
Abstract
Background
Laurasiatheria contains taxa with diverse diets, while the molecular basis and evolutionary history underlying their dietary diversification are less clear.
Results
In this study, we used the recently developed molecular phyloecological approach to examine the adaptive evolution of digestive system-related genes across both carnivorous and herbivorous mammals within Laurasiatheria. Our results show an intensified selection of fat and/or protein utilization across all examined carnivorous lineages, which is consistent with their high-protein and high-fat diets. Intriguingly, for herbivorous lineages (ungulates), which have a high-carbohydrate diet, they show a similar selection pattern as that of carnivorous lineages. Our results suggest that for the ungulates, which have a specialized digestive system, the selection intensity of their digestive system-related genes does not necessarily reflect loads of the nutrient components in their diets but appears to be positively related to the loads of the nutrient components that are capable of being directly utilized by the herbivores themselves. Based on these findings, we reconstructed the dietary evolution within Laurasiatheria, and our results reveal the dominant carnivory during the early diversification of Laurasiatheria. In particular, our results suggest that the ancestral bats and the common ancestor of ruminants and cetaceans may be carnivorous as well. We also found evidence of the convergent evolution of one fat utilization-related gene, APOB, across carnivorous taxa.
Conclusions
Our molecular phyloecological results suggest that digestive system-related genes can be used to determine the molecular basis of diet differentiations and to reconstruct ancestral diets.
Similar content being viewed by others

Background
Laurasiatheria includes typical carnivores (e.g., carnivorans and cetaceans) and herbivores (e.g., ungulates). These carnivorous and herbivorous lineages scatter the phylogeny of Laurasiatheria, suggesting the occurrence of dietary transitions among them, while the evolutionary history of their diets remains less clear. For instance, living bats contain both carnivores (e.g., insect-eaters) and herbivores (e.g., fruit-eaters), and the diet of ancestral bats is still unknown, with both insectivory and frugivory having been proposed [1]. Likewise, for typical herbivores, such as odd-toed ungulates and even-toed ungulates, they are deeply nested within several carnivorous lineages, including carnivorans, pangolins, bats, and Eulipotyphla; however, another carnivore, the cetacean, is deeply nested within even-toed ungulates. This may suggest that their diets must have changed; however, the evolutionary history of their diets remains largely unknown, and few relevant studies exist. One previous macroecological study infers the evolutionary history of the diets within mammals, including Laurasiatheria, and a high frequency of their dietary transitions from herbivory and carnivory to omnivory is reported [2], which is a pattern found in birds as well [3]. More studies, especially at the molecular level, may be needed as reconstructing ancestral diets is of importance to understanding the evolutionary origin of the dietarily specialized taxa.
The recent development of a molecular phyloecological (MPE) approach provides a new opportunity to investigate ancestral traits using molecular data [4,5,6,7]. The MPE approach mainly uses the adaptive evolutionary analyses of the molecular markers indicative of trait states to determine the molecular basis of phenotypic differentiation and to infer ancestral traits given a phylogeny, and it has been used to infer the diel activity patterns and diets of ancestral taxa [4,5,6,7,8]. Regarding diet reconstruction, the MPE approach employs digestive system-related genes as the molecular markers indicative of diets to infer ancestral diets. Accordingly, carnivores are characterized by the selection intensification of protein and fat utilization, while herbivores are normally characterized by the selection intensification of carbohydrates [7,8,9,10] because carnivore diets are high in proteins and fats and herbivore diets are normally high in carbohydrates [7, 9,10,11,12,13]. The MPE method has been used to infer ancestral diets in birds [7, 8], and its fitness to mammals remains to be explored. In this study, we used Laurasiatheria as an ideal clade to examine the molecular basis underlying the diet differentiations and to reconstruct their ancestral diets as it contains the taxa with highly specialized diets. Our results provide new insights into understanding their evolutionary history of diets.
Results
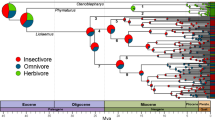
Following the MPE approach to examine the molecular evolution of diets [7, 8], we examined the adaptive evolution of 119 digestive system-related genes (Additional file 2: Table S1) in the given Laurasiatheria phylogeny (Fig. 1). These genes are involved in three KEGG pathways, and play important roles in the digestion and absorption of carbohydrates, proteins, and fats [7] (Fig. 2). The positive selection of these genes along particular branches (A–L in Fig. 1) was analyzed using branch and branch-site models implemented in PAML software [14]. Positively selected genes (PSGs) were mainly detected based on the branch-site model (Additional file 3: Table S2). We initially analyzed the positive selection along the lineages with highly specialized diets, including three primarily carnivorous lineages (Eulipotyphla, Pholidota, and Cetacea) and one typically herbivorous lineage (Ruminantia). Intriguingly, our results reveal a highly similar selection pattern across the four lineages and show the predominant selection of fat and protein utilization with relatively the weakest selection of carbohydrate utilization in terms of both the p values and the number of PSGs found. These results remain unchanged even after the Bonferroni multiple testing correction of the p values of PSGs (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4).

Laurasiatheria phylogeny and reconstructed ancestral dietary categories based on molecular data. The phylogenetic relationships among species follow published studies [94,95,96]. The branches under positive selection analyses are shown with letters (A–L). The dietary categories of each extant species and each mammalian order shown in the pet charts are based on one previous study [93]. Carnivores are shown in red, herbivores in green, and omnivores in violet. Black shows the species with no dietary categories available

Digestive system pathways and positively selected genes found in ruminants (green) and cetaceans (red). Three digestive system pathways (A, B, and C) were modified from that of KEGG, including carbohydrate digestion and absorption (map04973), protein digestion and absorption (map04974), and fat digestion and absorption (map04975). Positively selected genes are shown in parentheses, and their corresponding proteins are highlighted in red (cetaceans) and green (ruminants)
Eulipotyphla primarily eats invertebrate prey, and we found 10 PSGs along the branch leading to Eulipotyphla (branch L, Fig. 1), including four fat utilization-related genes (APOB, APOA1, LIPF, and NPC1L1), four protein utilization-related genes (MEP1B, CTRL, SLC3A2, and CPA1), and two carbohydrate utilization-related genes (HK1 and MGAM) (Additional files 3, 4, 5: Tables S2-4). Among these PSGs, APOB and APOA1 encode key apolipoproteins responsive to carrying fats and fat-like substances in the blood [15, 16]. LIPF encodes a gastric lipase, which plays an important role in the digestion of dietary triglycerides in the gastrointestinal tract [17]. NPC1L1 is responsible for the intestinal absorption of cholesterol and/or plant sterols [18]. MEP1B encodes metalloendopeptidases [19]. CTRL is considered to play a role in the digestion of proteins [20]. SLC3A2 encodes an amino acid transporter [21]. CPA1 encodes a pancreatic exopeptidase [22]. HK1 encodes hexokinase 1, which catalyzes the first step in glucose metabolism [23]. MGAM encodes maltase-glucoamylase, which is involved in the small intestinal digestion of starch to glucose [24].
