
Abstract
Lysosomes are acidic organelles involved in crucial intracellular functions, including the degradation of organelles and protein, membrane repair, phagocytosis, endocytosis, and nutrient sensing. Given these key roles of lysosomes, maintaining their homeostasis is essential for cell viability. Thus, to preserve lysosome integrity and functionality, cells have developed a complex intracellular system, called lysosome quality control (LQC). Several stressors may affect the integrity of lysosomes, causing Lysosomal membrane permeabilization (LMP), in which membrane rupture results in the leakage of luminal hydrolase enzymes into the cytosol. After sensing the damage, LQC either activates lysosome repair, or induces the degradation of the ruptured lysosomes through autophagy. In addition, LQC stimulates the de novo biogenesis of functional lysosomes and lysosome exocytosis. Alterations in LQC give rise to deleterious consequences for cellular homeostasis. Specifically, the persistence of impaired lysosomes or the malfunctioning of lysosomal processes leads to cellular toxicity and death, thereby contributing to the pathogenesis of different disorders, including neurodegenerative diseases (NDs). Recently, several pieces of evidence have underlined the importance of the role of lysosomes in NDs. In this review, we describe the elements of the LQC system, how they cooperate to maintain lysosome homeostasis, and their implication in the pathogenesis of different NDs.
Graphical Abstract

Similar content being viewed by others


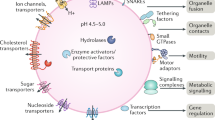
Lysosomal functions
Lysosomes are membrane-enclosed acidic organelles found in all eukaryotic cells, discovered by De Duve in the middle of the last century [1, 2]. Lysosomes contain a wide range of hydrolases capable of degrading all macromolecules present in cells: nucleic acids, lipids, carbohydrates, proteins, and cell debris. For decades lysosomes have been considered the “trash bin” of cells, without any other specific role [3]. In the last 10 years, however, research on lysosomal function has intensively increased and nowadays these organelles are considered an important hub for cell metabolism and nutrient sensing [4, 5]. Lysosomal functions are involved in: endocytosis, phagocytosis, autophagy [6], lysosome exocytosis and plasma membrane repair [7, 8], control of nutrient sensing [9, 10], and cell death processes [11]. This variety of functions implicates lysosomes in many human diseases. Notably, defects in genes coding for lysosomal enzymes are causative factors in a group of more than 50 inherited metabolic disorders. These disorders, named lysosomal storage disorders (LSDs), are characterized by the lysosomal accumulation of undigested substrates, and include Gaucher disease, Fabry disease, and Neimann–Pick disease [12,13,14]. Lysosomal dysfunctions have been also found to play important roles in cancer [15, 16] and neurodegenerative diseases (NDs) [17,18,19,20,21,22,23,24]. In most cases, these diseases show adulthood onset and a progressive decline. In addition, the decrease in lysosomal function observed during ageing may contribute to disease pathogenesis [25].
Lysosomal membrane composition
Extracellular material, intracellular molecules, and organelles are driven to lysosomes for degradation. Lysosomal catabolic functions require that these components are transported and delivered to the lumen of the organelle, where the acidic hydrolytic enzymes degrade the substrates. The integrity of the limiting membrane is crucial for the proper functionality of lysosomes, and this is ensured by a thick membrane (around 8 nm) composed of lipids and glycoproteins with a luminal glycosylated domain [26]. This lysosomal glycocalyx has a protective role in the acidic environment, and it is fundamental for the functionality of the lysosomal membrane proteins [i.e., lysosomal integral membrane proteins (LIMPs) and lysosomal associated membrane proteins (LAMPs)]. Alongside soluble lysosomal hydrolases, lysosomal membrane proteins play a pivotal role in organelle biogenesis and functionality [27, 28]. Among these, LAMP-1, LAMP-2, LIMP-1/CD63 and LIMP-2/SCARB2 are the most abundant, with the former two representing more than 50% of total lysosomal membrane proteins [29]. Since their discovery, LAMP-1 and LAMP-2 have been considered structural molecules committed to ensuring lysosome integrity by protecting the membrane from the acidic luminal compartment [30]. Recently, it has been demonstrated that these proteins perform functions beyond this preservation and that, despite their 37% sequence homology, they show important functional differences from one another. Experiments performed in mice show that the inactivation of the Lamp1 gene does not alter lysosomal morphology and function [31], while in Lamp2-deficient mice, increased cell mortality correlating with the accumulation of autophagic vacuoles (AVs) occurred in several tissues [32]. Lamp2-deficient mice model the symptoms of Danon disease in humans, an LSD caused by Lamp2 mutations characterized by abnormal accumulation of AVs in heart and in skeletal muscle [33, 34].
Another factor that contributes to the stability of the lysosomal membrane is lipid composition. Lysosomal membranes are enriched in sphingomyelin and are characterized by the presence of bis(monoacylglycerol)phosphate, a negatively charged lipid exclusively present in lysosomes [35,36,37]. Of note, cholesterol is an essential heterogeneously distributed membrane component, mainly present in the plasma membrane [38]; however, the lysosome represents a unique organelle in terms of cholesterol content, since its membrane cholesterol composition is intermediate between that of the plasma membrane and that of other intracellular organelles [39]. Indeed, lysosomes play a key role in maintaining cholesterol homeostasis and in cholesterol dynamics [39,40,41]. Thus, alterations in the cholesterol content affect the integrity and stability of the lysosomal membrane and a reduction in its levels induces lysosomal membrane permeabilization (LMP; see below for a detailed description) [42]. Conversely, the addition of cholesterol to isolated lysosomes or cell cultures increases lysosomal membrane stabilization [43,44,45].
Lysosomal quality control
Given the central role of lysosomes for cellular homeostasis, any stress or alteration that affects lysosomal integrity can critically impact cell viability. To maintain lysosomal functionality, lysosomal damage is recognized and resolved by lysosomal quality control (LQC). The LQC consists of multiple pathways dedicated to lysosomal repair, clearance (lysophagy), exocytosis, and biogenesis. LQC is generally activated in response to LMP, an event characterized by membrane damage, lysosomal swelling, and the release of lysosomal luminal content into the cytosol with possible uncontrolled breakdown of biomolecules [46, 47]. In addition to the previously mentioned changes in cholesterol content in the lysosomal membrane, a variety of exogenous and endogenous factors can cause LMP, including lysosomotropic agents, compounds entrapped in the lysosomes after protonation (l-leucyl-l-leucine methyl ester hydrobromide, glycyl-l-phenylalanine 2-naphthylamide, chloroquine, and the cationic amphiphilic drugs), reactive oxygen species, the apoptotic regulator Bcl-2-like protein 4, and infectious pathogens [48, 49]. LMP may also occur in response to neurotoxic events in NDs (see Lysosomal Damage in Neurodegeneration section for details) [50,51,52,53,54,55,56].
Lysosomal damage recognition and repair
To cope with lysosomal damage, cells have sensor proteins capable of recognizing lysosomal damage and activating intracellular responses (Fig. 1). These proteins are the galectins, a group of 15 proteins characterized by the presence of a common carbohydrate recognition domain, a beta-sandwich domain composed of 130–140 residues with high affinity for carbohydrates [57].

Lysosome repair. Lysosomal membrane permeabilization (LMP) and damage is recognized and repaired by mechanisms such as the PITT, the ESCRT pathways, and the sphingomyelin scrambling and turnover. (i) The PITT mechanisms consist of protein complexes that promote lysosome lipid membrane turnover by interacting with endoplasmic reticulum (ER). (ii) The ESCRT machinery is recruited by galectins and restores lysosome membrane integrity. (iii) The remodeling of damaged lysosomal membrane can be directed by sphingomyelin scrambling and turnover. Created with BioRender.com
Galectins are small, soluble, and dynamic cytosolic proteins, that can shuttle into the nucleus or be secreted into the extracellular environment by unconventional secretion processes [58, 59]. Multiple pathways seem to be implicated in galectin secretion; evidence suggests it can be mediated by direct release, by lysosome/endosome exocytosis, or by extracellular vesicle release [59]. In the extracellular space, secreted galectins may bind to specific beta-galactosides forming a cross-linked complex, a dynamic lattice [60], and play important roles in cell adhesion, cell migration, signaling, immune response, inflammation, and endocytosis [61,62,63,64]. At the intracellular level, galectins reside in the cytosol, but, when necessary, they can recognize and bind to damaged endosomal membranes [65, 66], with galectins-3, 8, and 9 being mostly involved. In normal conditions, glyco-conjugates are present in the lumen of lysosomes and endosomes, but upon membrane disruptions, they are exposed to the cytosol and may be recognized by galectins. Indeed, the accumulation of galectins in discrete cytosolic puncta is a hallmark of LMP [67]. In the presence of a small rupture in the lysosomal membrane, galectin-3 translocates to the lysosomes and recruits the programmed cell death 6 interacting protein (PDCD6IP/ALIX), tumor susceptibility 101 (TSG101), the Endosomal sorting complex required for transport III (ESCRT-III) component charged multivesicular body protein 4B (CHMP4B), and VPS4 essential proteins for lysosomal membrane repair [66, 68,69,70]. This process is followed by lysosomal calcium efflux and triggers the RAB29-mediated translocation of the leucine-rich repeat kinase 2 (LRRK2) to the damaged lysosomes. LRRK2 phosphorylates and engages RAB8A facilitating the recruitment of ESCRT components for membrane repair [17, 71, 72]. Concurrently, LRRK2 also recruits and activates both RAB10 and its protein interactor C-Jun-amino-terminal kinase-Interacting Protein 4 (JIP-4) promoting the lysosomal tubulation sorting, a process driven by LRRK2 and necessary for the release of vesicles from lysosomes [73, 74].
Evidence showing that ESCRT depletion was not fully capable counteracting lysosomal repair suggests that this is not the only lysosomal repair mechanism [68, 70].
Recently, two ESCRT-independent pathways for lysosomal membrane repair have been discovered.
