changes are of small magnitude (10% reduction on average) and affecting a small amount of the 450k array probes (2.1–15%).26, 57
A total of 700 DMCpGs in 467 genes were detected specifically in the ASD cohort, 13 per patient on average. Interestingly, 83.85% of these DMCpGs showed relative hypomethylation, in agreement with previous reports showing global hypomethylation in blood of ASD patients,58 although all clustered regions showed hypermethylation in ASD cases. The integrative study with blood transcriptome allowed the identification of cis-acting gene expression effects for 7.6% of the DMCpGs, more commonly overexpression associated to hypomethylation. In support of their potential pathogenic implications, 4.5% of the DMCpGs were also found in ASD patients’ brain,21 whereas 6.5% of the genes were ASD-related genes reported by mutation, copy number variant and/or association analyses. Most of the isolated DMCpGs identified were due to meSNVs, and were inherited in all tested cases. The high proportion of meSNVs among DMCpGs was due, at least in part, to the stringent filters applied to focus on the most severe methylation changes (50% or more), expected with mutations at CpG.30 Anyhow, heterozygous SNPs at CpGs are clearly implicated in the allele-specific methylation events, being an important class of cis-regulatory variants connecting genetics to epigenetics.59 The expression changes related to meSNVs can be due to the interference with trans-acting regulatory elements of either the methylation alteration itself, the underlying sequence variant or both factors together.
We identified and validated five DMRs in four ASD patients, with cis-acting expression effects on neighboring genes documented in four regions. Interestingly, all DMRs were inherited events also found in one of the parents. This germline transmission of epimutations could be explained by reprogramming evasion mechanisms60 or by undetected cis-acting genetic or genomic mutations affecting methylation of the region.61 The hypermethylation of the SMG7-antisense and SMG7 promoter region was paradoxically associated with SMG7 overexpression in a single patient (Supplementary Figure 6). SMG7 is involved in nonsense-mediated mRNA decay regulation, a pathway that has also been previously implicated in ASD and related syndromes.62 Other DMRs in single-ASD patients affected the regulation of genes involved in the development and function of the nervous system (PHACTR1,63 PIK3CD64 and PKD165). Methylation deregulation of PIK3CD has been shown in ASD brain,21 and the gene was associated to schizophrenia.66 Similarly, PKD1 expression was found downregulated in ASD67 and also associated to neuropsychiatric diseases, including ASD.68
Through an extended case/control association study, we identified six rare meSNVs significantly associated with ASD. The most significant association was detected with a CpG located close to an enhancer and DNAse hypersensitive site between the GALNT5 and ERMN genes (chr2q24.1). Although GALNT5 is not expressed in blood, ERMN was found overexpressed in one patient presenting the rare meSNV. Moreover, by targeted resequencing, we found that ERMN showed a higher load of rare damaging mutations in ASD patients. ERMN encodes an oligodendroglia-specific cytoskeletal protein (Ermin) involved in myelination, upregulated during active myelination periods in rat axons.69 The moesin/actin-binding domain has been shown to be essential for the ability of Ermin to promote arborization and morphological changes in cultured COS-7 cells.70 Correct myelination is crucial in ASD and mood disorders.71, 72, 73 Interestingly, ERMN is located within the boundaries of the AUTS5 locus, which has repeatedly shown linkage to autism,74 language impairment75 and IQ.76 Moreover, deletions including ERMN have been reported in patients with developmental delay and impaired communication,77 and ERMN expression is lower in epileptic patients brains suggesting also a role in the epileptogenic process.70 In fact, gain of function through overexpression and loss of function through deleterious mutations would suggest delicate gene dosage equilibrium.