Pholidota eats almost exclusively ants and termites. Our positive selection analyses along the Pholidota branch (branch J, Fig. 1) revealed 16 PSGs (Additional file 3: Table S2), including six fat utilization-related genes (APOB, PLPP2, APOA1, SLC27A4, PLA2G1B, and CLPS), six protein utilization-related genes (SLC8A2, SLC7A9, SLC3A1, DPP4, KCNN4, and SLC3A2), and four carbohydrate utilization-related genes (MGAM2, HK3, G6PC, and LCT). PLPP2 functions in phospholipid metabolism by converting phosphatidic acid to diacylglycerol [25]. SLC27A4 is known as an important fatty acid transporter in small intestinal enterocytes [26]. PLA2G1B encodes phospholipase A2 and catalyzes the release of fatty acids from glycero-3-phosphocholines [27]. CLPS encodes a pancreatic colipase [28]. SLC8A2 encodes a Na+-Ca2+ exchanger, which is widely expressed in different tissues [29]. SLC7A9 is involved in amino acid transport [30]. SLC3A1 encodes an amino acid transporter [31]. DPP4 codes a cell-surface protease [32]. KCNN4 codes for the calcium-activated potassium channels [33]. MGAM2 is involved in the degradation of starch or glycogen and is highly expressed in the small and large intestines [34]. HK3 is involved in glucose metabolism [35]. G6PC plays an important role in the homeostasis regulation of blood glucose concentrations, catalyzing the terminal step in gluconeogenesis and glycogenolysis [36]. LCT encodes a molecule with both lactase activity and phlorizin hydrolase activity [37].
Cetaceans are primarily carnivores, feeding on invertebrates and vertebrate prey. Our positive selection analyses detected six PSGs along the ancestral branch of Cetacea (branch F, Fig. 1), including four fat utilization-related genes (APOB, PNLIPRP2, PLA2G5, and SCARB1), one protein utilization-related gene (CPA3), and one carbohydrate utilization-related gene (SI) (Fig. 2; Additional file 3: Table S2). PNLIPRP2 is a pancreatic lipase-related protein [38]. PLA2G5 is a member of the phospholipase A2 gene family and plays a role in the hydrolysis of phospholipids [39]. SCARB1 mediates the uptake of cholesterol and a variety of lipids [40]. CPA3 is involved in the degradation of proteins [41]. SI encodes sucrase-isomerase and is essential for the digestion of dietary carbohydrates including starch, sucrose, and isomaltose [42].
Ruminantia is typically herbivorous. Our positive selection analyses revealed 12 PSGs along the ancestral branch leading to Ruminantia (branch G, Fig. 1), including four fat utilization-related genes (APOB, MOGAT2, MTTP, and FABP1), five protein utilization-related genes (SLC36A1, SLC6A19, SLC1A5, SLC7A8, and SLC15A1), one carbohydrate utilization-related gene (PRKCB), and two ionic homeostasis-related genes (ATP1B1 and ATP1B3) [43] involved in both protein and carbohydrate utilization (Fig. 2; Additional file 3: Table S2). MOGAT2 plays a role in dietary fat absorption from the small intestine [44]. MTTP catalyzes the transport of triglycerides, cholesteryl esters, and phospholipids [45]. FABP1 encodes a fatty acid-binding protein that regulates lipid transport and metabolism [46]. SLC36A1, SLC6A19, SLC1A5, and SLC7A8 mediate the transport of amino acids [47,48,49,50]. SLC15A1 encodes an intestinal transporter of peptides [51]. PRKCB encodes a protein kinase involved in many different cellular functions, including intestinal sugar absorption [52].
We also analyzed the positive selection of the digestive system-related genes in Chiroptera and Carnivora, both of which harbor dietary diverse species. Chiroptera contains both carnivores (e.g., insect-eaters) and herbivores (e.g., fruit-eaters), and our positive selection analyses along the ancestral Chiroptera branch (branch K, Fig. 1) revealed eight PSGs (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4). These eight PSGs include only protein utilization-related genes (SLC3A2, SLC1A5, CELA3B, and DPP4) and fat utilization-related genes (APOB, CD36, ABCG8, and PLPP2). CELA3B is a pancreatic serine proteinase that digests dietary protein substrates [53]. CD36 is mainly involved in the uptake and processing of fatty acids [54]. ABCG8 functions in the excretion of neutral sterols in the liver and intestines [55]. Like Chiroptera, for the ancestral branch of Carnivora (branch H, Fig. 1), only fat utilization-related genes (APOB and PIK3CD) and protein utilization-related genes (CPB2 and KCNK5) were found to be under positive selection (Additional file 3: Table S2). PIK3CD encodes phosphatidylinositol 3-kinase with a broad phosphoinositide lipid substrate specificity [56]. CPB2 encodes carboxypeptidase B2, cleaving C-terminal residues from peptides [57]. KCNK5 is considered to play an important role in potassium transport [58].
To determine the selection characterization of ancestral taxa, we subsequently examined the positive selection of the digestive system-related genes along other early branches of Laurasiatheria (branches A, B, C, D, E and I, Fig. 1) (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4). For the ancestral Laurasiatheria branch (branch A, Fig. 1), we found two fat utilization-related genes (APOB and NPC1L1), one protein utilization-related gene (MEP1B), and one glucose metabolism-related gene (HKDC1) [59] to be under positive selection. For branch B, only one fat utilization-related gene, AGPAT2, was found to be under positive selection. It plays a role in converting lysophosphatidic acid into phosphatidic acid [60]. For branch C, three PSGs (SLC7A8, MGAM2, and ATP1B3) were found. For branch I, one positively selected fat utilization-related gene, APOB, was detected. For branch D, two fat utilization-related genes (APOB and PLA2G2D) were found to be under positive selection, and notably, PLA2G2D is a member of lipolytic enzyme [61]. For branch E, two fat utilization-related genes (LIPF and PNLIP) and two protein utilization-related genes (SLC3A2 and MEP1B) were found to be under positive selection, of which PNLIP encodes a pancreatic lipase, also known as pancreatic triacylglycerol lipase. This pancreatic lipase hydrolyzes dietary triglycerides to free fatty acids and monoacylglycerols and is critical for the efficient digestion of dietary triglycerides in the intestines [62, 63].