Tan and Finkel identified phosphoinositide-initiated membrane tethering and lipid transport (PITT), a specific set of proteins involved in LMP beyond the known ESCRT components including the membrane-bound phosphatidyl-inositol-4 kinase type 2 alpha (PI4K2A) [75, 76]. This enzyme catalyzes the phosphorylation of phosphatidyl-inositol (PI) to phosphatidyl-inositol 4-phosphate (PI4P), a lipid essential for the endolysosome system, as well as its binding proteins, the oxysterol binding protein (OSBP) related 9, 10, and 11 (ORP9, ORP10 and ORP11). In this alternative repair pathway, LMP induces the activity of PI4K2A leading to the production of PI4P and the lysosomal accumulation of ORP9, ORP10, and ORP11; these are subsequently involved in the formation of inter-organelle membrane contact sites (MCS). ORP proteins dimerize with each other and establish endoplasmic reticulum (ER)-lysosome MCS via the interaction of the ER-resident VAMP-associated proteins A and B, favoring the exchange of PI4P with phosphatidyl-serine (PS) and transporting it to lysosomes. In a complementary way, OSBP transports cholesterol to damaged lysosomes for repair. The accumulation of PS in lysosomes stimulates the non-canonical activity of the autophagy-related 2 (ATG2) protein, involved in lipid transport [76]. The link between the ESCRT-pathway and PITT in lysosomal membrane repair remains to be elucidated.
The second ESCRT-independent mechanism for lysosomal repair has been uncovered by Niekamp and colleagues. The process is triggered by cytosolic exposure of sphingomyelin to the surface of damaged lysosomes catalyzed by the Ca2+-dependent scramblase. This is followed by the cleavage of sphingomyelin by neutral sphingomyelinase to produce ceramides facilitating membrane repair [77]. These repair pathways act in parallel to ensure lysosome integrity.
Interestingly, it has been shown that lysosomal damage inactivates mTOR, which normally functions to negatively regulate autophagy and catabolic pathways. This is mediated through a galectin-based system named GALTOR, suggesting a link between lysosomal damage and the regulation of cellular metabolism [78].The system is based on the interaction between galectin-8, the lysosomal aminoacidic transporter solute carrier family 38 member 9,and the ragulator–Rag complex. Concurrently, galectin-9 activates AMP-activated protein kinase (AMPK) increasing its phosphorylating activity via association with the AMPK upstream kinase mitogen-activated protein kinase 7 (MAP3K7/TAK1). The interaction between galectin-9 and the deubiquitinase ubiquitin-specific peptidase 9 X linked (USP9X) governs the lysine 63 ubiquitination (K63) rate of MAP3K7/TAK1, a process that regulates and activates the enzyme. In physiological conditions, USP9X negatively regulates MAP3K7/TAK1 activity by deubiquitination; instead, under LMP conditions, galectin-9 interferes with USP9X promoting MAP3K7/TAK1 ubiquitination and activation [66, 79].
Lysophagy
When the lysosomal membrane cannot be repaired, a complex mechanism is activated to promote the clearance of the whole organelle via selective autophagy, a process known as lysophagy (Fig. 2). Lysophagy is activated when repair mechanisms fail. To date, the exact mechanism responsible for the switch from lysosomal repair to clearance has not been characterized. Indeed, the degradation of damaged lysosomes is mainly triggered by the recruitment of galectins; galectins sense the LMP, bind exposed glycans, and recruit enzymes involved in lysosome ubiquitination (E2, E3 enzymes); these processes mainly involve galectin-3 and galectin-8. Galectin-3 recruits and binds the tripartite motif-containing 16 (TRIM16), an atypical E3 ubiquitin ligase that contributes to lysosome ubiquitination and serves as a platform to recruit autophagy-related proteins such as ULK1, ATG16L, and BECN-1 [52, 80]. These proteins are all part of complexes that together regulate autophagy initiation by mediating the formation of the phagophore, a double-membraned structure, and the activation of microtubule-associated protein 1 light chain 3 (MAP1LC3, or simply LC3). Thus, TRIM16 mediates the interaction between lysosomes and the forming phagophore, facilitating the engulfment of the damaged lysosomes. Moreover, TRIM16 regulates the activation of the transcription factor EB (TFEB), the master regulator of autophagy and lysosome biogenesis (see below for a detailed description) [80]. In parallel to galectin-3, galectin-8 directly binds a specific autophagy receptor (AR), the calcium-binding and coiled-coil domain 2 protein (CALCOCO2, also known as NDP52). NDP52 recruits the forming phagophore and interacts with the ragulator–Rag complex inhibiting mTOR and therefore activating TFEB [66]. Hence, TFEB activation induces the transcription of autophagic genes responsible for lysophagy and lysosomal biogenesis (see below for a detailed description of TFEB activity).

Lysophagy. Lysosome clearance through autophagy is activated when the repair mechanisms fail or the damage persists. Lysosome clearance can occur by marking the damaged lysosomes and recruiting them to the forming phagophore via various galectin-dependent or independent mechanisms. Created with BioRender.com
Ubiquitination of damaged lysosomes is also regulated through galectin-independent mechanisms by E2 and E3 enzymes, such as the SKP1/CUL1/F-box (SCF) protein ubiquitin ligase complex, Cullin-4A—DNA damage-binding protein—WD repeat and FYVE domain-containing 1 complex, and UBE2QL1. Ubiquitination can occur on K63- or K48- linked ubiquitin chains. ARs recognize K63-linked ubiquitin conjugates, while K48-linked chains generally are associated with proteasome degradation. K63- and K48-ubiquitinations on lysosomal membrane proteins occur with a different timing and play different roles. K63-ubiquitination arises quickly after the damage, together with the recruitment of the AR sequestosome-1 (SQSTM1/p62). Different ligases can mediate K63 ubiquitination: F-box protein 27, the substrate recognition subunit of the SCF complex, directly binds to glycans and damaged lysosome membranes promoting LAMP-1 and LAMP-2 K63 ubiquitination; ITCH ubiquitylates membrane-associated proteins to initiate lysophagy. ITCH is recruited and activated by SPART/SPG20, a galectin/LMP-independent detector of lipid-packing defects on the lysosome membrane. SPART/SPG20 binds to IST1, a repair factor, and senses membrane defects that precede LMP. When lipid membrane alterations are unacceptable, SPART/SPG20 recruits ITCH initiating autophagy [81, 82].
K48-ubiquitination occurs later on and is mediated by E2 or E3 enzymes such as UBE2QL1 or Cullin-4A [83,84,85]. The K48 conjugates targeted by UBE2QL1 are recognized by the heat shock protein B1 (HSPB1), which favors their segregation by valosin-containing protein (VCP) and their clearance by the proteasome [86, 87]. The first set of proteins targeted by ubiquitination is ARs, which promote phagophore engulfment [81]. Subsequently, the second set of ubiquitinated proteins is membrane trafficking regulators, such as soluble n-ethylmaleimide-sensitive factor attachment protein receptors, which suppress the fusion of damaged lysosomes with autophagosomes or late endosomes. Finally, the third set of proteins targeted by ubiquitination are proteins that orchestrate the cytoskeleton in lysophagy dynamics, such as cellular communication network factor 2. The clearance of this last set of proteins is regulated by VCP and is necessary for lysosomal degradation [86].
Damaged lysosomes, marked with ubiquitin-chains, are linked to autophagic membranes by ARs [e.g.: SQSTM1/p62, optineurin (OPTN), NDP52, NBR1 autophagy cargo receptor (NBR1), Tax1 binding protein 1]. They possess an ubiquitin-associated domain, which recognizes ubiquitin chains, and an LC3-interacting region, which directly binds LC3 present on the forming phagophore. AR activation is regulated by TANK-binding kinase 1 (TBK1), which by phosphorylating them, increases their affinity to ubiquitin chains [88, 89]. SQSTM1/p62 is the major actor in lysophagy; in fact, it is consistently found on damaged lysosomes and its depletion prevents lysosome clearance [81, 83, 90]. The recruitment of SQSTM1/p62 upon lysosomal damage is regulated by HSPB1. During phagophore formation, HSPB1 is recruited to lysosomes and is phosphorylated to allow its inclusion in the SQSTM1/p62 condensates (also known as p62 bodies) formed by liquid–liquid phase separation (LLPS), favoring the maintenance of its liquid–liquid phase properties, and thus promoting lysophagy [90].
Besides the canonical pathway just described, lysosomal damage also induces non-canonical lysophagy. In this process, the ATG12/ATG5/ATG16 complex is recruited to the lysosomal membrane through a V-ATPase-mediated process. Consequently, activated LC3 is directly attached to the lysosomal membrane by non-canonical autophagy and conjugation of ATG8s to single membranes (CASM). This is likely followed by (i) recruitment of the lipid transfer protein ATG2, which is involved in PITT-dependent lysosome repair, and (ii) the fusion of LC3-labeled vesicles with other intact lysosomes [91,92,93].
Lysosomal biogenesis and exocytosis
Lysosomal biogenesis and replacement are adaptive mechanisms that maintain the functional pool of lysosomes needed for cellular homeostasis (Fig. 3). These processes depend both on the endocytic pathway and on the biosynthesis of new lysosomal proteins; they further require the coordinated transcription of genes coding for lysosomal and autophagic proteins regulated by TFEB and by its cognate transcription factor E3 (TFE3). These two proteins belong to the microphthalmia (MiT/TFE) family of basic helix-loop-helix-leucin zipper transcription factors, a class of evolutionarily conserved and structurally related proteins. This family includes four members: the microphthalmia transcription factor, TFEB, TFE3, and the transcription factor EC. TFEB and TFE3 recognize a 10 bp palindromic responsive element (GTCACGTGAC), termed the coordinated lysosomal expression and regulation, present in genes controlling the integrated expression of networks regulating autophagy and lysosomal biogenesis, exocytosis [94,95,96,97].

Lysosome biogenesis and exocytosis. To maintain the pool of functional lysosomes the damage of lysosomes activates galectin-dependent mechanisms that induce the transcription factors TFEB and TFE3. Exocytosis of damaged lysosomes with a Ca2+-dependent process is induced as an alternative mechanism to lysophagy. Created with BioRender.com
TFEB and TFE3 continuously shuttle between cytosol and nucleus, and different stimuli can modify the dynamics of this mechanism. When sufficient nutrients are available, the mTOR complex 1 (mTORC1) inhibits TFEB/TFE3 activity through their phosphorylation (at Ser138, Ser142, or Ser211 for TFEB; at Ser321 for TFE3) [98]. Conversely, starvation or cellular stress conditions promote TFEB activation and nuclear translocation with two parallel mechanisms: by switching off mTORC1 activity and by inducing lysosomal calcium efflux via the mucolipin TRP cation channel 1 (MCOLN1), an event that triggers the activation of the calcium-dependent serine/threonine phosphatase calcineurin that dephosphorylates TFEB/TFE3, thus activating them. [94, 99]. The mitogen-activated protein kinase 1 (MAPK1/ERK2) and the protein kinase C type beta are also involved in TFEB phosphorylation and regulation [100].
How TFEB/TFE3 phosphorylation inhibits their function has been elucidated. Specific phosphorylated serine residues allow both the recognition and the binding of the chaperone YWHA/14-3-3 that retains TFEB/TFE3 in the cytosol [101,102,103,104] and mediates their nuclear export via exportin1 (known as chromosomal maintenance 1) [105, 106], thus preventing their nuclear localization.