The other five rare meSNVs significantly associated are also strong ASD candidates affecting relevant genes. (1) USP24, encodes a deubiquitinating enzyme that has been associated to Parkinson’s physiopathology.78 (2) PDE10A, a paternally imprinted gene79 highly expressed in specific brain regions, encodes a cyclic nucleotide phosphodiesterase that regulates signal transduction; the gene was found disrupted by a chromosomal translocation in a patient with neurodevelopmental disorder and was proposed as a therapeutic target for cognitive impairment in schizophrenia.80 (3) METTL21C, encoding a lysine methyltransferase for non-histone proteins, regulates molecular chaperones related to neuroinflamation, such as the Hsp70 found altered in the plasma of some ASD patients.81 (4) The meSNV on TRM13 associated with overexpression of the nearby DBT, coding for the transacylase E2 subunit of the branched-chain alpha-keto-acid dehydrogenase complex mutated in patients with autosomal recessive urine disease82 and ASD with epilepsy phenotype.83 (5) A meSNV showing nominal association was associated with overexpression of STX16, which is involved in synaptic vesicle transport, a pathway known to be impaired in ASD.84
The remaining rare DMCpGs/meSNVs found in single or few ASD patients need additional studies in larger data sets to confirm a potential association with the phenotype. Some of them have been related to ASD by point mutations (C15orf62/DNAJC17 and ADAP1), linkage studies (TLE3), aberrant brain expression (PRCD, LCP1, RXRB and SETD1A)67 and copy number variants (n=11 genes). Other genes were involved in relevant functions as neuron differentiation (SERPINF1, TCF12 and KAL1), neuron signaling (C1QL3) and ubiquitinization (DNAJC17 and RAB40B). Pathways enrichment analysis showed significant results in mechanisms previously related to ASD including axonogenesis,71 cell adhesion,85 immune chemokine signaling86 and PIP signaling.87
In summary, this epigenome-wide association study integrated with transcriptome in blood samples revealed significant association of ASD with hypomethylation caused by rare meSNVs at several loci, as well as a few clustered epimutations in single-ASD patients. Thus, methylation alterations detectable in blood DNA, due to either genetic or epigenetic defects, can contribute to ASD susceptibility. Given that most probands harbor more than one methylation variant and all of them are inherited from unaffected parents, their contribution to the phenotype is more likely to be additive, in an oligogenic manner. The study also reveals that ERMN is a strong ASD susceptibility gene that can be altered by both rare meSNV and mutations. Additional studies of genome-wide DNA methylation in larger ASD cohorts, integrated with genomic, transcriptomic, proteomic as well as the phenotypic data, are needed to further unravel the complex etiopathogeny of this multifactorial disorder.
Accession codes
References
Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention (CDC). Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ 2014; 63: 1–21.
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A . The familial risk of autism. JAMA 2014; 311: 1770–1777.
Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995; 25: 63–77.
Devlin B, Scherer SW, Emanuel B, Warren S . Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev 2012; 22: 229–237.
O’Roak BJ, State MW . Autism genetics: strategies, challenges, and opportunities. Autism Res 2008; 1: 4–17.
Veenstra-VanderWeele J, Cook EH Jr . Molecular genetics of autism spectrum disorder. Mol Psychiatry 2004; 9: 819–832.
Persico AM, Bourgeron T . Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 2006; 29: 349–358.
Jones JR, Skinner C, Friez MJ, Schwartz CE, Stevenson RE . Hypothesis: dysregulation of methylation of brain-expressed genes on the X chromosome and autism spectrum disorders. Am J Med Genet A 2008; 146A: 2213–2220.
Schumacher A, Petronis A . Epigenetics of complex diseases: from general theory to laboratory experiments. Curr Top Microbiol Immunol 2006; 310: 81–115.
Petronis A . Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 2010; 465: 721–727.
Belmonte MK, Bourgeron T . Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci 2006; 9: 1221–1225.
Nagarajan RP, Patzel KA, Martin M, Yasui DH, Swanberg SE, Hertz-Picciotto I et al. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res 2008; 1: 169–178.
Schanen NC . Epigenetics of autism spectrum disorders. Hum Mol Genet 2006; 15 Spec No 2: R138–R150.
