
Abstract
Background
Silkie is a traditional Chinese chicken breed characterized by its unique combination of specialized morphological traits. While previous studies have focused on the genetic basis of these traits, the overall genomic characteristics of the Silkie breed remain largely unexplored. In this study, we employed whole genome resequencing data to examine the genetic diversity, selective signals and demographic history of the Silkie breed through comparative analyses with seven other Chinese indigenous breeds (IDGBs), a commercial breed, and the wild ancestor Red Jungle Fowl.
Results
In total, 20.8 million high-quality single nucleotide polymorphisms and 86 large structural variations were obtained. We discovered that Silkie exhibits a relatively high level of inbreeding and is genetically distinct from other IDGBs. Furthermore, our analysis indicated that Silkie has experienced a stronger historical population bottleneck and has a smaller effective population size compared with other IDGBs. We identified 45 putatively selected genes that are enriched in the melanogenesis pathway, which probably is related to the feather color. Among these genes, LMBR1 and PDSS2 have been previously associated with the extra toe and the hookless feathers, respectively. Six of the selected genes (KITLG, GSK3B, SOBP, CTBP1, ELMO2, SNRPN) are known to be associated with neurodevelopment and mental diseases in human, and are possibly related to the distinct behavior of Silkie. We further identified structural variants in Silkie and found previously reported variants linked to hyperpigmentation (END3), muff and beard (HOXB8), and Rose-comb phenotype (MNR2). Additionally, we found a 0.61 Mb inversion overlapping with the GMDS gene, which was previously linked to neurodevelopmental defects in zebrafish and humans. This may also be related to the behavior distinctiveness of Silkie.
Conclusions
Our study revealed that Silkie is genetically distinct and relatively highly inbred compared to other IDGB chicken populations, possibly attributed to more prolong population bottlenecks and selective breeding practice. These results enhance our understanding of how domestication and selective breeding have shaped the genome of Silkie. These findings contribute to the broader field of domestication and avian genomics, and have implications for the future conservation and breeding efforts.
Similar content being viewed by others

Introduction
The modern chicken was domesticated from Red Jungle Fowls (Gallus gallus) in the mid-early Holocene with genomic contributions from other Gallus species [1, 2]. The domestic chicken is probably the most widely distributed bird on Earth and exhibits an incredible scale of phenotypic variations due to different population histories, adaptation to diverse geographic conditions, and artificial selection for different purposes, including meat and egg production, companionship, ornamentation and gaming. Commercial breeds of boilers and layers were established in the last century through intensified artificial selection. Over a thousand breeds around the world provide valuable resources for studying phenotype-genotype relationships, as well as how artificial selection could shape the patterns of genomic diversity [3,4,5]. These insights could, in turn, help inform future breeding programs, build breed standards, and prioritize conservation of important genomic resources.
Silkie (SK) is one of the most well-known Chinese breeds with a long breeding history. It is reported to have originated in Jiangxi Province of China [6] and was described by Marco Polo in the thirteenth century for its peculiar appearance that is not often seen in other chicken breeds [7]. A typical Silkie chicken has distinctive characteristics, including black skin, bones and sheaths of internal organs, five toes on each foot, turquoise earlobes, feathered shanks, muffs, beards, and hookless white feathers that make their plumage furry (Fig. 1a). These unique qualities contribute to their importance in the poultry market of China, where they are valued both for their meat and as traditional Chinese medicine. In addition to its distinctive morphological traits, Silkie is renowned for its docile nature, which has contributed to its global popularity as a pet chicken breed. Due to its uniqueness, Silkie has been officially recognized and listed as a chicken breed with high conservation priority in China [6], and represents one of the three chicken breeds used to generate the first genome-wide genetic variation map as early as 2004 [8].

Genetic diversity and levels of inbreeding. a Adult male Silkies. b Genetic diversity \(\theta\)w and \(\pi\) for each population. c F and total ROH length for each population. Letters indicate significance from Tukey-Kramer tests. Abbreviations as in Table 1
Several studies have examined the underlying genetic basis of distinct morphological traits of Silkie through QTL analyses using single nucleotide polymorphism (SNP) arrays. For example, hyperpigmentation in exterior skin and internal connective tissue is found to be associated with EDN3 duplication [9]. The loss of feather hooklets is caused by a substitution in the 5' UTR of PDSS2 [10]. Facial feathers and rose comb phenotypes are the results of complex structural rearrangements [11, 12].