Beyond phosphorylation, other post-translational modifications control TFEB/TFE3 activity, including acetylation, a process regulated by histone deacetylases (HDAC2, HDAC5, HDAC6, HDAC9) and acetyltransferases (ACAT1, ELP3, CREBP). Treatment with pan-HDAC inhibitors, such as suberoylanilide hydroxamic acid or trichostatin, induces TFEB acetylation and accumulation into the nucleus, promoting lysosomal biogenesis and autophagy [107, 108]. Finally, TFEB/TFE3 are also involved in the redox signaling mediated by the KEAP1/NRF2 pathway, suggesting that lysosomal biogenesis might also be sensitive to the intracellular redox state [109].
In the nucleus, both TFEB and TFE3 exert their transcriptional activity through an LLPS-dependent mechanism involving the formation of physiological protein condensates, which also regulate their activity. It has been demonstrated that the inositol polyphosphate multikinase (IPMK) does not influence TFEB phosphorylation or nuclear translocation, but IPMK can associate with TFEB suppressing its LLPS. IPMK knockdown induces the formation of TFEB condensates promoting its transcriptional activity and leading to autophagy induction and lysosomal biogenesis [110, 111].
Lysosomal damage and LMP have been shown to induce TFEB activation and lysosomal biogenesis to replace the pool of damaged lysosomes cleared by lysophagy [112, 113]. Besides the role of galectin-8 described above, other proteins might be involved in the activation of TFEB following LMP. In kidney injury, it has been observed that lysosomal damage triggers the recruitment of LC3 by the activation of ATG conjugation system. LC3 interacts with MCOLN1 leading to calcium efflux, which induces TFEB nuclear translocation through the activation of calcineurin [114]. Interestingly, it has been recently observed that calcineurin is also indirectly regulated by galectin-3: galectin-3 recruits the SMAD specific E3 ubiquitin-protein ligase SMURF1 to damaged lysosomes, which in turn binds and controls calcineurin promoting its phosphatase activity on TFEB [115].
A peculiar activity of TFEB is its involvement in the regulation of lysosome exocytosis; this process requires that lysosomes fuse directly with the plasma membrane to release their content into the extracellular environment. Lysosomal exocytosis was shown to be involved in the restoration or remodeling of the plasma membrane [116,117,118,119], as well as in neurite outgrowth processes and axonal myelinization [120,121,122].
Lysosomal exocytosis is regulated by lysosomal calcium efflux through the MCOLN1 channel. Interestingly, TFEB overexpression stimulates the activation of MCOLN1 and calcium release from lysosomes mediating lysosomal exocytosis. TFEB and MCOLN1 act in a feedback loop, where TFEB triggers MCOLN1 gene transcription, being a TFEB target gene, while MCOLN1 stimulates TFEB activation via calcineurin, as described above [95].
Recent findings suggest lysosomal exocytosis as a mechanism for the secretion of protein aggregates from neurons, contributing to the maintenance of cellular proteostasis when intracellular degradative systems are impaired [123].
Lysosome reformation
Beside lysosomal biogenesis, cells can provide a new pool of lysosomes via reformation processes. Lysosomes can also originate through the recycling of the autolysosome membrane via a mechanism known as autophagic lysosome reformation (ALR). ALR involves the protrusion of tubules from autolysosomes, giving rise to small vesicles named proto-lysosomes, which subsequently mature into functional lysosomes [124]. The initiation of ALR is dependent on the reactivation of mTOR. The mechanisms governing mTOR reactivation and the subsequent initiation of ALR during autophagy are still mostly unclear. An important process implicated in this context is the increased production of amino acids, which activates mTOR [124]. Moreover, the release of calcium by lysosomes also plays a role in mTOR activation through a calmodulin-dependent mechanism [125].
The remodeling of autolysosome membranes during ALR is finely regulated by the transient and reversible formation of a specific set of membrane-bound phosphoinositides, following a precise spatiotemporal pattern. Notably, PtdIns P2 recruits clathrin to the autolysosome membrane, which, in turn, stimulates membrane budding [126]. Clathrin additionally serves as a membrane platform to facilitate the accumulation of AP-2-PtdIns P2, which, in turn, promotes the recruitment and clustering of the kinesin family member 5B (KIF5B) protein. KIF5B is a kinesin motor protein that binds to autolysosome membranes and microtubule filaments, thereby facilitating the formation of autolysosome membrane tubules. Finally, the mechanism that promotes the final scission of the lysosome is still unclear [127]. Interestingly, it has been recently demonstrated that, in the presence of severe LMP, the ALR machinery is recruited to damaged lysosomes by TBC1 domain family member 15 to regenerate functional lysosomal membranes. This mechanism represents a prompt cellular response to compensate for the reduction of functional lysosomes before the activation of lysosomal biogenesis mediated by TFEB/TFE3 [128].
Alternatively, lysosomes are also regenerated by the endocytic pathway. Transient “kiss and run” interactions between late endosomes and lysosomes occur to deliver the endocytosed cargoes into the lysosomes. These events result in the formation of endolysosomes, hybrid and heterogeneous organelles from which lysosomes are regenerated with an analogous pathway to that occurring in ALR: characterized by tubulation of endolysosomes, scission, and maturation [129,130,131,132,133].
Lysosomal damage in neurodegeneration
NDs are fatal progressive disorders characterized by the loss of functionality and/or the death of specific subpopulations of neurons controlling cognitive or motor functions. Different pathological mechanisms induce neuronal death, among which alteration in proteostasis is of great relevance. Proteostasis dysfunction coincides with the generation of damaged organelles involved in the protein quality control (PQC), variation in the expression of contributors to PQC, and the formation of protein aggregates which may ultimately lead to cell death through different mechanisms [134,135,136,137]. The most common NDs, depicted in Fig. 2, include Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Alzheimer’s disease (AD). Their classification is based on primary clinical features, anatomical distribution of neuronal degeneration, and the main molecular alterations that characterize each of them. Although these NDs differ significantly in etiopathogenesis and clinical aspects, they generally present certain common cellular and molecular alterations such as protein aggregation, impairment of degradative systems, and damage to degradation-related organelles such as lysosomes. Indeed, emerging evidence suggests that lysosomal dysfunction is strictly correlated with the pathogenesis of these diseases, either as a trigger or a consequence of neuronal or microglial cell dysfunction. LMP and leakage of lysosomal contents, including cathepsin B and calcium, have been observed in various NDs [138,139,140]. As shown in Table 1, lysosomal damage is associated with ND-related mutations in genes encoding proteins directly involved in lysosomal membrane integrity and lysosomal functionality. These include lysosomal transmembrane proteins such as transmembrane protein (TMEM) 106B and ATPase cation transporting 13A2 (ATP13A2) [141, 142]; or proteins implicated in lysosome repair or degradation, such as LRRK2 and VCP, SQSTM1/p62, OPTN, or TBK1 [143,144,145,146] (see below for further details). Of note, neurons can uptake extracellular aggregates through endocytosis; depending upon their nature and biophysical properties, these aggregates can induce lysosomal membrane rupture [55]. Proteins implicated in this mechanism can be found in Table 2. The lysosomal toxicity of protein aggregates has been directly demonstrated for various ND-related proteins, including alpha-synuclein (SNCA), β-amyloid (Aβ), tau, and superoxide dismutase 1 [83, 140, 147,148,149]. Together, these findings show a strong interaction between lysosomal alterations and NDs.
Parkinson’s disease
PD is the most common ND, affecting 1% of the population over 65 years old. The clinical manifestations of the disease can vary among individuals. The most recurrent symptoms and signs include bradykinesia, tremors, muscular rigidity, and speech and cognitive impairments [150]. The histopathological hallmark of PD is the presence of Lewy body (LBs) inclusions, which result from intracellular accumulation of SNCA. LBs are associated with the death of dopaminergic neurons present in the substantia nigra [151]. Other hallmarks of PD include a correlation of the disease with lysosome alterations, such as increased galectin-3 plasma levels, which has been proposed as a potential biomarker to monitor PD-related neurodegeneration [152]. Moreover, patients exhibit an overactivation of microglia that leads to an inflammatory response. The activation of microglia is also associated with lysosomal alterations mediated by galectin-3 [153, 154].
Only 10% of PD cases occur in familiar forms, while 90% are sporadic. To date, a large part of the genetic causes of PD has still to be identified. Approximately 5–10% of hereditary PD cases are associated with identified mutations in genes such as SNCA, LRRK2, and PRKN [155, 156]. Conversely, in most cases, PD etiology is multifactorial and involves an interplay between environmental and genetic factors. Genome-wide association studies have identified various risk genes and loci linked to PD [157, 158]; several have a strong link to lysosomes (as reviewed in [159]). In particular, LRRK2 encodes for a protein involved in lysosome repair thanks to its phosphorylation and interaction with RAB29, which also interacts directly with the a1 subunit of the vacuolar-type ATPase H+ pump that maintains lysosomal pH [72, 160, 161]. Other relevant genes are ATP13A2, encoding for a cation transporter that maintains the proper pH in lysosomes [162]; GBA, encoding for glucocerebrosidase, a lysosomal enzyme that converts glucosylceramide and is involved in lysosome activity [163]; TMEM175, encoding for a lysosome channel regulator of potassium in lysosomes [164]; and SCARB2, encoding for a structural transmembrane lysosomal protein, which acts as a regulator of cholesterol-membrane composition and a receptor of β-glucocerebrosidase, which in turn controls the clearance of SNCA [165,166,167]. This long list of genes supports the notion that alterations of lysosome function and dynamics may contribute to PD onset and disease progression. Additionally, other elements correlate PD and lysosome disruption, such as the close relationship between lysosomes misfunctioning and SNCA. This is evidenced by several facts: that lysosomes are essential for SNCA degradation [168] and their alterations or the accumulation of lysosomal substrates result in increased SNCA cytoplasmatic levels, triggering the pathological aggregation [169,170,171]; that other key players in lysosome repair and clearance, such as galectin-3 and TRIM16, have been shown to promote SNCA release and its spreading into the extracellular environment upon lysosomal damage and to promote SNCA conversion into fibrils [54, 152, 153]; and that SNCA aggregation can alter the autophagic-lysosomal pathway either by directly disrupting lysosomal components or by inhibiting trafficking events [147, 168, 172]. Altogether, these findings show a dual interaction between SNCA aggregation and lysosome functionality, underlying a crucial correlation between them.