Cook EH Jr., Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C et al. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 1997; 60: 928–934.
Jiang Y-HH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet A 2004; 131: 1–10.
Gregory SG, Connelly JJ, Towers AJ, Johnson J, Biscocho D, Markunas CA et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med 2009; 7: 62.
Nguyen A, Rauch Ta, Pfeifer GP, Hu VW . Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J 2010; 24: 3036–3051.
James SJ, Shpyleva S, Melnyk S, Pavliv O, Pogribny IP . Complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry 2013; 3: e232.
Wong CCY, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry 2013; 19: 495–503.
Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP . Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry 2013; 19: 862–871.
Nardone S, Sharan Sams D, Reuveni E, Getselter D, Oron O, Karpuj M et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry 2014; 4: e433.
Berko ER, Suzuki M, Beren F, Lemetre C, Alaimo CM, Calder RB et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet 2014; 10: e1004402.
Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H et al. A DNA methylation fingerprint of 1628 human samples. Genome Res 2012; 22: 407–419.
Wang Y, Fang Y, Zhang F, Xu M, Zhang J, Yan J et al. Hypermethylation of the enolase gene (ENO2) in autism. Eur J Pediatr 2014; 173: 1233–1244.
Salas LA, Bustamante M, Gonzalez JR, Gracia-Lavedan E, Moreno V, Kogevinas M et al. DNA methylation levels and long-term trihalomethane exposure in drinking water: an epigenome-wide association study. Epigenetics 2015; 10: 650–661.
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA 2012; 109: 10522–10527.
Mancuso FM, Montfort M, Carreras A, Alibés A, Roma G . HumMeth27QCReport: an R package for quality control and primary analysis of Illumina Infinium methylation data. BMC Res Notes 2011; 4: 546.
Du P, Kibbe Wa, Lin SM . lumi: a pipeline for processing Illumina microarray. Bioinformatics 2008; 24: 1547–1548.
Smyth GK . Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3: 1–25.
Chen Y, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8: 203–209.
Bell JT, Pai AA, Pickrell JK, Gaffney DJ, Degner JF, Gilad Y et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 2011; 12: R10.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ . Comb-p: Software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 2012; 28: 2986–2988.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014; 30: 1363–1369.
Jiang M, Zhang Y, Fei J, Chang X, Fan W, Qian X et al. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab Invest 2010; 90: 282–290.
Storey J qvalue: Q-value estimation for false discovery rate control 2015; Available at http://qvalue.princeton.edu/, http://github.com/jdstorey/qvalue.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL . TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013; 14: R36.
Langmead B, Trapnell C, Pop M, Salzberg SL . Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10: R25.
Anders S, Pyl PT, Huber W . A Python framework to work with high-throughput sequencing data. bioRxiv Serv 2014; 31: 166–169.
Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD . The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012; 28: 882–883.
Portela A, Esteller M . Epigenetic modifications and human disease. Nat Biotechnol 2010; 28: 1057–1068.
Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 2012; 8: e1002629.
Low D, Mizoguchi A, Mizoguchi E . DNA methylation in inflammatory bowel disease and beyond. World J Gastroenterol 2013; 19: 5238–5249.
Cheadle C, Vawter MP, Freed WJ, Becker KG . Analysis of microarray data using Z score transformation. J Mol Diagn 2003; 5: 73–81.
Informatics B. FastQC. 2010; Available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Informatics B. Trim Galore 2012; Available at http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
Wang K, Li M, Hakonarson H . ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
González-Pérez A, López-Bigas N . Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 2011; 88: 440–449.
Exome Aggregation Consortium (ExAC), Cambridge MA, USA. Available at http://exac.broadinstitute.org (accessed on 1 January 2015).
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery Genome Biol 2003; 4: P3.
Kamburov A, Wierling C, Lehrach H, Herwig R . ConsensusPathDB—a database for integrating human functional interaction networks. Nucleic Acids Res 2009; 37: D623–D628.
O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012; 485: 246–250.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–215.