Apart from its unique phenotypes, Silkie is also genetically distinctive from other Chinese indigenous chicken breeds (IDGBs), or even those from the same province [13,14,15]. Several studies that have focused on other Chinese IDGBs have included Silkie as a representative breed in their sample panels for comparisons [16,17,18]. These studies have reported that Silkie exhibits one of the lowest levels of genetic diversity and heterozygosity among Chinese IDGBs, suggesting that it might undergo more intensive artificial selection compared to most IDGBs [16]. Additionally, Silkie appears as a population with a relatively low level of genetic admixture, although widespread genetic introgression from commercial chickens and Gallus species and subspecies into IDGBs has been suggested [14, 16].
These findings emphasized the uniqueness and the conservation importance of the Silke breed. However, high-coverage genome resequencing data for Silkie are still limited, and a focused investigation into the domestication history, genomic diversity, and artificial selection of this special breed has not been performed. In this study, we generated high-coverage whole genome sequencing (WGS) data for 10 Silkie individuals. Combined with public data of Silkie and other breeds, we examined the demographic history, genomic characteristics, and selective signals of the Silkie breed.
Results
Genomic data
Ten male Silkie chickens were sampled in a poultry farm in Taihe, Jiangxi Province, China (Fig. 1a, Table S1). Sequencing libraries were prepared using DNA extracted from blood samples, and 840 million pairs of clean reads was obtained in total. The average sequencing depth for each sample was 19.2× , ranging from 16.1× to 21.1× (PRJNA896380; Table S1). To examine the genomic features of Silkie in comparison with other breeds, we also selected publicly available sequencing data from 98 samples with a mean depth of 18.25× , including 13 additional Silkie samples [16, 19, 20], chicken's wild ancestor Red Jungle Fowl (RJF) [16, 17], a highly commercialized breed White Leghorn (WLH) [18, 21], and seven other representative Chinese IDGBs, including village chicken (Yunnan village chicken,YVC) [17], local chicken (Dulong chicken, DULO; Tibetan chicken, TBC; Wuhua yellow chicken,WHYC) [17, 19, 22, 23] and gamecocks (Tulufan gamecock, TLF; Luxi gamecock, LX; Xishuangbanna gamecock, YNLC) [16, 17] (see Methods for selection criteria; Table 1; Table S2). After trial runs of SNP calling, one RJF were removed due to an extremely low rate of polymorphism (see SNP discovery, Method). Together, we detected over 22.38 million biallelic autosomal variants, including 1,561,952 small IDNELs and 20,826,932 SNPs, that passed our quality filters and were genotyped in more than 90% of the 107 samples from 10 populations.
Relatively high inbreeding level of Silkie
Among the sampled populations, genome-wide nucleotide diversity (π) is estimated to be 0.00318 – 0.00531, which is comparable with previous estimates [23, 24]. Watterson’s \(\theta\)(\(\theta\)w) ranges from 0.00240 to 0.00532 (Fig. 1b). For all domestic populations, genome-wide \(\theta\)w is smaller than π, indicating an excess of rare variants possibly resulting from a quick population expansion after the domestication bottleneck. As expected, both π and \(\theta\)w are highest for RJF and lowest for WLH. Among IDGBs, Silkie has the lowest π and \(\theta\)w, suggesting that this breed is more inbred compared to the rest of the IDGB chickens (Fig. 1b).
We quantified and compared inbreeding levels of Silkie and other populations using the inbreeding coefficient (F) and total length of runs of homozygosity (ROH). Silkie has one of the highest F among IDGB populations, not significantly different from TBC and LX, but significantly higher than the rest of IDGB populations (Fig. 1c). One TBC individual shows an extremely negative F due to a large number of heterozygous sites. As expected, WLH has the highest F, and RJF is among the populations that have the lowest F.
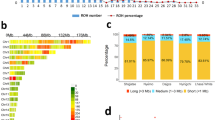
The average total length of ROH in each population ranges from 53.3 Mb to 415.8 Mb (Table 1; Table S3). WLH has the longest total length of ROH (415.8 \(\pm\) 140.8 Mb). We found that Silkie has significantly longer ROH (202.5 ± 60.1 Mb) than the rest of the IDGB populations and RJF (Fig. 1c). We further divided ROH into three categories by length, i.e. short (100 kb ~ 300 kb), long (300 kb ~ 1 Mb) and mega (> 1 Mb) ROH. Silkie has the greatest number of ROH in all three categories among IDGBs (Table S3). Both F and ROH suggest that Silkie has a relatively higher inbreeding level and possibly a more prolonged inbreeding compared to other Chinese IDGB populations.