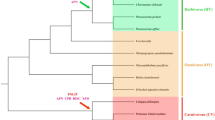
Among the PSGs found, one gene APOB showed a particularly strong positive selection with hundreds of positively selected amino acid sites found in most taxa examined, including those carnivorous taxa (Additional file 3: Table S2). To test whether the APOB gene was subject to convergent evolution among the carnivorous taxa, we subsequently examined the convergent and/or parallel amino acid substitutions along the branches related to those carnivores by reconstructing ancestral sequences using PAML [14], and many parallel amino acid substitutions were detected among them with high statistical significance (Additional file 1: Fig. S1; Additional file 6: Table S5). For instance, nine parallel substitutions each were found between the branches of Eulipotyphla and Pholidota, and between the branches of Chiroptera and Pholidota. Eight parallel substitutions were found between the branch of Carnivora and two other branches of Eulipotyphla and Chiroptera. These parallel amino acid substitutions may have led to their sequence convergence and thus to their phylogenetic affinity. To test this, we then constructed the maximum likelihood phylogeny based on the protein sequence of the gene APOB. Intriguingly, our results showed that the APOB tree (Fig. 3) was largely different from their species tree (Fig. 1). In particular, we found that four carnivory-dominant taxa (Carnivora, Pholidota, Chiroptera, and Eulipotyphla) were grouped into the same clade with bootstrap values ranging from 39 to 43 upon three independent runs, indicating their sequence convergence.

Maximum-likelihood phylogeny of the gene APOB. The phylogeny is based on 4550 amino-acid sites with the best-fit substitution model of the HIVb+F+R4 used. Red shows the clustering of four carnivory-dominant taxa
Discussion
We in this study examined the adaptive evolution of digestive system-related genes to determine the diet evolution within Laurasiatheria. Consistent with previous studies that demonstrate the evolutionary enhancement of protein and fat utilization in carnivores [7, 9, 10], all three primarily carnivorous mammal taxa (Eulipotyphla, Pholidota, and Cetacea) examined in this study showed a particularly intensified selection of fat and protein utilization with relatively the weakest positive selection of carbohydrate utilization (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4). This is consistent with their high-protein and high-fat diets. Unexpectedly, for the typical herbivores, the ruminants, which have a high-carbohydrate diet, we detected an intensified selection of fat and protein utilization rather than carbohydrate utilization (Additional file 3: Table S2), resembling that of carnivores. This may suggest that convergent evolution may occur between the carnivores and the herbivores studied. The convergent evolution of diet-related genes is often considered to be resulted from the utilization of similar food [12, 64,65,66], while its occurrence in the ruminants, as evidenced previously [67,68,69], may largely attribute to their specialized digestive system rather than similar food. As we know, ruminants primarily consume plant materials rich in carbohydrates, but they have no enzymes to digest the refractory materials (e.g., cellulose) in their diets. These refractory materials are transferred through microbial fermentation in their guts to volatile fatty acids and microbe proteins, constituting the major sources of energy and amino acids for subsequent utilization by ruminants [70,71,72,73]. This suggests that though their diets are rich in carbohydrates, the main nutritional substrates that ruminants are capable of directly utilizing are actually fats and proteins that are generated through microbial fermentation. Therefore, the intensified selection of fat and protein utilization found in the ruminants may be mainly due to their specialized digestive system.
Our results show that the carnivorous mammals studied are consistently characterized by an intensified selection of fat and protein utilization, which is in line with their high-protein and high-fat diets. For herbivorous mammals, because fermentation through their gut microbes can transfer dietary carbohydrates to other nutritional substrates, such as volatile fatty acids and microbial proteins, for their subsequent use [70,71,72,73], the selection characterization of their digestive system-related genes do not necessarily reflect the amounts of nutritional substrates in their diets. Previous studies suggest that microbial fermentation widely occurs in animals, while the contribution of microbial fermentation to energy production is largely different among animals. Its importance seems to be limited to particular taxa (e.g., ungulates), possibly due to high amounts of refractory materials (e.g., cellulose) in their diets, but is relatively trivial to other animals [74]. Consequently, the adaptive evolution of digestive system–related genes of animals is considered to be generally positively related to loads of their dietary substrates [10, 12, 13, 74,75,76]. Accordingly, carnivores are characterized by the selection intensification of protein and fat utilization, while herbivores are normally characterized by the selection intensification of carbohydrates [7, 9, 10]. Thus, we could reconstruct the diets of ancestral taxa based on the selection characterization of digestive system-related genes [7, 8].
To determine ancestral diets, we analyzed the positive selection of the digestive system-related genes along the ancestral branches of the living animals studied (Fig. 1). For the ancestral branches of bats (branch K) and of carnivorans (branch H), we detected their evolutionary enhancements mainly in fat and protein utilization, as found in the ancestral branches leading to other primarily carnivorous mammals (Eulipotyphla, Pholidota, and Cetacea) (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4). This may suggest that all the ancestral taxa including ancestral bats and ancestral carnivorans were largely carnivorous (Fig. 1). Similarly, an evolutionary enhancement of fat and/or protein utilization was also found in other ancestral branches (branches A, B, and I) (Additional file 3: Table S2). This may suggest that the early evolutionary diversification of Laurasiatheria was mainly characterized by carnivory (Fig. 1), which is largely consistent with one previous study [2]. Nonetheless, for the ungulates examined in this study, we unexpectedly found an intensified selection of fat utilization along branch D, leading to ancestral ungulates, and an evolutionary enhancement of fat and protein utilization along branch G, leading to ruminants (Additional file 3: Table S2). This may reflect their high fat and/or protein nutrition generated by nutritional transformation through microbial fermentation, and hence it may suggest that the ancestral ungulate and the ancestral ruminant may be herbivorous (Fig. 1). For branch C, we detected the positive selection of three PSGs involved in carbohydrate and protein utilization (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4), suggesting a high-carbohydrate and high-protein diet. A high-carbohydrate and high-protein diet may suggest a combination of herbivory and carnivory, hence implying that the common ancestor of carnivorans and ungulates was possibly omnivorous (Fig. 1), which may be derived from its carnivorous progenitor (branch B). If this is the case, it may suggest that the herbivory of ungulates and the carnivory of carnivorans were secondarily evolved. This is consistent with fossil evidence showing that the earliest stem carnivorans, such as Ravenictis and Pristinictis, exhibit relatively unspecialized molars, indicating an omnivorous diet with only limited specialization to true carnivory [77]. Fossil evidence indicates the resemblance of some primitive ungulates to carnivores. For instance, Phenacodus, which is considered a stem Perissodactyla [78], lived during late Palaeocene and early Eocene, and was a plant-eater yet shows some characteristics (e.g., large canine teeth) resembling a primitive carnivore [79]. These lines of evidence may suggest the herbivory of the ungulates, and the pure carnivory found in modern carnivorans may be secondarily evolved from an omnivorous ancestor.