Frontotemporal dementia and amyotrophic lateral sclerosis
FTD and ALS are two distinct NDs that display overlapping clinical signs and pathological mechanisms [173]. FTD primarily affects the frontal and temporal lobes of the brain, leading to changes in behavior, personality, and language skills accompanied by a decline in social cognition, emotional regulation, and executive functions [174]. ALS mainly affects motor neurons, responsible for voluntary muscle control, leading to muscle weakness and paralysis, and impairs the ability to speak, swallow, and breathe [175]. Both FTD and ALS present familial (fFTD and fALS) and sporadic (sFTD and sALS) forms. Although FTD and ALS are distinct diseases, they belong to a spectrum of diseases known as FTD/ALS, which highlights their overlapping nature.
FTD and ALS exhibit common molecular pathological features, including the mislocalization and aggregation of TAR DNA-binding protein 43 (TDP-43), a ribonucleotide protein that regulates mRNA metabolism, the accumulation of FTD/ALS-associated mutated proteins in inclusions, and the failure of the PQC system [173, 176, 177]. FTD/ALS are also associated with alterations to the autophagy–lysosomal pathway, detectable in postmortem tissue of FTD/ALS patients [87, 178] and evidenced by increased levels of galectin-3 in the spinal cord and cerebrospinal fluid, suggesting changes in lysosome dynamics [178, 179].
FTD and ALS also overlap at the genetic level; roughly 30% of fFTD cases, 5–10% of sALS cases, and approximately 50% of fALS cases are linked to a mutation in the C9ORF72 gene [173, 180]. This mutation involves the abnormal expansion of a hexanucleotide sequence (G4C2) localized in the first C9ORF72 intron. This mutation triggers three pathological mechanisms, one related to a loss of function due to haploinsufficiency and the other two involving a gain of toxicity. The toxicity may be caused either by the formation of aberrant RNA foci in the nucleus or by an unconventional repeat-associated ATG-independent (RAN) translation, which leads to the production of five different dipeptide repeat proteins. C9ORF72, known as a regulator of autophagic flux, plays a crucial role in maintaining the proper functionality of the autophagy-lysosomal pathway [181]. Haploinsufficiency of C9ORF72 causes the impairment of autophagy and lysosome functions, resulting in the accumulation of lysosome-like organelles that precede neurodegeneration, thereby contributing to the pathogenesis of FTD and ALS (reviewed in [182]). These phenotypes are partially caused by a decreased TFEB expression and by its cytoplasmic retention [183].
Other genes associated with FTD and ALS cases, including SQSTM1/p62, UBQLN2, DCTN1, TBK1, OPTN, and VCP [184,185,186,187,188,189], have been previously described to play a role in autophagy. Moreover, other genes associated exclusively with FTD are implicated in lysosomal trafficking, including TMEM106B and the major facilitator superfamily domain containing 8 (MFSD8) or in lysosomal activity, such as Cathepsin F (Ctsf) and granulin precursor (GRN) [190,191,192,193]. Thus, mutations in genes associated with lysosomal stability, functioning, or degradation underline an important implication of lysosomes and autophagy in pathological neurodegenerative mechanisms.
Lysosomal alterations in FTD/ALS can also be caused by a gain of toxicity associated with an increased toxic aggregation of TDP-43, mutated proteins such as tau, a microtubule-associated protein, or fused in sarcoma (FUS), a protein involved in regulating RNA metabolism. The aggregation of TDP-43 alters a specific autophagic pathway, chaperone-mediated autophagy (CMA), and disrupts lysosome function, which in turn exacerbates TDP-43 toxicity and loss of function [194, 195]. Indeed, TDP-43 aggregation and functional loss are associated with the activation of autophagosome and lysosome biogenesis through the inhibition of mTORC1 and activation of TFEB. Simultaneously, TDP-43 loss of function causes impairment in the fusion of autophagosomes with lysosomes, via an mTORC1-independent mechanism. Consequently, the buildup of AVs contributes to the aggregation of TDP-43 and neurodegeneration [196]. The aggregation of tau also impairs lysosomal functions through various mechanisms. In physiological conditions, tau stabilizes microtubules, facilitating the proper trafficking and maintenance of lysosomes [197, 198]. However, FTD-associated tau mutants are prone to aggregate, leading to hyperphosphorylation, ubiquitination, and destabilization of microtubules [199]. Moreover, tau aggregates also inhibit IST1, a member of the ESCRT complex, block CMA, and impair lysosome function. This results in the formation of enlarged dysfunctional lysosomes and even their rupture [200, 201]. Similarly, to tau, mutated FUS forms protein aggregates. These aggregates may sequester LAMP-1-positive structures, leading to the aberrant accumulation of functional lysosomes around the abnormal FUS aggregates [202].
Huntington’s disease
HD is characterized by the progressive deterioration of cognitive, motor, and psychiatric functions. As the disease progresses, HD symptoms include involuntary movements (chorea), cognitive decline, psychiatric disturbances, and difficulties with speech and swallowing [203]. HD is an inherited condition caused by an expansion of CAG trinucleotide repeats in the Huntingtin (HTT) gene, resulting in the production of an abnormal form of the HTT protein containing an elongated polyglutamine tract. The mutation leads to the accumulation of toxic protein aggregates in the brain, particularly in the basal ganglia and cortex (Fig. 2). HD displays several hallmarks of impairments in lysosomal function and dynamics. For instance, galectin-3 levels increase in the brains of HD mice and patients, suggesting alterations to lysosome activity. Galectin-3 levels increase in microglia before the onset of the disease and mediate the initiation of the inflammatory response which contribute to HD pathogenesis [204]. Moreover, an increase in the perinuclear accumulation of lysosomes is visible in HD models and it is normalized upon the overexpression of wild-type HTT. Mutant HTT (mHTT)-induced lysosome accumulation is associated with an increase in mTORC1 basal activity and the autophagic flux, resulting in a premature fusion of lysosomes with autophagosomes [205]. To further emphasize the autophagy–lysosome connection with HD, mHTT is recruited to vesicle-rich organelles that resemble multivesicular bodies or autolysosomes, suggesting a lysosome-dependent degradation of mHTT [206]. In addition, lysosomes are implicated in mHTT removal through an unconventional lysosome-dependent secretion mechanism [207].
These findings underscore the importance of an autophagy-lysosome role in HD and provide insights into potential therapeutic targets for the disease.
Alzheimer’s disease
AD is the most common cause of dementia and primarily affects memory, cognitive abilities, and behavior, gradually impairing daily functioning. AD typically starts with mild memory loss and progresses to severe cognitive decline and loss of independence [208]. The exact cause of AD is not fully understood, but age, genetic factors (such as the apolipoprotein E ε4 allele), and certain lifestyle and environmental factors are believed to play a role in AD pathogenesis [209]. AD is characterized by the accumulation of abnormal protein aggregates in the brain, such as Aβ plaques and tau tangles. Like in the previously discussed NDs, AD also presents signs of altered autophagy–lysosome pathways. This evidence includes galectin-3 accumulation in Aβ plaques in microglia, mediating the maladaptive activation of the inflammatory response [210]; dysregulation in endosome– and lysosome–ER contact sites due to amyloid precursor protein (APP) [211]; increased alkalinization in neuronal lysosomes which appears before Aβ deposition outside the cells. Lysosomal pH alteration is caused by decreased v-ATPase activity, associated with presenilin-1 mutations, and the accumulation of Aβ within enlarged autolysosomes that have lost their acidity. In line with this, in vitro studies have shown that the reacidification of lysosomes rescues lysosome dysfunction and accumulation [212]. Moreover, in affected neurons, AVs containing Aβ accumulate in a tightly packed manner within large membrane protrusions [213,214,215]. Similar observations have been described in the brains of AD patients. Additional AVs merge to form networks of membrane tubules surrounding the nucleus, where fibrillar Aβ accumulates within the lumens. This leads to the disruption of lysosomal membranes, the release of cathepsins, and ultimately cell death, accompanied by the invasion of microglial cells [215]. Recently it was found that positive modulation of TRIM16-mediated lysophagy decreases the accumulation of Aβ/tau, further underlying the tight connection between lysosome alterations and AD pathology [216].
Conclusions
Lysosomes are essential organelles for cell viability and alterations in their function are associated with several diseases, including LSDs and NDs. Indeed, accumulating evidence suggests that maintaining lysosomal integrity and efficient lysosomal degradation processes is crucial for neuronal protection and the prevention of NDs. To maintain homeostasis, cells activate different complex mechanisms to repair damaged lysosomes or, when this is not possible, to clear them away through lysophagy or exocytosis. These crucial processes are finely regulated by various proteins and complexes. However, many aspects of these processes remain unknown or not fully understood. Thus, unraveling the complex interplay between lysosomal dysfunction, aggregates accumulation, inflammation, and neuronal cells death holds promise for identifying novel therapeutic targets and developing strategies to counteract or slow down ND progression.
Some steps in this direction have already been taken, and therapeutic approaches and molecules that facilitate lysosomal clearance or biogenesis have been identified. Notably, certain molecules activate lysosome biogenesis by promoting the nuclear localization of TFEB. For example, compounds such as PP 242 and LY 294002 activate TFEB by inhibiting mTOR [217, 218]. Other substances, such as trehalose and its analogs lactulose and melibiose, enhance TFEB activity through an mTOR-independent pathway, as described in ref. [219]. The use of these compounds in disease models has shown promising results, encouraging research in this direction [220,221,222,223].
Altogether, this review highlights the intricate nature of the mechanisms governing lysosomal function and dynamics, as well as the consequence of their dysfunction in the development of pathological conditions. The complexity and significance of the described mechanisms underline the necessity of further investigation to enhance our understanding of pathological processes and development of therapeutic strategies.