Loke YJ, Hannan AJ, Craig JM . The role of epigenetic change in autism spectrum disorders. Front Neurol 2015; 6: 107.
Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR . Brain transcriptional and epigenetic associations with autism. PLoS ONE 2012; 7: e44736.
Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49: 359–367.
Melnyk S, Fuchs GJ, Schulz E, Lopez M, Kahler SG, Fussell JJ et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J Autism Dev Disord 2012; 42: 367–377.
Shoemaker R, Deng J, Wang W, Zhang K . Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res 2010; 20: 883–889.
Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet 2010; 42: 1093–1100.
Tycko B . Allele-specific DNA methylation: beyond imprinting. Hum Mol Genet 2010; 19: R210–R220.
Jolly LA, Homan CC, Jacob R, Barry S, Gecz J . The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet 2013; 22: 4673–4687.
Allen PB, Greenfield AT, Svenningsson P, Haspeslagh DC, Greengard P . Phactrs 1-4: A family of protein phosphatase 1 and actin regulatory proteins. Proc Natl Acad Sci USA 2004; 101: 7187–7192.
Eickholt BJ, Ahmed AI, Davies M, Papakonstanti EA, Pearce W, Starkey ML et al. Control of axonal growth and regeneration of sensory neurons by the p110delta PI 3-kinase. PLoS ONE 2007; 2: e869.
Harris PC, Torres VE . Polycystic kidney disease. Annu Rev Med 2009; 60: 321–337.
Law AJ, Wang Y, Sei Y, O’Donnell P, Piantadosi P, Papaleo F et al. Neuregulin 1-ErbB4-PI3K signaling in schizophrenia and phosphoinositide 3-kinase-p110δ inhibition as a potential therapeutic strategy. Proc Natl Acad Sci USA 2012; 109: 12165–12170.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011; 474: 380–384.
Daniels RJ . Sequence, structure and pathology of the fully annotated terminal 2 Mb of the short arm of human chromosome 16. Hum Mol Genet 2001; 10: 339–352.
Brockschnieder D, Sabanay H, Riethmacher D, Peles E . Ermin, a myelinating oligodendrocyte-specific protein that regulates cell morphology. J Neurosci 2006; 26: 757–762.
Wang T, Jia L, Lv B, Liu B, Wang W, Wang F et al. Human Ermin (hErmin), a new oligodendrocyte-specific cytoskeletal protein related to epileptic seizure. Brain Res 2011; 1367: 77–84.
Zikopoulos B, Barbas H . Changes in prefrontal axons may disrupt the network in autism. J Neurosci 2010; 30: 14595–14609.
Edgar N, Sibille E . A putative functional role for oligodendrocytes in mood regulation. Transl Psychiatry 2012; 2: e109.
Broek JA, Guest PC, Rahmoune H, Bahn S . Proteomic analysis of post mortem brain tissue from autism patients: evidence for opposite changes in prefrontal cortex and cerebellum in synaptic connectivity-related proteins. Mol Autism 2014; 5: 41.
Maestrini E, Pagnamenta AT, Lamb JA, Bacchelli E, Sykes NH, Sousa I et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol Psychiatry 2010; 15: 954–968.
Bartlett CW, Flax JF, Logue MW, Smith BJ, Vieland VJ, Tallal P et al. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum Hered 2004; 57: 10–20.
Posthuma D, Luciano M, Geus EJC, de, Wright MJ, Slagboom PE, Montgomery GW et al. A genomewide scan for intelligence identifies quantitative trait loci on 2q and 6p. Am J Hum Genet 2005; 77: 318–326.
Newbury DF, Warburton PC, Wilson N, Bacchelli E, Carone S, Lamb JA et al. Mapping of partially overlapping de novo deletions across an autism susceptibility region (AUTS5) in two unrelated individuals affected by developmental delays with communication impairment. Am J Med Genet Part A 2009; 149A: 588–597.