For the branch leading to the common ancestor of ruminants and cetaceans (CARC), we detected the enhanced selection of fat and protein utilization (Additional file 3: Table S2), which is similar to that found in both of its two derived taxa, the ruminants and the cetaceans. This seems to make the reconstruction of the diet of the CARC unresolved; however, our finding of the positive selection of the two fat digestion-related genes (PNLIP and LIPF) along the branch leading to the CARC (Additional file 3: Table S2) may suggest that the CARC was more likely carnivorous (Fig. 1). This is because: i) PNLIP and LIPF are both critical lipases mainly involved in digesting dietary triglycerides in the digestive system [17, 62, 63, 80], and the selection enhancement of the digestion of dietary triglycerides may suggest a lipid-rich diet of the CARC. A lipid-rich diet most often characterizes carnivores rather than herbivores because carnivore diets are relatively rich in lipids, while herbivore diets are normally rich in carbohydrates [9,10,11,12,13]. ii) The evolutionary enhancement of digesting dietary triglycerides found in the CARC may suggest that the CARC itself may have the capability to digest dietary fats (e.g., triglycerides). This is consistent with carnivorous mammals (e.g., cetaceans), which normally use their own lipases to digest fats [81], but is substantially different from ruminants, from which their dietary lipids (e.g., triglycerides) are predominantly hydrolyzed by the lipases of rumen bacteria in their guts [82,83,84,85]. iii) The detected positive selection of PNLIP and LIPF in the CARC has been found in carnivores (e.g., cetaceans) [68, 76] but not in ruminants, which is evidenced in this study and one previous study [68], suggesting the resemblance of the digestion ability of the CARC with that of carnivores (cetaceans). In addition to the molecular evidence, fossil evidence shows that early ruminant ancestors were omnivores and did not ruminate until about 40 Ma based on dental morphology [86]. This indicates that the herbivory and rumination observed in modern ruminants may be secondarily evolved, which is consistent with the possible carnivory of the CARC. These four lines of evidence may suggest that the CARC was more likely a carnivore, though its existence in fossils remains to be explored. Considering that the CARC is phylogenetically deeply nested within the ungulates, it is thus more likely a carnivorous ungulate closely related to cetaceans and/or ruminants. Among carnivorous mammals known, previous studies have long considered one extinct carnivorous ungulate, mesonychians, as early members of cetaceans or Cetartiodactyla [87,88,89,90,91], though there is uncertainty regarding the phylogenetic position of mesonychians [77, 92]. Mesonychians are considered secondary carnivores derived from archaic ungulates (Condylarthra) [88]. If this is the case, mesonychians might be the candidate of the CARC from which cetaceans and ruminants derived, though this requires further investigation.
Conclusions
Our molecular phyloecological results show that the carnivorous mammals consistently exhibit the evolutionary enhancement of fat and protein utilization, which is in line with their high-protein and high-fat diets. This is previously found in birds and crabs as well. For herbivores, previous studies on birds and crabs suggest that they tend to show an evolutionary enhancement of carbohydrates; however, the ungulates with a high-carbohydrate diet examined in this study present an evolutionary enhancement of fat and protein utilization, resembling that of carnivores. Apparently, this is largely due to their specialized digestive system that transfers abundant carbohydrates to volatile fatty acids and microbial proteins for their use. Our results suggest that the adaptive evolution of digestive system-related genes do not necessarily reflect the nutritional loads in the diets of the herbivorous animals (e.g., ungulates) that mainly rely on nutritional transformation before utilization but appear to be positively related to the loads of the nutrient substrates that can be directly utilized by the herbivores themselves. Based on these findings, we reconstructed ancestral diets, and our results revealed predominant carnivory during the early diversification of Laurasiatheria. More importantly, our reconstructed results suggest that the ungulates and carnivorans may have been derived from an omnivorous ancestor, and ancestral bats and the common ancestor of ruminants and cetaceans may be carnivorous. We also found evidence of the convergent evolution of one fat utilization-related gene, APOB, across carnivorous lineages, suggesting the resemblance of nutritional utilization in carnivorous mammals. Further studies incorporating information about the gene duplications and losses besides positive selection may be helpful to understand the molecular bases underlying the diet evolution of the herbivorous and carnivorous mammals.
Materials and methods
Taxa used
Ninety species within Laurasiatheria were included (Fig. 1). These 90 species covered all known main clades of Laurasiatheria, including four species of Eulipotyphla, two pangolin species of Pholidota, three species of odd-toed ungulates (Perissodactyla), and 14 species of even-toed ungulates (Artiodactyla), of which nine species belong to Ruminantia. We also included 25 bat species from both two suborders (Yangochiroptera and Yinpterochiroptera) of Chiroptera. For Carnivora, 28 species from its two suborders (Feliformia and Caniformia) were included. For Cetacea, 14 species from its two suborders (Mysticeti and Odontoceti) were included. In addition to these 90 Laurasiatheria species, we included two species from the sister taxa (Euarchontoglires) of Laurasiatheria as outgroups. For the two outgroup species, Homo sapiens and Mus musculus were primarily used, while the two relatives (Rattus rattus and Rattus norvegicus) of Mus musculus were also considered if some gene sequences of Mus musculus were unavailable.
Diet data
The dietary categories of each species used in this study were based on one previously published dataset, EltonTraits 1.0 [93], in which the dietary information of a total of 5400 extant mammal species from diverse published literature is compiled and the dietary composition of each species is recorded in 10% dietary categories. To determine the dietary categories of the species used in our study, we converted EltonTraits’ 10% dietary categories into our three dietary categories (carnivore, herbivore, and omnivore). Carnivore = Diet-Inv + Diet-Vend + Diet-Vect + Diet-Vfish + Diet-Vunk + Diet-Scav, herbivore = Diet-Fruit + Diet-Nect + Diet-Seed + Diet-PlantO, and omnivores were referred to as the animals that contain a percentage of dietary categories of both the carnivore and the herbivore.