Data availability
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
amyloid β
- ALR:
-
Autophagic lysosome reformation
- ALS:
-
Amyotrophic lateral sclerosis
- AMPK:
-
AMP-activated Protein Kinase
- APP:
-
Amyloid precursor protein
- AR:
-
Autophagy receptor
- ATG:
-
Autophagy-related
- ATP13A2:
-
ATPase cation transporting 13A2
- AV:
-
Autophagic vacuole
- CALCOCO2/NDP52:
-
Calcium-binding and coiled-coil domain 2 protein
- CASM:
-
Conjugation of ATG8s to single membranes
- CHMP4B:
-
Charged multivesicular body protein 4B
- CLEAR:
-
Coordinated lysosomal expression and regulation
- CMA:
-
Chaperone-mediated autophagy
- ESCRT:
-
Endosomal sorting complex required for transport
- ER:
-
Endoplasmic reticulum
- FTD:
-
Frontotemporal dementia
- FUS:
-
Fused in sarcoma
- GBA:
-
Glucocerebrosidase
- GRN:
-
Granulin precursor
- HD:
-
Huntington’s disease
- HDAC:
-
Histone deacetylases
- HSPA8:
-
Heat shock protein family A (Hsp70) member 8
- HTT:
-
Huntingtin
- ILVs:
-
Intraluminal vesicles
- IPMK:
-
Inositol polyphosphate multikinase
- KIF5B:
-
Kinesin family member 5B
- LAMP1:
-
Lysosomal associated membrane protein 1
- LAMP2A:
-
Lysosome-associated membrane protein 2A
- LB:
-
Lewy body
- LGALS3:
-
Galectin 3
- LIMP:
-
Lysosomal integral membrane proteins
- LLPS:
-
Liquid–liquid phase separation
- LMP:
-
Lysosomal membrane permeabilization
- LQC:
-
Lysosomal quality control
- LRRK2:
-
Leucine-rich repeat kinase 2
- LSDs:
-
Lysosomal storage disorders
- LYTL:
-
Lysosomal tubulation sorting
- MAP1LC3:
-
Microtubule Associated Protein 1 Light Chain 3 Beta
- MAP3K7/TAK1:
-
Mitogen-activated protein kinase kinase kinase 7
- MAPK1:
-
Mitogen-activated protein kinase 1
- MCOLN1:
-
Mucolipin TRP cation channel 1
- MCS:
-
Membrane contact sites
- MFSD8:
-
Major facilitator superfamily domain containing 8
- mTOR:
-
Mechanistic target of rapamycin
- NDs:
-
Neurodegenerative diseases
- OPTN:
-
Optineurin
- OSBP:
-
Oxysterol binding protein
- PD:
-
Parkinson’s disease
- PDCD6IP/ALIX:
-
Programmed cell death 6 interacting protein
- PI:
-
Phosphatidyl-inositol
- PI4K2A:
-
Phosphatidyl-inositol-4 kinase type 2 alpha
- PI4P:
-
Phosphatidyl-inositol 4-phosphate
- PITT:
-
Phosphoinositide-initiated membrane tethering and lipid transport
- PPP3CB:
-
Protein phosphatase 3 catalytic subunit beta
- PQC:
-
Protein quality control
- PS:
-
Phosphatidyl-serine
- RAN:
-
Repeat-associated ATG independent
- SMURF1:
-
SMAD specific E3 ubiquitin-protein ligase
- SNCA:
-
Alpha-synuclein
- SQSTM1/p62:
-
Sequestosome 1
- TARDBP/TDP-43:
-
TAR DNA binding protein
- TBK1:
-
TANK-binding kinase 1
- TFE3:
-
Transcription factor E3
- TFEB:
-
Transcription factor EB
- TMEM:
-
Transmembrane protein
- TRIM16:
-
Tripartite motif-containing 16
- UBA:
-
Ubiquitin-associated
- USP9X:
-
Ubiquitin-specific peptidase 9 X linked
- VCP:
-
Valosin containing protein
References
De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92.
De Duve C, Pressman BC, Gianetto R, Wattiaux R, Applemans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60(4):604–17. https://doi.org/10.1042/bj0600604.
Seglen PO, Gordon PBHI. Non-selective autophagy. Semin Cell Biol. 1990;1(6):441–8.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H±ATPase. Science. 2011;334(6056):678–83.
Gray MA, Choy CH, Dayam RM, Escobar EO, Somerville A, Xiao X, et al. Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB Matthew. Curr Biol. 2017;26(15):1955–64.
Reddy A, Caler EV, Andrews NW, Haven N. Plasma membrane repair is mediated by Ca 2 ± regulated exocytosis of lysosomes. Cell. 2001;106:157–69.
Encarnação M, Espada L, Escrevente C, Mateus D, Ramalho J, Michelet X, et al. A Rab3a-dependent complex essential for lysosome positioning and plasma membrane repair. J Cell Biol. 2016. https://doi.org/10.1083/jcb.201511093.
Lim CY, Zoncu R. The lysosome as a command-and-control center for cellular metabolism. J Cell Biol. 2016;214(6):653–64.
Lawrence RE, Zoncu R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat Cell Biol. 2019;21(2):133–42.
Repnik U, Česen MH, Turk B. The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol. 2013;5(1): a008755.
Sun A. Lysosomal storage disease overview. Ann Transl Med. 2018. https://doi.org/10.21037/atm.2018.11.39.
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases : from pathophysiology to therapy. Annu Rev Med. 2015. https://doi.org/10.1146/annurev-med-122313-085916.
Marques ARA, Saftig P. Lysosomal storage disorders—challenges, concepts and avenues for therapy: beyond rare diseases. J Cell Sci. 2019. https://doi.org/10.1242/jcs.221739.
Kirkegaard T, Jäättelä M. Biochimica et biophysica acta lysosomal involvement in cell death and cancer. BBA Mol Cell Res. 2009;1793(4):746–54.
Davidson SM, Vander HMG. Critical functions of the lysosome in cancer biology. Annu Rev Pharmacol Toxicol. 2017;57:481–510.
Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, et al. LRRK 2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020;39(18):1–14.
Feng T, Mai S, Roscoe JM, Sheng RR, Ullah M, Zhang J, et al. Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep. 2020;21(10): e50219.
Tedeschi V, Petrozziello T, Secondo A. Calcium dyshomeostasis and lysosomal Ca2+ dysfunction in amyotrophic lateral sclerosis. Cells. 2019. https://doi.org/10.3390/cells8101216.
Cortes CJ, La Spada AR. TFEB dysregulation as a driver of autophagy dysfunction in neurodegenerative disease: molecular mechanisms, cellular processes, and emerging therapeutic opportunities. Neurobiol Dis. 2018. https://doi.org/10.1016/j.nbd.2018.05.012.
Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18(21):4153–70.
Dodson MW, Zhang T, Jiang C, Chen S, Guo M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum Mol Genet. 2012;21(6):1350–63.
Tohnai G, Adachi H, Katsuno M, Doi H, Matsumoto S, Kondo N, et al. Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum Mol Genet. 2014. https://doi.org/10.1093/hmg/ddu066.
Mathis S, Goizet C, Soulages A, Vallat JM, Le MG. Genetics of amyotrophic lateral sclerosis: a review. J Neurol Sci. 2019;399(February):217–26.
Carmona-gutierrez D, Hughes AL, Madeo F, Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Age Res Rev. 2016;32:2–12.
Peters C, von Figura K. Biogenesis of lysosomal membranes. FEBS Lett. 1994;346(1):108–14.
Schwake M, Schröder B, Saftig P. Lysosomal membrane proteins and their central role in physiology. Traffic. 2013;14(7):739–48.
Schröder B, Wrocklage C, Pan C, Jäger R, Kösters B, Schäfer H, et al. Integral and associated lysosomal membrane proteins. Traffic. 2007;8(12):1676–86.
Schröder BA, Wrocklage C, Hasilik A, Saftig P. The proteome of lysosomes. Proteomics. 2010;10(22):4053–76.
Lewis V, Green SA, Marsh M, Vlhko P, Helenius A, Mellman I. Glycoproteins of the lysosomal membrane. J Cell Biol. 1985;100(6):1839–47.
Andrejewski N, Punnonen EL, Guhde G, Tanaka Y, Lüllmann-Rauch R, Hartmann D, et al. Normal lysosomal morphology and function in LAMP-1-deficient mice. J Biol Chem. 1999;274(18):12692–701.
Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, et al. Accumulation of autophagic vacuoles and cardiomyopathy LAMP-2-deficient mice. Nature. 2000;406(6798):902–6.
Nishino I. Autophagic vacuolar myopathy. Semin Pediatr Neurol. 2006;13(2):90–5.
Xu J, Wang L, Liu X, Dai Q. A novel LAMP2 p.G93R mutation associated with mild Danon disease presenting with familial hypertrophic cardiomyopathy. Mol Genet Genomic Med. 2019;7(10):e00941.
Kolter T, Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol. 2005;21(1):81–103.
Rudnik S, Damme M. The lysosomal membrane—export of metabolites and beyond. FEBS J. 2021;288(14):4168–82.
Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392(6672):193–7.
van Meer G, Voelker DRFG. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–24.
Schoer JK, Gallegos AM, McIntosh AL, Starodub O, Kier AB, Billheimer JT, et al. Lysosomal membrane cholesterol dynamics. Biochemistry. 2000;39(26):7662–77.
Lim CY, Davis OB, Shin HR, Zhang J, Berdan CA, Jiang X, et al. ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in niemann-pick type C. Nat Cell Biol. 2019;21(10):1206–18.
Heybrock S, Kanerva K, Meng Y, Ing C, Liang A, Xiong ZJ, et al. Lysosomal integral membrane protein-2 (LIMP-2/SCARB2) is involved in lysosomal cholesterol export. Nat Commun. 2019;10(1):3521.
Jadot M, Andrianaivo F, Dubois F, Wattiaux R. Effects of methylcyclodextrin on lysosomes. Eur J Biochem. 2001;268(5):1392–9.
Appelqvist H, Nilsson C, Garner B, Brown AJ, Kågedal K, Öllinger K. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol. 2011;178(2):629–39.
Appelqvist H, Sandin L, Björnström K, Saftig P, Garner B, Öllinger K, et al. Sensitivity to lysosome-dependent cell death is directly regulated by lysosomal cholesterol content. PLoS ONE. 2012;7(11):1–11.
Fouchier F, Mego JL, Dang JSC. Thyroid lysosomes: the stability of the lysosomal membrane. Eur J Cell Biol. 1983;30(2):272–8.
Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–51.
Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 2010;17(7):1167–78.
Funk RS, Krise JP. Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Mol Pharm. 2012;9(5):1384–95.
Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2011;17(5):766–74.
Kågedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, et al. Lysosomal membrane permeabilization during apoptosis—involvement of bax? Int J Exp Pathol. 2005;86(5):309–21.
Eriksson I, Wäster P, Öllinger K. Restoration of lysosomal function after damage is accompanied by recycling of lysosomal membrane proteins. Cell Death Dis. 2020. https://doi.org/10.1038/s41419-020-2527-8.
Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013;32(17):2336–47.
Martins WK, Costa ÉT, Cruz MC, Stolf BS, Miotto R, Cordeiro RM, et al. Parallel damage in mitochondrial and lysosomal compartments promotes efficient cell death with autophagy: the case of the pentacyclic triterpenoids. Sci Rep. 2015;5(June):1–17.
Burbidge K, Rademacher DJ, Mattick J, Zack S, Grillini A, Bousset L, et al. LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/α-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons. Autophagy. 2021;6:1–29.
Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, et al. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017;134(4):629–53.
Jadot M, Colmant C, Wattiaux-De Coninck S, Wattiaux R. Intralysosomal hydrolysis of glycyl-L-phenylalanine 2-naphthylamide. Biochem J. 1984;219(3):965–70.
Barondes SH, Cooper DN, Gitt MA, Leffler H. Galectins. structure and function of a large family of animal lectins. J Biol Chem. 1994;269(33):20807–10.
Haudek KC, Spronk KJ, Voss PG, Patterson RJ, Wang JLAE. Dynamics of galectin-3 in the nucleus and cytoplasm. Biochim Biophys Acta. 2011;23(1):1–7.
Popa SJ, Stewart SE, Moreau K. Unconventional secretion of annexins and galectins. Semin Cell Dev Biol. 2018;83:42–50.
Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci. 2015;128(13):2213–9.
Stegmayr J, Zetterberg F, Carlsson MC, Huang X, Sharma G, Kahl-Knutson B, et al. Extracellular and intracellular small-molecule galectin-3 inhibitors. Sci Rep. 2019;9(1):1–12.
Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006. https://doi.org/10.1093/glycob/cwl025.
Lindgreen A, Lindgreen A. Extracellular functions of galectin-3. Glycoconj J. 2004;19(1):527–35.
Garner OB, Baum LG. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem Soc Trans. 2008;36(6):1472–7.
Weng IC, Chen HL, Lo TH, Lin WH, Chen HY, Hsu DK, et al. Cytosolic galectin-3 and -8 regulate antibacterial autophagy through differential recognition of host glycans on damaged phagosomes. Glycobiology. 2018;28(6):392–405.
Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, et al. Galectins control mTOR in response to endomembrane damage. Mol Cell. 2018;70(1):120-135.e8.
Aits S, Kricker J, Liu B, Ellegaard AM, Hämälistö S, Tvingsholm S, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11(8):1408–24.
Radulovic M, Schink KO, Wenzel EM, Lafont F, Stenmark H, Nähse V, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018;37(21):e99753.
Jia J, Claude-Taupin A, Gu Y, Choi SW, Peters R, Bissa B, et al. Galectin-3 coordinates a cellular system for lysosomal repair and removal. Dev Cell. 2020;52(1):69-87.e8.
Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360(6384):eaar5078.
Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci U S A. 2018;115(39):E9115–24.
Kuwahara T, Funakawa K, Komori T, Sakurai M, Yoshii G, Eguchi T, et al. Roles of lysosomotropic agents on LRRK2 activation and Rab10 phosphorylation. Neurobiol Dis. 2020;145(May): 105081.
Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv. 2022;6(46):eabb2454.
Kluss JH, Bonet-Ponce L, Lewis PA, Cookson MR. Directing LRRK2 to membranes of the endolysosomal pathway triggers RAB phosphorylation and JIP4 recruitment. Neurobiol Dis. 2022;170(April): 105769.
Goul CS, Zoncu R. PITTching in for lysosome repair. Dev Cell. 2022;57(20):2347–9.
Tan JX, Finkel T. A phosphoinositide signalling pathway mediates rapid lysosomal repair. Nature. 2022;609(7928):815–21.
Niekamp P, Scharte F, Sokoya T, Vittadello L, Kim Y, Deng Y, et al. Ca2+-activated sphingomyelin scrambling and turnover mediate ESCRT-independent lysosomal repair. Nat Commun. 2022. https://doi.org/10.1038/s41467-022-29481-4.
Shin HR, Zoncu R. Finding sugar in the pantry: how galectins detect and signal lysosomal damage. Mol Cell. 2018;70(1):5–7.
Jia J, Bissa B, Brecht L, Allers L, Choi SW, Gu Y, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020;77(5):951-969.e9.
Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39(1):13–27.
Yoshida Y, Yasuda S, Fujita T, Hamasaki M, Murakami A, Kawawaki J, et al. Ubiquitination of exposed glycoproteins by SCFFBXO27 directs damaged lysosomes for autophagy. Proc Natl Acad Sci U S A. 2017;114(32):8574–9.
Gahlot P, Kravic B, Rota G, van den Boom J, Levantovsky S, Schulze N, et al. Lysosomal damage sensing and lysophagy initiation by SPG20-ITCH. Mol Cell. 2024. https://doi.org/10.1016/j.molcel.2024.02.029.
Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017;36(2):135–50.
Koerver L, Papadopoulos C, Liu B, Kravic B, Rota G, Brecht L, et al. The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep. 2019;20(10): e48014.
Teranishi H, Tabata K, Saeki M, Umemoto T, Hatta T, Otomo T, et al. Identification of CUL4A-DDB1-WDFY1 as an E3 ubiquitin ligase complex involved in initiation of lysophagy. Cell Rep. 2022;40(11): 111349.
Kravić B, Bionda T, Siebert A, Gahlot P, Levantovsky S, Behrends C, et al. Ubiquitin profiling of lysophagy identifies actin stabilizer CNN2 as a target of VCP/p97 and uncovers a link to HSPB1. Mol Cell. 2022;82(14):2633-2649.e7.
Papadopoulos C, Kirchner P, Bug M, Grum D, Koerver L, Schulze N, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017;36(2):135–50.
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci. 2016;113(15):4039–44.
Thurston TLM, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–21.
Gallagher ER, Holzbaur ELF. The selective autophagy adaptor p62/SQSTM1 forms phase condensates regulated by HSP27 that facilitate the clearance of damaged lysosomes via lysophagy. Cell Rep. 2023;42(2): 112037.
Corkery DP, Castro-Gonzalez S, Knyazeva A, Herzog LK, Wu Y-W. An ATG12-ATG5-TECPR1 E3-like complex regulates unconventional LC3 lipidation at damaged lysosomes. EMBO Rep. 2023;24(9): e56841.
Cross J, Durgan J, McEwan DG, Tayler M, Ryan KM, Florey O. Lysosome damage triggers direct ATG8 conjugation and ATG2 engagement via non-canonical autophagy. J Cell Biol. 2023;222(12): e202303078.
Hooper KM, Jacquin E, Li T, Goodwin JM, Brumell JH, Durgan J, et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J Cell Biol. 2022;221(6): e202105112.
Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32(1):255–78.
Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011;21(3):421–30.
Martina JA, Diab HI, Lishu L, Jeong LA, Patange S, Raben N, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014;7(309):ra9.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–7.
Cui Z, Napolitano G, de Araujo MEG, Esposito A, Monfregola J, Huber LA, et al. Structure of the lysosomal mTORC1–TFEB–Rag–ragulator megacomplex. Nature. 2023;614(7948):572–9.
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17(3):288–99.
Ferron M, Settembre C, Shimazu J, Lacombe J, Kato S, Rawlings DJ, et al. A RANKL–PKCβ–TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013;27(8):955–69.
Xu Y, Ren J, He X, Chen H, Wei T, Feng W. YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization. Autophagy. 2019;15(6):1017–30.
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–14.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–108.
Martina JA, Puertollano R. Rag GTPases mediate amino acid–dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol. 2013;200(4):475–91.
Silvestrini MJ, Johnson JR, Kumar AV, Thakurta TG, Blais K, Neill ZA, et al. Nuclear export inhibition enhances HLH-30/TFEB activity, autophagy, and lifespan. Cell Rep. 2018;23(7):1915–21.
Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, et al. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat Commun. 2018;9(1):3312.
Zhang J, Wang J, Zhou Z, Park J-E, Wang L, Wu S, et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy. 2018;14(6):1043–59.
Li T, Yin L, Kang X, Xue W, Wang N, Zhang J, et al. TFEB acetylation promotes lysosome biogenesis and ameliorates Alzheimer’s disease–relevant phenotypes in mice. J Biol Chem. 2022;298(12): 102649.
Ong AJS, Bladen CE, Tigani TA, Karamalakis AP, Evason KJ, Brown KK, et al. The KEAP1–NRF2 pathway regulates TFEB/TFE3-dependent lysosomal biogenesis. Proc Natl Acad Sci. 2023;120(22): e2217425120.
Chen D, Wang Z, Zhao YG, Zheng H, Zhao H, Liu N, et al. Inositol polyphosphate multikinase inhibits liquid-liquid phase separation of TFEB to negatively regulate autophagy activity. Dev Cell. 2020;55(5):588-602.e7.
Wang Z, Chen D, Guan D, Liang X, Xue J, Zhao H, et al. Material properties of phase-separated TFEB condensates regulate the autophagy-lysosome pathway. J Cell Biol. 2022;221(5): e202112024.
Chauhan S, Kumar S, Jain A, Ponpuak M, Mudd MH, Kimura T, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016;39(1):13–27.
Leow SM, Serene Chua SX, Venkatachalam G, Shen L, Luo L, Clement MV. Sub-lethal oxidative stress induces lysosome biogenesis via a lysosomal membrane permeabilization-cathepsin-caspase 3-transcription factor EB-dependent pathway. Oncotarget. 2016;8(10):16170.
Nakamura S, Shigeyama S, Minami S, Shima T, Akayama S, Matsuda T, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. 2020;22(10):1252–63.
Xia Q, Zheng H, Li Y, Xu W, Wu C, Xu J, et al. SMURF1 controls the PPP3/calcineurin complex and TFEB at a regulatory node for lysosomal biogenesis. Autophagy. 2023;20(4):1–17.
Reddy A, Caler EV, Andrews NW. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell. 2001;106(2):157–69.
Castro-Gomes T, Corrotte M, Tam C, Andrews NW. Plasma membrane repair is regulated extracellularly by proteases released from lysosomes. PLoS ONE. 2016;11(3): e0152583.
Andrews NW. Regulated secretion of conventional lysosomes. Trend Cell Biol. 2000;10(8):316–21.
Lachuer H, Le L, Lévêque-Fort S, Goud B, Schauer K. Spatial organization of lysosomal exocytosis relies on membrane tension gradients. Proc Natl Acad Sci. 2023;120(8): e2207425120.
Arantes RME, Andrews NW. A role for synaptotagmin VII-regulated exocytosis of lysosomes in neurite outgrowth from primary sympathetic neurons. J Neurosci. 2006;26(17):4630–7.
Chen G, Zhang Z, Wei Z, Cheng Q, Li X, Li W, et al. Lysosomal exocytosis in Schwann cells contributes to axon remyelination. Glia. 2012;60(2):295–305.
Grochowska KM, Sperveslage M, Raman R, Failla AV, Głów D, Schulze C, et al. Chaperone-mediated autophagy in neuronal dendrites utilizes activity-dependent lysosomal exocytosis for protein disposal. Cell Rep. 2023;42(8):112998.