Genetic distinctiveness of Silkie
Principal component analysis (PCA) was performed to identify population structure [25]. The first two principal components account for 8.35% and 4.85% of the total genetic variation and reveal clear clustering patterns. WLH individuals are first separated from all IDGB populations and RJF on PC1 (Fig. 2a). Silkie samples, including 10 samples that we sequenced and 13 publicly available samples, form a distinct cluster and are separated on PC2 from other IDGB chicken populations and RJF (Fig. 2a), indicating that Silkie is a highly differentiated breed. The remaining IDGB populations and RJF cluster together, suggesting a higher level of genetic similarity to each other than to WLH or Silkie. Breeds from different geographic locations all form distinct groups, such as WHYC (Guangdong Province), LX (Shangdong Province), and TLF (Xinjiang Province). Most samples from Yunnan Province (YNLC, DULO, and YVC) and some RJF and TBC individuals do not form clearly separated clusters, which is consistent with the previous study [16] and possibly due to their close geographical proximity. PCA without Silkie and WLH samples shows that all populations form separated clusters, except for one TBC individual that overlaps with the YVC cluster (Fig. 2a).

Population structure of the 10 populations. a Principal component analysis (PCA). Insert shows PCA without Silkie and WLH, corresponding to the individuals in the dashed-circle. b Pairwise FST between populations. c ADMIXURE analysis with K = 2 to K = 5. K = 3 has the lowest cross-validate error. Each column represents an individual, populations were separated by black lines with abbreviated breed names at the bottom. Abbreviations as in Table 1
We also used ADMIXUTRE analysis (v1.3.0) to infer population structure (Fig. 2c; Figure S1) [26]. When K = 2, WLH is first identified as a distinct group, and when K = 3, Silkie is separated from all other populations (Fig. 2c), consistent with the PCA results. The best K is determined to be 3 with the smallest cross-validation error rate, where WLH and Silkie are distinctive from all other populations. This pattern is consistent with K from 3 to 8 (Figure S1). With the increase of K, the three gamecock breeds (LX, TLF, and YNLC) show high similarity until K = 7, although they are from geographically distant regions (Figure S1). Two RJF individuals seem to resemble individuals in the YVC population, consistent with the PCA result and possibly due to recent hybridization.
Pairwise FST between populations ranges from 0.028 to 0.301 (Fig. 2b). The comparisons between WLH and other chicken populations exhibit the highest FST (0.209 – 0.301). When considering only IDGBs, FST between Silkie and any other IDGB are the highest among all pairwise comparisons. Together with the population structure analysis, our results suggest that the Silkie breed is genetically the most differentiated compared to other Chinese IDGBs.
A population bottleneck in Silkie
Genome-wide linkage disequilibrium (LD) decreases as the physical distance between SNPs increases (Fig. 3a). LD decays the fastest in RJF compared to all domesticated chicken populations, which corresponds with its larger effective population size (Ne) as the wild progenitor of domesticated chicken. In contrast, LD decays the slowest in WLH compared to all IDGBs, which is consistent with intensive commercial inbreeding of this breed and a persistently small Ne. Among IDGBs, TLF has the slowest decay of LD. Silkie shows a faster decay of LD than TLF but slower decay of LD compared to the rest of IDGBs.

Levels of linkage disequilibrium and inferred demographic history. a LD decays over distance. b SMC++ analysis showing changes in Ne of each population over time. c TreeMix analysis with two migration events for nine chicken populations (no WLH). The inferred migration events are shown as arrows, with color indicating the weight of migration. d) Residual and model fitting for models with different numbers of migration events. Mutaiton rate was set as 1.8 × 10–9 per site per year with the generation time being one year [27]. Abbreviations as in Table 1
We used SMC++ [28], which takes a hidden Markov Model approach, to infer the demographic history of Silkie and other populations. Our results show that the ancestral population of chicken and RJF experienced a decline in effective population size (Ne) starting at about 150 kya,which has been reported by previous studies and coincides with the Last Glacial Period (~125 – 10 kya) [17]. Ne of RJF declined at a slower rate and eventually recovered around 10 kya (Fig. 3b). At the same time, the Ne for other chicken populations continued to decrease, possibly reflecting the domestication event as a separated demographic path from their wild ancestor. YNLC and YVC populations lack sufficient data points to infer their Ne in more recent history. Among these nine domesticated chicken populations, the bottleneck is much more severe and lasts longer for Silkie and WLH.