Genes and sequence alignment
We included the digestive system-related genes that have been recently used to determine the diet evolution in birds [7]. These genes were from three KEGG pathways, including carbohydrate digestion and absorption (map04973), protein digestion and absorption (map04974), and fat digestion and absorption (map04975). For these genes, we downloaded their coding sequences of our focal species from GenBank (Additional file 2: Table S1). We excluded genes with sequences unavailable or available for only a few species from our analyses, and ultimately, 119 genes were retained for subsequent analyses. We aligned gene sequences using webPRANK with default parameters (http://www.ebi.ac.uk/goldman-srv/webprank/), and individual species sequences with lengths that were too short were removed. The sequence alignments were checked by eye and the sequence gaps that lead to incorrect protein translation were cut. After this pruning, we blasted the translated protein sequences of these genes against the non-redundant protein sequence database to confirm the correctness of the sequence cutting.
Positive selection analyses
For the positive selection analyses, we initially constructed a Laurasiatheria phylogeny of the 90 species used in this study, as shown in Fig. 1. Our Laurasiatheria phylogeny was based on published studies [94,95,96]. In particular, the phylogenetic relationships used among taxonomical orders within Laurasiatheria, which have received increasing support for the past 20 years [95,96,97,98], are the same as those used in one previous diet study of mammals [2]. Based on the Laurasiatheria phylogeny, we analyzed the positive selection of our target genes using branch and branch-site models implemented in the Codeml program of PAML [14]. The ratio of non-synonymous to synonymous substitutions per site (dN/dS or ω) was evaluated, and likelihood ratio tests (LRT) were employed to determine the statistical significance. Positive selection is determined by the value of ω > 1 with statistical significance. The Bonferroni multiple testing correction was used to adjust the p values.
Branch model Branch model allows for the variation of the ω ratio among branches in a given phylogeny, and it is used to detect the positive selection of genes on a particular branch. For the branch model analyses, we used a two-rate branch model by labeling our focal branches as foreground branches and the others as background branches. During the analyses, the goodness of fit of the two-rate branch model relative to the null model—that is one-rate branch model— was analyzed using the LRT. When a statistically significant value of ω > 1 was found in our foreground branches, to determine whether the value of ω > 1 of the foreground branch was further supported with statistical significance, we then compared the two-ratio branch model with the two-ratio branch model with ω = 1 fixed in our foreground branches.
Branch-site model The branch-site model allows for the variation of ω among sites in the protein and across phylogenetic branches, and it is used to detect positive selection affecting some sites along a particular branch. For the branch-site model analyses, we employed a branch-site test of positive selection (Test 2), which compares a modified model A with its corresponding null model with ω = 1 fixed. The modified model A assumes four classes of sites, and site class 0 and site class 1, respectively, represent evolutionarily conserved (0 < ω0 < 1) and neutral codons (ω1 = 1) for both background and foreground branches. Site classes 2a and 2b, respectively, represent evolutionarily conserved (0 < ω0 < 1) and neutral (ω1 = 1) codons for background branches, but allowed to be under positive selection (ω2 > 1) for the foreground branches. The goodness of fit of the modified model A was evaluated using the LRT by comparing it with a null model with ω = 1 fixed. Positively selected sites were identified by employing an empirical Bayes method.
Ancestral sequence reconstruction and convergent evolution analyses
Amino acid-based marginal reconstruction implemented in the empirical Bayes approach in PAML [14] was used to reconstruct the ancestral sequence. For the marginal reconstruction, we employed two different substitution models (JTT and Poisson) of amino acids to examine the consistency of our results. For the model JTT, different substitution rates of different amino acids were assumed, and for the Poisson model, the same substitution rate of all amino acids was assumed. The analyses based on the two substitution models generated similar results, and for convenience, we only showed the results based on the JTT model. Based on the reconstructed ancestral sequences of internal nodes, convergent and/or parallel amino acid substitutions along branches could then be identified. To further estimate the probabilities that the observed convergent and/or parallel substitutions are attributable to random chance, the program converg2 implemented in the software Convergent and Parallel Evolution at the Amino Acid Sequence Level (CAPE) [99] was used.
Phylogenetic analyses
Phylogenetic analyses were conducted using the IQ-TREE, a fast and effective stochastic algorithm for inferring maximum-likelihood (ML) phylogeny [100, 101]. The IQ-TREE is considered to have high performance for ML inference compared to other popular software, such as RAxML [102] and PhyML [103]. This is considered to result from its efficient integration of fast model selection, an effective tree search algorithm, and a novel ultrafast bootstrap approximation [100]. Especially, the effective tree search algorithm was believed to overcome the problem of local optima and thus to help to achieve ML phylogeny with higher likelihoods than RAxML or PhyML. For our phylogenetic analyses, 4550 amino-acid sites of the gene APOB were used. Among 546 protein models examined by ModelFinder implemented in the IQ-TREE, two models, HIVb+F+R4 and HIVb+F+R5, were recommended as the best-fit substitution models according to Bayesian and Akaike information criteria, respectively. For result robustness, the two substitution models were both used and almost identical results were obtained. For convenience, only the ML phylogeny based on the substitution model of the HIVb+F+R4 was presented with the bootstrap value of 10,000 used.
Availability of data and materials
The sequence alignment and tree file of each gene used in this study are deposited in the Dryad repository (doi:https://doi.org/10.5061/dryad.3tx95x6gz).
Abbreviations
- CAPE:
-
Convergent and parallel evolution at the amino acid sequence level
- CARC:
-
Common ancestor of ruminants and cetaceans
- JTT:
-
Jones–Taylor–Thornton
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LRT:
-
Likelihood ratio tests
- ML:
-
Maximum-likelihood
- MPE:
-
Molecular phyloecological
- PAML:
-
Phylogenetic analysis by maximum likelihood
- PhyML:
-
Phylogenetic estimation using maximum likelihood
- PSGs:
-
Positively selected genes
- RAxML:
-
Randomized axelerated maximum likelihood
References
Speakman JR. The evolution of flight and echolocation in bats: another leap in the dark. Mamm Rev. 2001; 31:111–130.
Price SA, Hopkins SSB, Smith KK, Roth VL. Tempo of trophic evolution and its impact on mammalian diversification. Proc Natl Acad Sci USA. 2012; 109:7008–7012.