Tsunemi T, Perez-Rosello T, Ishiguro Y, Yoroisaka A, Jeon S, Hamada K, et al. Increased lysosomal exocytosis induced by lysosomal Ca2+ channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J Neurosci. 2019;39(29):5760–72.
Yu L, Mcphee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010. https://doi.org/10.1038/nature09076.
Sun X, Yang Y, Zhong XZ, Cao Q, Zhu X-H, Zhu X, et al. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy. 2018;14(1):38–52.
Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012;14(9):924–34.
Du W, Su QP, Chen Y, Zhu Y, Jiang D, Rong Y, et al. Kinesin 1 drives autolysosome tubulation. Dev Cell. 2016;37(4):326–36.
Bhattacharya A, Mukherjee R, Kuncha SK, Brunstein ME, Rathore R, Junek S, et al. A lysosome membrane regeneration pathway depends on TBC1D15 and autophagic lysosomal reformation proteins. Nat Cell Biol. 2023;25(5):685–98.
Bright NA, Reaves BJ, Mullock BM, Luzio JP. Dense core lysosomes can fuse with late endosomes and are re-formed from the resultant hybrid organelles. J Cell Sci. 1997;110(17):2027–40.
Bissig C, Hurbain I, Raposo G, van Niel G. PIKfyve activity regulates reformation of terminal storage lysosomes from endolysosomes. Traffic. 2017;18(11):747–57.
Sava I, Davis LJ, Gray SR, Bright NA, Luzio JP. Reversible assembly and disassembly of V-ATPase during the lysosome regeneration cycle. Mol Biol Cell. 2024;35(5):ar63.
Luzio JP, Gray SR, Bright NA. Endosome–lysosome fusion. Biochem Soc Trans. 2010;38(6):1413–6.
Duclos S, Corsini R, Desjardins M. Remodeling of endosomes during lysosome biogenesis involves `kiss and run’ fusion events regulated by rab5. J Cell Sci. 2003;116(5):907–18.
Lim KH. Diverse misfolded conformational strains and cross-seeding of misfolded proteins implicated in neurodegenerative diseases. Front Mol Neurosci. 2019;12:158.
Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40(1):189–210.
Boi L, Pisanu A, Palmas MF, Fusco G, Carboni E, Casu MA, et al. Modeling parkinson’s disease neuropathology and symptoms by intranigral inoculation of preformed human α-synuclein oligomers. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21228535.
Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer’s disease. IJMS. 2018;29(6):613–27.
Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30(37):12535–44.
McBrayer M, Nixon RA. Lysosome and calcium dysregulation in Alzheimer’s disease: partners in crime. Biochem Soc Trans. 2013;41(6):1495–502.
Ferrari V, Cristofani R, Cicardi ME, Tedesco B, Crippa V, Chierichetti M, et al. Pathogenic variants of valosin-containing protein induce lysosomal damage and transcriptional activation of autophagy regulators in neuronal cells. Neuropathol Appl Neurobiol. 2022;48(5):1–22.
Stagi M, Klein ZA, Gould TJ, Bewersdorf J, Strittmatter SM. Lysosome size, motility and stress response regulated by fronto-temporal dementia modifier TMEM106B. Mol Cell Neurosci. 2014;61:226–40.
Dehay B, Martinez-Vicente M, Ramirez A, Perier C, Klein C, Vila M, et al. Lysosomal dysfunction in Parkinson disease. Autophagy. 2012;8(9):1389–91.
Bhattacharya J, Edwards J, Mamelak AN, Schuman EM. Long-range temporal correlations in the spontaneous spiking of neurons in the hippocampal-amygdala complex of humans. Neuroscience. 2005;131(2):547–55.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–6.
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–6.
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–64.
Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8(4):e62143.
Oku Y, Murakami K, Irie K, Hoseki J, Sakai Y. Synthesized Aβ42 caused intracellular oxidative damage, leading to cell death, via lysosome rupture. Cell Struct Funct. 2017;42(1):71–9.
Zeineddine R, Pundavela JF, Corcoran L, Stewart EM, Do-Ha D, Bax M, et al. SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol Neurodegener. 2015;10(1):57.
Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm. 2017;124(8):901–5.
Braak H, Del Tredici K. Neuropathological staging of brain pathology in sporadic Parkinson’s disease: separating the wheat from the chaff. J Parkinsons Dis. 2017;7:S71-85.
Wu HC, Chang KH, Chiang MC, Chen CM. Alterations of plasma galectin-3 and C3 levels in patients with parkinson’s disease. Brain Sci. 2021. https://doi.org/10.3390/brainsci11111515.
García-Revilla J, Boza-Serrano A, Jin Y, Vadukul DM, Soldán-Hidalgo J, Camprubí-Ferrer L, et al. Galectin-3 shapes toxic alpha-synuclein strains in Parkinson’s disease. Acta Neuropathol. 2023. https://doi.org/10.1016/j.ibneur.2023.08.786.
Boza-Serrano A, Reyes JF, Rey NL, Leffler H, Bousset L, Nilsson U, et al. The role of Galectin-3 in α-synuclein-induced microglial activation. Acta Neuropathol Commun. 2014;2(1):156.
Day JO, Mullin S. The genetics of parkinson’s disease and implications for clinical practice. Genes. 2021. https://doi.org/10.3390/genes12071006.
Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson’s disease: an introspection of its journey towards precision medicine. Neurobiol Dis. 2020;137: 104782.
Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49(10):1511–6.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Zunke F. The function of lysosomes and their role in Parkinson’s disease. Neuroforum. 2020;26(1):43–51.
Wallings R, Connor-Robson N, Wade-Martins R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 2019;28(16):2696–710.
Komori T, Kuwahara T, Fujimoto T, Sakurai M, Koyama-Honda I, Fukuda M, et al. Phosphorylation of Rab29 at Ser185 regulates its localization and role in the lysosomal stress response in concert with LRRK2. J Cell Sci. 2023;136(14):jcs261003.
van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. 2020;578(7795):419–24.
Yun SP, Kim D, Kim S, Kim S, Karuppagounder SS, Kwon S-H, et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol Neurodegener. 2018;13(1):1.
Hu M, Li P, Wang C, Feng X, Geng Q, Chen W, et al. Parkinson’s disease-risk protein TMEM175 is a proton-activated proton channel in lysosomes. Cell. 2022;185(13):2292-2308.e20.
Usenko TS, Bezrukova AI, Bogdanova DA, Kopytova AE, Senkevich KA, Gracheva EV, et al. Genetics variants and expression of the SCARB2 gene in the pathogenesis of Parkinson’s disease in Russia. Neurosci Lett. 2021;741: 135509.
Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lüllmann-Rauch R, et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci. 2014;111(43):15573–8.
Reczek D, Schwake M, Schröder J, Hughes H, Blanz J, Jin X, et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of β-glucocerebrosidase. Cell. 2007;131(4):770–83.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–5.
Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated parkinson’s disease. J Neurosci. 2017;37(40):9617LP – 9631.
Suzuki M, Fujikake N, Takeuchi T, Kohyama-Koganeya A, Nakajima K, Hirabayashi Y, et al. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet. 2015;24(23):6675–86.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of α-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97(1):92-107.e10.
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci. 2016;113(7):1931–6.
Burrell JR, Halliday GM, Kril JJ, Ittner LM, Götz J, Kiernan MC, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388(10047):919–31.
Younes K, Miller BL. Frontotemporal dementia: neuropathology, genetics, neuroimaging, and treatments. Psychiatr Clin North Am. 2020;43(2):331–44.
Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918–29.
Mandrioli J, Mediani L, Alberti S, Carra S. ALS and FTD: where RNA metabolism meets protein quality control. Semin Cell Dev Biol. 2020;99:183–92.
Parobkova E, Matej R. Amyotrophic lateral sclerosis and frontotemporal lobar degenerations: similarities in genetic background. Diagnostics. 2021;11(3):1–21.
Zhou JY, Afjehi-Sadat L, Asress S, Duong DM, Cudkowicz M, Glass JD, et al. Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J Proteom Res. 2010;9(10):5133–41.
Borrego-Écija S, Pérez-Millan A, Antonell A, Fort-Aznar L, Kaya-Tilki E, León-Halcón A, et al. Galectin-3 is upregulated in frontotemporal dementia patients with subtype specificity. Alzheimer Dement. 2023;20(3):1515–26.
Zampatti S, Peconi C, Campopiano R, Gambardella S, Caltagirone C, Giardina E. C9orf72-related neurodegenerative diseases: from clinical diagnosis to therapeutic strategies. Front Aging Neurosci. 2022;14(June):1–9.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016;35(15):1656–76.
Cozzi M, Ferrari V. Autophagy dysfunction in ALS: from transport to protein degradation. J Mol Neurosci. 2022;72(7):1456–81.
Cunningham KM, Maulding K, Ruan K, Senturk M, Grima JC, Sung H, et al. TFEB/Mitf links impaired nuclear import to autophagolysosomal dysfunction in C9-ALS. Elife. 2020;9:e59419.
Hardy J, Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp Neurol. 2014;262:75–83.
Wu JJ, Cai A, Greenslade JE, Higgins NR, Fan C, Le NTT, et al. ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci. 2020;117(26):15230–41.
Borg R, Herrera P, Purkiss A, Cacciottolo R, Cauchi RJ. Reduced levels of ALS gene DCTN1 induce motor defects in Drosophila. Front Neurosci. 2023;17(June):1–10.
Harding O, Evans CS, Ye J, Cheung J, Maniatis T, Holzbaur ELF. ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc Natl Acad Sci. 2021;118(24): e2025053118.
Dominguez J, Yu JT, Tan YJ, Ng A, De Guzman MF, Natividad B, et al. Novel optineurin frameshift insertion in a family with frontotemporal dementia and parkinsonism without amyotrophic lateral sclerosis. Front Neurol. 2021;12(May):1–8.
Scarian E, Fiamingo G, Diamanti L, Palmieri I, Gagliardi S, Pansarasa O. The role of VCP mutations in the spectrum of amyotrophic lateral sclerosis—frontotemporal dementia. Front Neurol. 2022;13(February):1–15.
Bauer CS, Webster CP, Shaw AC, Kok JR, Castelli LM, Lin YH, et al. Loss of TMEM106B exacerbates C9ALS/FTD DPR pathology by disrupting autophagosome maturation. Front Cell Neurosci. 2022;16(December):1–18.
Huang L, Liu Z, Yuan Y, Shen L, Jiang H, Tang B, et al. Mutation analysis of MFSD8 in an amyotrophic lateral sclerosis cohort from mainland China. Eur J Neurosci. 2021;53(4):1197–206.