To investigate gene flow across populations, we applied a maximum likelihood (ML) approach with different numbers of migration events using TreeMix (v1.13) [29] (Fig. 3; Figure S2). RJF served as the outgroup for all IDGBs, and WLH was excluded from this analysis due to its distant origin. The model without migration demonstrated a poor fit when compared to models incorporating migration events (Fig. 3d). The model with one migration event inferred gene flow from Silkie to LX, with 96.92% of total genetic variation explained (Fig. 3c; Figure S2). When increasing the number of migration events, model fit improved and residuals decreased in general. The model with two migratory events accounted for 98.67% of total genetic variation, and models with more than two migratory edges showed only marginal improvements in fit (Fig. 3d). In the model with two migration events, the three gamecock populations (YNLC, LX and TLF) were grouped together while Silkie and WHYC formed the closest sister groups. Gene flow was supported from Silkie to LX, and from the common ancestor of LX and TLF to the common ancestor of WHYC and Silkie (Fig. 3c).
Taken together, our results suggest that Silkie has experienced a relatively more pronounced population bottleneck in comparison to other IDGB populations. Furthermore, there is evidence of possibly genetic introgression from Silkie to other IDGB populations, but no discernible introgression from other populations to Silkie.
Identification of loci under selection in Silkie
We employed a combination of three statistics, namely XP-EHH, pairwise FST and the ratio of nucleotide diversity (πratio), to detect selective signals in Silkie in comparison to other domestic chicken populations. These statistics were calculated in sliding windows of 20 kb, and genomic windows with more than 50% missing sites or fewer than five SNPs were excluded from further analyses. Of the remaining 45,553 windows, we identified candidate windows with the most extreme values of each statistic, using thresholds of top 1% of FST, top 1% of XP-EHH, and bottom 1% of πratio (ZFST ≥ 3.41, Zπ_ratio ≤ -3.05, ZXP-EHH ≥ 3.07; Fig. 4a; Figure S3). We obtained a final set of 62 windows (Table S4), which contain 45 genes that are significantly enriched in the melanogenesis pathway (adjusted p-value = 1.20 × 10–3) (Table S5).

Identification of putative genomic regions under selection. a Detecting selective sweeps combining signals from FST, \(\pi\)ratio and XP-EHH. Each dot represents a non-overlapping 20 kb window in chicken genome that passes quality control. The dashed lines are the 1% cutoff Z-scores for FST and \(\pi\)ratio. Candidate windows with selective signals are highlighted by black circles, with candidate genes within them noted. b A putative selected genomic region in Silkie on Chromosome 3. The dashed box contains two consecutive 20kb windows both identified as being under selection and is at the 5' regions of both SOBP and PDSS2 genes. c The haplotypes of all 107 samples. Each row represents the haplotype of one sample, and populations are separated using black lines
Three of the 45 candidate genes, PDSS2, LMBR1 and TYR, were prevsiously associated with Silkie phenotypes. Specifically, a point mutation upstream of PDSS2 leads to hookless feathers [10]. A mutation in the ZRS (zone of polarising activity regulatory sequence) region of LMBR1 has been linked to the polydactyly phenotype [30, 31]. TYR codes for a key enzyme tyrosinase in melanogenesis, and a homozygous retroviral insertion in the TYR intron leads to white plumage [32]. One candidate region with strong signatures of selection contains both PDSS2 and SOBP (Fig. 4b). The window with the most extreme statistics is in closer proximity to SOBP. The haplotype of this region in Silkie is distinctive from other populations, and three upstream variants of both SOBP and PDSS2 are fixed in all Silkie samples (Fig. 4c). Interestingly, there are several candidate genes associated with human neurological and psychological diseases, including SOBP, KITLG (receptor tyrosine kinase ligand), GSK3B (Glycogen synthase kinase-3 beta), ELMO2 (Engulfment And Cell Motility 2), CTBP1 (C-Terminal Binding Protein 1) and SNRPN (Small Nuclear Ribonucleoprotein Polypeptide N) (Table 2).