Burin G, Kissling WD, Guimarães PR, Şekercioğlu ÇH, Quental TB. Omnivory in birds is a macroevolutionary sink. Nat Comm. 2016; 7:11250.
Wu Y, Wang H. Convergent evolution of bird-mammal shared characteristics for adapting to nocturnality. Proc R Soc B. 2019;286:20182185.
Wu Y, Wang H, Wang H, Feng J. Arms race of temporal partitioning between carnivorous and herbivorous mammals. Sci Rep. 2018; 8:1713.
Wu Y, Wang H, Hadly EA. Invasion of ancestral mammals into dim-light environments inferred from adaptive evolution of the phototransduction genes. Sci Rep. 2017; 7:46542.
Wu Y. Molecular phyloecology suggests a trophic shift concurrent with the evolution of the first birds. Commun Biol. 2021; 4:547.
Wu Y, Yan Y, Zhao Y, Gu L, Wang S, Johnson DH. Genomic bases underlying the adaptive radiation of core landbirds. BMC Evol Biol. 2021; 21:162.
Chen Y-H, Zhao H. Evolution of digestive enzymes and dietary diversification in birds. PeerJ. 2019; 7:e6840.
Wang Z, Tang D, Guo H, Shen C, Wu L, Luo Y. Evolution of digestive enzyme genes associated with dietary diversity of crabs. Genetica. 2020; 148:87–99.
Clinic M. Encyclopedia of foods: a guide to healthy nutrition. San Diego: Academic Press; 2002.
Hecker N, Sharma V, Hiller M. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. Proc Natl Acad Sci USA. 2019; 116:3036–3041.
Karasov WH, del Rio MC, Caviedes-Vidal E. Ecological physiology of diet and digestive systems. Annu Rev Physiol. 2011; 73:69–93.
Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007; 24:1586–1591.
Mangaraj M, Nanda R, Panda S. Apolipoprotein A-I: a molecule of diverse function. Indian J Clin Biochem. 2016; 31:253–259.
Qu J, Ko C-W, Tso P, Bhargava A. Apolipoprotein A-IV: a multifunctional protein involved in protection against atherosclerosis and diabetes. Cells. 2019; 8:319.
Bodmer MW, Angal S, Yarranton GT, Harris TJ, Lyons A, King DJ, Pieroni G, Riviere C, Verger R, Lowe PA. Molecular cloning of a human gastric lipase and expression of the enzyme in yeast. Biochim Biophys Acta Gene Struct Expr. 1987; 909:237–244.
Izar MC, Tegani DM, Kasmas SH, Fonseca FA. Phytosterols and phytosterolemia: gene–diet interactions. Gene Nutr. 2011; 6:17–26.
Crisman JM, Zhang B, Norman LP, Bond JS. Deletion of the mouse meprin β metalloprotease gene diminishes the ability of leukocytes to disseminate through extracellular matrix. J Immunol. 2004; 172:4510–4519.
Reseland JE, Larsen F, Solheim J, Eriksen JA, Hanssen LE, Prydz H. A novel human chymotrypsin-like digestive enzyme. J Biol Chem. 1997; 272:8099–8104.
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999; 274:11455–11458.
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995; 270:484–487.
Bianchi M, Crinelli R, Serafini G, Giammarini C, Magnani M. Molecular bases of hexokinase deficiency. Biochim Biophys Acta Mol Basis Dis. 1997; 1360:211–221.
Naim HY, Sterchi EE, Lentze MJ. Structure, biosynthesis, and glycosylation of human small intestinal maltase-glucoamylase. J Biol Chem. 1988; 263:19709–19717.
Hooks SB, Ragan SP, Lynch KR. Identification of a novel human phosphatidic acid phosphatase type 2 isoform. FEBS Lett. 1998; 427:188–192.
Stahl A, Hirsch DJ, Gimeno RE, Punreddy S, Ge P, Watson N, Patel S, Kotler M, Raimondi A, Tartaglia LA et al. Identification of the major intestinal fatty acid transport protein. Mol Cell. 1999; 4:299–308.
Cash JG, Kuhel DG, Goodin C, Hui DY. Pancreatic acinar cell-specific overexpression of group 1B phospholipase A 2 exacerbates diet-induced obesity and insulin resistance in mice. Int J Obes. 2011; 35:877–881.
Davis RC, Xia Y, Mohandas T, Schotz MC, Lusis AJ. Assignment of the human pancreatic colipase gene to chromosome 6p21. 1 to pter. Genomics. 1991; 10:262–265.
Li Z, Matsuoka S, Hryshko LV, Nicoll DA, Bersohn MM, Burke EP, Lifton RP, Philipson KD. Cloning of the NCX2 isoform of the plasma membrane Na+-Ca2+ exchanger. J Biol Chem. 1994; 269:17434–17439.
Font M, Feliubadaló Ld, Estivill X, Nunes V, Golomb E, Kreiss Y, Pras E, Bisceglia L, d’Adamo AP, Zelante L et al. Functional analysis of mutations in SLC7A9, and genotype–phenotype correlation in non-Type I cystinuria. Hum Mol Genet. 2001; 10:305–316.
Pras E, Raben N, Golomb E, Arber N, Aksentijevich I, Schapiro JM, Harel D, Katz G, Liberman U, Pras M et al. Mutations in the SLC3A1 transporter gene in cystinuria. Am J Hum Genet. 1995; 56:1297–1303.
Lambeir A-M, Durinx C, Scharpé S, De Meester I. Dipeptidyl-peptidase IV from bench to bedside: an update on structural properties, functions, and clinical aspects of the enzyme DPP IV. Crit Rev Clin Lab Sci. 2003; 40:209–294.
Joiner WJ, Wang L-Y, Tang MD, Kaczmarek LK. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc Natl Acad Sci USA. 1997; 94:11013–11018.
Xiang R, Oddy VH, Archibald AL, Vercoe PE, Dalrymple BP. Epithelial, metabolic and innate immunity transcriptomic signatures differentiating the rumen from other sheep and mammalian gastrointestinal tract tissues. PeerJ. 2016; 4:e1762.
Furuta H, Nishi S, Le Beau MM, Fernald AA, Yano H, Bell GI. Sequence of human hexokinase III cDNA and assignment of the human hexokinase III gene (HK3) to chromosome band 5q35.2 by fluorescence in situ hybridization. Genomics. 1996; 36:206–209.
Lei K-J, Shelly LL, Pan C-J, Sidbury JB, Chou JY. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science. 1993; 262:580–583.