Simon MJ, Logan T, DeVos SL, Di Paolo G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. 2023;33(4):324–39.
Van Der Zee J, Mariën P, Crols R, Van Mossevelde S, Dillen L, Perrone F, et al. Mutated CTSF in adult-onset neuronal ceroid lipofuscinosis and FTD. Neurol Genet. 2016;2(5):1–6.
Ormeño F, Hormazabal J, Moreno J, Riquelme F, Rios J, Criollo A, et al. Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front Mol Neurosci. 2020;13(February):1–17.
Leibiger C, Deisel J, Aufschnaiter A, Ambros S, Tereshchenko M, Verheijen BM, et al. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet. 2018;27(9):1593–607.
Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016;35(2):121–42.
Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, et al. Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci. 2019;10(JUL):1–14.
Balabanian L, Lessard DV, Swaminathan K, Yaninska P, Sébastien M, Wang S, et al. Tau differentially regulates the transport of early endosomes and lysosomes. Mol Biol Cell. 2022;33(13):ar128.
Bodea L-G, Eckert A, Ittner LM, Piguet O, Götz J. Tau physiology and pathomechanisms in frontotemporal lobar degeneration. J Neurochem. 2016;138(S1):71–94.
Feng Q, Luo Y, Zhang XN, Yang XF, Hong XY, Sun DS, et al. MAPT/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy. 2020;16(4):641–58.
Lim F, Hernández F, Lucas JJ, Gómez-Ramos P, Morán MA, Ávila J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and tau filaments in forebrain. Mol Cell Neurosci. 2001;18(6):702–14.
Trnka F, Hoffmann C, Wang H, Sansevrino R, Rankovic B, Rost BR, et al. Aberrant phase separation of FUS leads to lysosome sequestering and acidification. Front Cell Dev Biol. 2021;9(October):1–13.
Kim A, Lalonde K, Truesdell A, Welter PG, Brocardo PS, Rosenstock TR, et al. New avenues for the treatment of huntington’s disease. Int J Mol Sci. 2021;22(16):1–50.
Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, et al. Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’s disease. Nat Commun. 2019;10(1):1–18.
Erie C, Sacino M, Houle L, Lu ML, Wei J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur J Neurosci. 2015;42(3):1941–51.
Zhou Y, Peskett TR, Landles C, Warner JB, Sathasivam K, Smith EJ, et al. Correlative light and electron microscopy suggests that mutant huntingtin dysregulates the endolysosomal pathway in presymptomatic Huntington’s disease. Acta Neuropathol Commun. 2021;9(1):70.
Trajkovic K, Jeong H, Krainc D. Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J Neurosci. 2017;37(37):9000–12.
Miao J, Ma H, Yang Y, Liao Y, Lin C, Zheng J, et al. Microglia in Alzheimer’s disease: pathogenesis, mechanisms, and therapeutic potentials. Front Aging Neurosci. 2023;15(June):1–35.
Sun YY, Wang Z, Huang HC. Roles of ApoE4 on the pathogenesis in Alzheimer’s disease and the potential therapeutic approaches. Cell Mol Neurobiol. 2023;43(7):3115–36.
Boza-Serrano A, Vrillon A, Minta K, Paulus A, Camprubí-Ferrer L, Garcia M, et al. Galectin-3 is elevated in CSF and is associated with Aβ deposits and tau aggregates in brain tissue in Alzheimer’s disease. Acta Neuropathol. 2022;144(5):843–59.
Bretou M, Sannerud R, Escamilla-Ayala A, Leroy T, Vrancx C, Van Acker ZP, et al. Accumulation of APP C-terminal fragments causes endolysosomal dysfunction through the dysregulation of late endosome to lysosome-ER contact sites. Dev Cell. 2024;59(12):1571-1592.e9.
Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience. 2014;263:111–24.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(23):4081–91.
Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–97.
Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022;25(6):688–701.
Chae CW, Yoon JH, Lim JR, Park JY, Cho JH, Jung YH, et al. TRIM16-mediated lysophagy suppresses high-glucose-accumulated neuronal Aβ. Autophagy. 2023;19(10):2752–68.
Chin MY, Ang KH, Davies J, Alquezar C, Garda VG, Rooney B, et al. Phenotypic screening using high-content imaging to identify lysosomal pH modulators in a neuronal cell model. ACS Chem Neurosci. 2022;13(10):1505–16.
Zhang Z, Yue P, Lu T, Wang Y, Wei Y, Wei X. Role of lysosomes in physiological activities, diseases, and therapy. J Hematol Oncol. 2021;14(1):79.
Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019;15(4):631–51.
Massenzio F, Peña-Altamira E, Petralla S, Virgili M, Zuccheri G, Miti A, et al. Microglial overexpression of fALS-linked mutant SOD1 induces SOD1 processing impairment, activation and neurotoxicity and is counteracted by the autophagy inducer trehalose. Biochim Biophys Acta Mol Basis Dis. 2018;1864(12):3771–85.
Perucho J, Gómez A, Muñoz MP, de Yébenes JG, Mena MÁ, Casarejos MJ. Trehalose rescues glial cell dysfunction in striatal cultures from HD R6/1 mice at early postnatal development. Mol Cell Neurosci. 2016;74:128–45.
Pupyshev AB, Tikhonova MA, Akopyan AA, Tenditnik MV, Dubrovina NI, Korolenko TA. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol Biochem Behav. 2019;177:1–11.
Galbiati M, Meroni M, Boido M, Cescon M, Rusmini P, Crippa V, et al. Bicalutamide and trehalose ameliorate spinal and bulbar muscular atrophy pathology in mice. Neurotherapeutics. 2023;20(2):524–45.
Farrer M, Stone J, Mata IF, Lincoln S, Kachergus J, Hulihan M, et al. LRRK2 mutations in Parkinson disease. Neurology. 2005;65(5):738–40.
Bonet-Ponce L, Cookson MR. LRRK2 recruitment, activity, and function in organelles. FEBS J. 2022;289(22):6871–90.
Djarmati A, Hagenah J, Reetz K, Winkler S, Behrens MI, Pawlack H, et al. ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord. 2009;24(14):2104–11.
Greuel A, Trezzi JP, Glaab E, Ruppert MC, Maier F, Jäger C, et al. GBA variants in Parkinson’s disease: clinical, metabolomic, and multimodal neuroimaging phenotypes. Mov Disord. 2020;35(12):2201–10.
Palomba NP, Fortunato G, Pepe G, Modugno N, Pietracupa S, Damiano I, et al. Common and rare variants in TMEM175 gene concur to the pathogenesis of parkinson’s disease in italian patients. Mol Neurobiol. 2023;60(4):2150–73.
Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, et al. SQSTM1 mutations in french patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013;70(11):1403–10.
Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset alS and ALS/dementia. Nature. 2011;477(7363):211–5.
Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trend Genet. 2018;34(6):404–23.
Münch C, Rosenbohm A, Sperfeld A-D, Uttner I, Reske S, Krause BJ, et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58(5):777–80.
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–41.
Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Philtjens S, Heeman B, et al. Loss of TBK1 is a frequent cause of frontotemporal dementia in a Belgian cohort. Neurology. 2015;85(24):2116–25.
Wong TH, Pottier C, Hondius DC, Meeter LHH, van Rooij JGJ, Melhem S, et al. Three VCP mutations in patients with frontotemporal dementia. J Alzheimer Dis. 2018;65:1139–46.
Jun MH, Han JH, Lee YK, Jang DJ, Kaang BK, Lee JA. TMEM106B, a frontotemporal lobar dementia (FTLD) modifier, associates with FTD-3-linked CHMP2B, a complex of ESCRT-III. Mol Brain. 2015;8(1):85.
Geier EG, Bourdenx M, Storm NJ, Cochran JN, Sirkis DW, Hwang J-H, et al. Rare variants in the neuronal ceroid lipofuscinosis gene MFSD8 are candidate risk factors for frontotemporal dementia. Acta Neuropathol. 2019;137(1):71–88.
Mackenzie IRA. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007;114(1):49–54.
Kelleher RJ, Shen J. Presenilin-1 mutations and Alzheimer’s disease. Proc Natl Acad Sci. 2017;114(4):629–31.
Beckers J, Tharkeshwar AK, Van Damme P. C9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy. 2021;17(11):3306–22.
Yang J, Zhang C. Targeting the autophagy-lysosomal pathway in huntington disease: a pharmacological perspective. Front Aging Neurosci. 2023;15:1175598.
Funding
This study was supported by: Fondazione Telethon—Italy (n. GGP19128 to A.P.); Fondazione Cariplo—Italy (n. 2021–1544 to R.C.); Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA)—Italy (n. MLOpathy to A.P.; Target-RAN to A.P.); Association Française contre les Myopathies—France (AFM Telethon n. 23236 to A.P.); Kennedy’s Disease Association—USA (2018 grant to RC; 2020 grant to M.G.); Ministero dell’Università e della Ricerca (MIUR)—Italy [PRIN—Progetti di ricerca di interesse nazionale (n. 2017F2A2C5 to A.P.; n. 2022EFLFL8 to A.P.; n. 2020PBS5MJ to V.C.; n.2022KSJZF5 to V.C.); PRIN—Progetti di ricerca di interesse nazionale—bando 2022, PNRR finanziato dall’Unione europea—Next Generation EU, componente M4C2, investimento 1.1 (n. P2022B5J32 to R.C. and n. P20225R4Y5 to V.C.); CN3: RNA—Codice Proposta: CN_00000041; Tematica Sviluppo di terapia genica e farmaci con tecnologia a RNA (Centro Nazionale di Ricerca—CN3 National Center for Gene Therapy and Drugs based on RNA Technology to A.P.); Progetto Dipartimenti di Eccellenza to Di.S.Fe.B.]; Ministero della Salute, Agenzia Italiana del Farmaco (AIFA)—Italy (Co_ALS to A.P.); Università degli Studi di Milano [piano di sviluppo della ricerca (PSR) UNIMI—linea B to R.C., B.T., and V.F.]; the University of Milan through the APC initiative.
Author information
Authors and Affiliations
Contributions
V.F. and P.R. drafted and supervised the manuscript. A.P. supervised and revised the manuscript. B.T., M.Co., M.Ch., and E.C. performed literature search. P.P., L.C., G.P., A.M., and M.P. prepared the figures and the tables. R.C., M.G., and V.C. revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferrari, V., Tedesco, B., Cozzi, M. et al. Lysosome quality control in health and neurodegenerative diseases. Cell Mol Biol Lett 29, 116 (2024). https://doi.org/10.1186/s11658-024-00633-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s11658-024-00633-2