Wang K, Liu S, Wang J, Wu Y, Cai F, Song W . Transcriptional regulation of human USP24 gene expression by NF-kappa B. J Neurochem 2014; 128: 818–828.
Wang X, Soloway PD, Clark AG . A survey for novel imprinted genes in the mouse placenta by mRNA-seq. Genetics 2011; 189: 109–122.
Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012; 149: 525–537.
El-Ansary A, Al-Ayadhi L . Neuroinflammation in autism spectrum disorders. J Neuroinflammation 2012; 9: 265.
MENKES JH, HURST PL, CRAIG JM . A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 1954; 14: 462–467.
Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science 2012; 338: 394–397.
Pescosolido MF, Gamsiz ED, Nagpal S, Morrow EM . Distribution of disease-associated copy number variants across distinct disorders of cognitive development. J Am Acad Child Adolesc Psychiatry 2013; 52: 414–430, e14.
Stewart LT . Cell adhesion proteins and the pathogenesis of autism spectrum disorders. J Neurophysiol 2014; 113: 1283–1286.
Onore C, Careaga M, Ashwood P . The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun 2012; 26: 383–392.
Chen J, Alberts I, Li X . Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int J Dev Neurosci 2014; 35: 35–41.
Acknowledgements
We thank ASD patients and their families for their support. We are also grateful to Armand Gutiérrez-Arumí and Marcos López-Sánchez for their help with the data acquisition and handling, Dr Francesco Mancuso for his help with R programming, and Dr Alejandro Martín from the IDIBELL for his contribution to the pyrosequencing analyses. The funding source for the study was received from the Spanish Ministry of Economy and Competitivity (FIS PI1002512, PI1302481 and PI1300823 co-financed by FEDER), Fundación Alicia Koplowitz, Generalitat de Catalunya (2014SGR1468), and Fundación Ramón Areces. AH had a predoctoral fellowship of Ministry of Education, Culture and Sport (FPU AP2009-4795).
DATA DEPOSITION
The complete methylation data have been deposited in NCBI’s Gene Expression Omnibus under accession number GSE83424.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
BR-S and LAP-J are currently employee and scientific advisor, respectively, of qGenomics SL. The remaining authors declare no conflict of interest.
Additional information
Data of this study were published in two poster contributions: A. Homs, I. Cusco, L. A. Pérez-Jurado. Epigenetic and related transcriptional alterations affecting chromatin remodeling and synaptic genes in Autism Spectrum disorders. International Meeting for Autism Research (IMFAR); 2–5 May 2013; San Sebastián, Spain; A. Homs, I. Cusco, B. Rodríguez-Santiago, C. M. Villanueva and L. A. Pérez-Jurado. Genomic methylation profile in peripheral blood DNA of autism spectrum disorders: a case–control study. 12th International Congress on Human Genetics; 11–15 October 2011; Montreal, Canada.
Supplementary Information accompanies the paper on the Translational Psychiatry website
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Homs, A., Codina-Solà, M., Rodríguez-Santiago, B. et al. Genetic and epigenetic methylation defects and implication of the ERMN gene in autism spectrum disorders. Transl Psychiatry 6, e855 (2016). https://doi.org/10.1038/tp.2016.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2016.120
- Springer Nature Limited
This article is cited by
-
Association of Relative Telomere Length and LINE-1 Methylation with Autism but not with Severity
Journal of Autism and Developmental Disorders (2024)
-
Identification and validation of cuproptosis related genes and signature markers in bronchopulmonary dysplasia disease using bioinformatics analysis and machine learning
BMC Medical Informatics and Decision Making (2023)
-
Hair Trace Elements and Mineral Content in Moroccan Children with Autism Spectrum Disorder: a Case–Control Study
Biological Trace Element Research (2023)
-
De novo mutations within metabolism networks of amino acid/protein/energy in Chinese autistic children with intellectual disability
Human Genomics (2022)
-
DNA methylation signatures in autism spectrum disorders
The Nucleus (2021)