Boll W, Wagner P, Mantei N. Structure of the chromosomal gene and cDNAs coding for lactase-phlorizin hydrolase in humans with adult-type hypolactasia or persistence of lactase. Am J Hum Genet. 1991; 48:889–902.
Giller T, Buchwald P, Blum-Kaelin D, Hunziker W. Two novel human pancreatic lipase related proteins, hPLRP1 and hPLRP2. Differences in colipase dependence and in lipase activity. J Biol Chem. 1992; 267:16509–16516.
Chen J, Engle SJ, Seilhamer JJ, Tischfield JA. Cloning and recombinant expression of a novel human low molecular weight Ca (2+)-dependent phospholipase A2. J Biol Chem. 1994; 269:2365–2368.
Shen W-J, Azhar S, Kraemer FB. SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. 2018; 80:95–116.
Reynolds DS, Gurley DS, Austen KF. Cloning and characterization of the novel gene for mast cell carboxypeptidase A. J Clin Investig. 1992;89:273–82.
Naim HY, Sterchi EE, Lentze MJ. Biosynthesis of the human sucrase-isomaltase complex. Differential O-glycosylation of the sucrase subunit correlates with its position within the enzyme complex. J Biol Chem. 1988; 263:7242–7253.
Li Y, Yang J, Li S, Zhang J, Zheng J, Hou W, Zhao H, Guo Y, Liu X, Dou K et al. N-myc downstream-regulated gene 2, a novel estrogen-targeted gene, is involved in the regulation of Na+/K+-ATPase. J Biol Chem. 2011; 286:32289–32299.
Yen C-LE, Farese RV. MGAT2, a monoacylglycerol acyltransferase expressed in the small intestine. J Biol Chem. 2003; 278:18532–18537.
Hussain MM, Rava P, Walsh M, Rana M, Iqbal J. Multiple functions of microsomal triglyceride transfer protein. Nutr Metabol. 2012; 9:14.
Wang G, Bonkovsky HL, de Lemos A, Burczynski FJ. Recent insights into the biological functions of liver fatty acid binding protein 1. J Lipid Res. 2015; 56:2238–2247.
Bröer A, Klingel K, Kowalczuk S, Rasko JEJ, Cavanaugh J, Bröer S. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J Biol Chem. 2004; 279:24467–24476.
Frølund S, Holm R, Brodin B, Nielsen CU. The proton-coupled amino acid transporter, SLC36A1 (hPAT1), transports Gly‐Gly, Gly‐Sar and other Gly‐Gly mimetics. Br J Pharmacol. 2010; 161:589–600.
Larriba S, Sumoy L, Ramos MD, Giménez J, Estivill X, Casals T, Nunes V. ATB0/SLC1A5 gene. Fine localisation and exclusion of association with the intestinal phenotype of cystic fibrosis. Eur J Hum Genet. 2001; 9:860-866.
Pineda M, Fernández E, Torrents D, Estévez R, López C, Camps M, Lloberas J, Zorzano A, Palacín M. Identification of a membrane protein, LAT-2, that co-expresses with 4F2 heavy chain, an L-type amino acid transport activity with broad specificity for small and large zwitterionic amino acids. J Biol Chem. 1999; 274:19738–19744.
Liang R, Fei Y-J, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, Hediger MA, Ganapathy V, Leibach FH. Human intestinal H+/peptide cotransporter cloning, functional expression, and chromosomal localization. J Biol Chem. 1995; 270:6456–6463.
Sheng Y-J, Gao J-P, Li J, Han J-W, Xu Q, Hu W-L, Pan T-M, Cheng Y-L, Yu Z-Y, Ni C et al. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11. 21 for systemic lupus erythematosus. Rheumatology. 2011; 50:682–688.
Szabó A, Pilsak C, Bence M, Witt H, Sahin-Tóth M. Complex formation of human proelastases with procarboxypeptidases A1 and A2. J Biol Chem. 2016; 291:17706–17716.
Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014; 34:281–303.
Lee J-Y, Kinch LN, Borek DM, Wang J, Wang J, Urbatsch IL, Xie X-S, Grishin NV, Cohen JC, Otwinowski Z. Crystal structure of the human sterol transporter ABCG5/ABCG8. Nature. 2016; 533:561–564.
Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD. P110δ, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997; 94:4330–4335.
Zhao L, Morser J, Bajzar L, Nesheim M, Nagashima M. Identification and characterization of two thrombin-activatable fibrinolysis inhibitor isoforms. Thromb Haemost. 1998; 80:949–955.
Bittner S, Bobak N, Herrmann AM, Göbel K, Meuth P, Höhn KG, Stenner MP, Budde T, Wiendl H, Meuth SG. Upregulation of K2P5.1 potassium channels in multiple sclerosis. Ann Neurol. 2010; 68:58–69.
Ludvik AE, Pusec CM, Priyadarshini M, Angueira AR, Guo C, Lo A, Hershenhouse KS, Yang G-Y, Ding X, Reddy TE et al. HKDC1 is a novel hexokinase involved in whole-body glucose use. Endocrinology. 2016; 157:3452–3461.
Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296:E1195–209.
Ishizaki J, Suzuki N, Higashino K-i, Yokota Y, Ono T, Kawamoto K, Fujii N, Arita H, Hanasaki K. Cloning and characterization of novel mouse and human secretory phospholipase A2s. J Biol Chem. 1999; 274:24973–24979.
Davis RC, Diep A, Hunziker W, Klisak I, Mohandas T, Schotz MC, Sparkes RS, Lusis AJ. Assignment of human pancreatic lipase gene (PNLIP) to chromosome 10q24–q26. Genomics. 1991; 11:1164–1166.
Lowe ME, Rosenblum JL, Strauss AW. Cloning and characterization of human pancreatic lipase cDNA. J Biol Chem. 1989; 264:20042–20048.
Hu Y, Wu Q, Ma S, Ma T, Shan L, Wang X, Nie Y, Ning Z, Yan L, Xiu Y et al. Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc Natl Acad Sci USA. 2017; 114:1081–1086.
Malenke JR, Skopec MM, Dearing MD. Evidence for functional convergence in genes upregulated by herbivores ingesting plant secondary compounds. BMC Ecol. 2014; 14:1–16.
Pajic P, Pavlidis P, Dean K, Neznanova L, Romano R-A, Garneau D, Daugherity E, Globig A, Ruhl S, Gokcumen O. Independent amylase gene copy number bursts correlate with dietary preferences in mammals. Elife. 2019; 8:e44628.
Chen L, Qiu Q, Jiang Y, Wang K, Lin Z, Li Z, Bibi F, Yang Y, Wang J, Nie W et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science. 2019; 364:eaav6202.
Endo Y, Kamei Ki, Inoue-Murayama M. Genetic signatures of lipid metabolism evolution in Cetacea since the divergence from terrestrial ancestor. J Evol Biol. 2018; 31:1655–1665.
Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, Wu C, Muzny DM, Li Y, Zhang W et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014; 344:1168–1173.
Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990; 70:567–590.
Geor RJ, Coenen M, Harris P. Equine applied and clinical nutrition. New York: Elsevier; 2013.
Shackleton DM. Hoofed mammals of British Columbia. Vancouver: UBC Press; 1999.
Wattiaux MA. Dairy essentials: nutrition and feeding, reproduction and genetic selection, lactation and milking, raising dairy heifers. Madison: Babcock Institute for International Dairy Research and Development; 1994.
Karasov WH, Douglas AE. Comparative digestive physiology. Compr Physiol. 2013; 3:741–783.
Schondube JE, Herrera-M LG, del Rio CM. Diet and the evolution of digestion and renal function in phyllostomid bats. Zoology. 2001; 104:59–73.
Wang Z, Xu S, Du K, Huang F, Chen Z, Zhou K, Ren W, Yang G. Evolution of digestive enzymes and RNASE1 provides insights into dietary switch of cetaceans. Mol Biol Evol. 2016; 33:3144–3157.
Halliday TJD, Upchurch P, Goswami A. Resolving the relationships of Paleocene placental mammals. Biol Rev. 2017; 92:521–550.
Cooper LN, Seiffert ER, Clementz M, Madar SI, Bajpai S, Hussain ST, Thewissen JG. Anthracobunids from the middle Eocene of India and Pakistan are stem perissodactyls. PloS One. 2014; 9:e109232.
Colbert EH, Morales M, Minkoff EC. Colbert’s evolution of the vertebrates. New York: Wiley-Liss; 2001.
Kong Y, Zheng Y, Jia Y, Li P, Wang Y. Decreased LIPF expression is correlated with DGKA and predicts poor outcome of gastric cancer. Oncol Rep. 2016; 36:1852–1860.
Davis RW. Marine Mammals: adaptations for an aquatic life. Switzerland: Springer; 2019.
Jenkins TC. Lipid metabolism in the rumen. J Dairy Sci. 1993; 76:3851–3863.
Jenkins TC, Wallace RJ, Moate PJ, Mosley EE. Recent advances in biohydrogenation of unsaturated fatty acids within the rumen microbial ecosystem. J Anim Sci. 2008; 86:397–412.
Lock AL, Harvatine KJ, Drackley JK, Bauman DE. Concepts in fat and fatty acid digestion in ruminants. In: Proceedings Intermountain Nutrition Conference: 2006; Logan. Utah State University: 85-100.
Wright DE. Fermentation of glycerol by rumen micro-organisms. N Zeal J Agric Res. 1969;12:281–6.
Hackmann TJ, Spain JN. Ruminant ecology and evolution: perspectives useful to ruminant livestock research and production. J Dairy Sci. 2010; 93:1320–1334.
Benton MJ. Vertebrate Palaeontology. West Sussex: Wiley Blackwell; 2015.
Kemp TS. The origin and evolution of mammals. Oxford: Oxford University Press; 2005.
O’Leary MA. Phylogenetic and morphometric reassessment of the dental evidence for a mesonychian and cetacean clade. In: Thewissen J, editor. The emergence of whales: patterns in the origin of Cetacea. New York: Springer; 1998. p. 133–62.
O’Leary MA. Parsimony analysis of total evidence from extinct and extant taxa and the cetacean-artiodactyl question (Mammalia, Ungulata). Cladistics. 1999; 15:315–330.
O’Leary MA, Geisler JH. The position of Cetacea within Mammalia: phylogenetic analysis of morphological data from extinct and extant taxa. Syst Biol. 1999; 48:455–490.
Spaulding M, O’Leary MA, Gatesy J. Relationships of Cetacea (Artiodactyla) among mammals: increased taxon sampling alters interpretations of key fossils and character evolution. PloS One. 2009; 4:e7062.
Wilman H, Belmaker J, Simpson J, de la Rosa C, Rivadeneira MM, Jetz W. EltonTraits 1.0: Species-level foraging attributes of the world’s birds and mammals: Ecological Archives E095‐178. Ecology. 2014; 95:2027–2027.
Cucchi T, Mohaseb A, Peigné S, Debue K, Orlando L, Mashkour M. Detecting taxonomic and phylogenetic signals in equid cheek teeth: towards new palaeontological and archaeological proxies. R Soc Open Sci. 2017; 4:160997.
Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O’Brien SJ. Molecular phylogenetics and the origins of placental mammals. Nature. 2001; 409:614–618.
Upham NS, Esselstyn JA, Jetz W. Inferring the mammal tree: species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLOS Biol. 2019; 17:e3000494.
Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RDE, Beck RMD, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. The delayed rise of present-day mammals. Nature. 2007; 446:507–512.
Liu L, Zhang J, Rheindt FE, Lei F, Qu Y, Wang Y, Zhang Y, Sullivan C, Nie W, Wang J et al. Genomic evidence reveals a radiation of placental mammals uninterrupted by the KPg boundary. Proc Natl Acad Sci USA. 2017; 114:E7282-E7290.
Zhang J, Kumar S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol Biol Evol. 1997; 14:527–536.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, Von Haeseler A, Lanfear R. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020; 37:1530–1534.
Nguyen L-T, Schmidt HA, Von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015; 32:268–274.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006; 22:2688–2690.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010; 59:307–321.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural Science Foundation of China (grant numbers, 32171604 and 31770401) and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
Y. W. designed research, performed analyses, and wrote the paper. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
We have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Amino acid substitutions along carnivorous lineages.
Additional file 2: Table S1.
Gene sequences with GenBank accession numbers used in this study.
Additional file 3: Table S2.
Positively selected genes identified by branch-site model. P values were corrected by multiply them by the number of branches tested of each gene (Bonferroni multiple testing correction).
Additional file 4: Table S3.
Positively selected genes identified by branch model.
Additional file 5: Table S4.
Positively selected genes identified by branch model.
Additional file 6: Table S5.
Tests of parallel evolution of the gene APOB between branches using software CAPE.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wu, Y. Diet evolution of carnivorous and herbivorous mammals in Laurasiatheria. BMC Ecol Evo 22, 82 (2022). https://doi.org/10.1186/s12862-022-02033-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12862-022-02033-6