High frequency structural variants in Silkie
Several structural variants (SV) have been previously related to Silkie-specific phenotypes. These include the inversion on Chr7 and the MNR gene for the rose comb [11], the inversion duplication on Chr20 and the EDN3 gene for skin hyperpigmentation [9, 50], the complex duplication on Chr27 and the HOX8 gene for the muff and beard [12], and a 17.7 kb deletion on Chr13 and the PITX1 gene attributed to the feathered shank phenotype [51]. To detect additional SVs in Silkie, we chose three software, LUMPY [52], Manta [53] and GRIDSS [54], based on their performance in calling SV using short-reads data [55]. Only SVs detected by at least two of the three methods were further investigated.
In total, 86 large SVs were discovered with high allele frequencies of at least 0.70 in the Silkie breed but not in other populations (Table S6). We manually examined alignments to confirm each SV and identified genes whose functions may be influenced by each SV. We successfully recovered all four aforementioned SVs in the Silkie breed. All but four of the remaining SVs are deletions. Forty-one deletions contain at least one gene, and three deletions overlap with exons of protein-coding genes UBASH3A, TNFRSF19, and HOXB7 (Table S6). Breakends of a possibly 600 kb inversion on Chr2 (67,174,958 – 67,785,951) overlap with genes GMDS and VPS4B. Twenty-two of 23 Silkie samples carry this inversion while only 24.4% of IDGB samples share this SV.
Discussion
In this study, we generated WGS data and investigated how domestication and selective breeding have shaped the genome of Silkie, a Chinese chicken breed with distinct phenotypical traits. Silkie samples sequenced in our study cluster with those published in previous studies, distinct from other IDGB populations. Compared to other IDGBs, the Silkie genome has lower genetic diversity and a higher level of inbreeding. Demographic analysis indicates a sustained decline in the effective population size, and a more pronounced bottleneck in their history of domestication, in comparison to other IDGB chicken. This decline in Ne more closely resembles that of WLH which is highly commercialized. This pattern may be attributed to the intense artificial selection and the strict breeding program aimed at selecting and preserving the special traits of Silkie. A genome-wide scan of selective signals and high-frequency SV in Silkie identified several candidates that may be associated with artificial selection during the domestication process of Silkie, including ones that have previously been reported to underlie Silkie-specific phenotypes.
In population structure analysis using all samples, Silkie and WLH form distict clusters that are well separated from their wild progenitor RJF and other Chinese IDGBs examined, while clusters of RJF and other other Chinese IDGBs are closer (Fig. 2a). This is probably due to the long-term breeding and intense artificial selection in Silkie and WLH. Similar PCA pattern had been observed in several previous studies [4, 14]. This pattern in PCA is also supported by the stronger correlation of θπ between RJF and other native chicken breed compared to that between RJF and Silkie (Figure S4).
Compared with other IDGBs, Silkie exhibits lower genetic diversity and higher inbreeding level characterized by significant portions of its genome being ROH of various lengths (Fig. 1c; Table S3). ROH refers to contiguous genome segments with homozygous genotypes due to haplotype identity by descent. Since ROH are slowly broken down by recombination, the length distribution of ROH reflects the timing of historical inbreeding events. Longer ROH are usually results of recent inbreeding events, while shorter ROH indicate relatively ancient inbreeding [56]. Among the Chinese IDGBs we included, Silkie possesses the longest average ROH segment length, the largest average ROH segment number, and as a result, the largest average total ROH length. Interestingly, Silkie has higher average segment numbers for both mega and short ROH compared to all other IDGBs, implying a long and persistent inbreeding process (Table S3).
As suggested by many studies and anecdotal references, hybridization between domestic species and their wild ancestor was common [16, 57, 58], and a recent study has discovered significant introgression from Gallus gallus jabouillei to many Chinese chicken populations [1]. Chicken, being small-sized farm animals, can be easily transported by humans during long-distance migration, thus gene flows between chicken or wild populations were not unusual. At the same time, hybridization is also a common practice in the farming industry for obtaining advantageous traits or creating novel breeds [14, 59]. Looking at a closer time frame within chicken populations, a possible gene flow from Silkie to LX is inferred by introgression analysis, but not from other sampled populations to Silkie. Silkie has a long breeding history. One possibility is that this breed has been used to bring preferred trait to other IDGB populations. Another possibility is that because we only include a limited number of IDGB populations, the precise direction of gene flow may be influenced by unsampled sister breeds.
Several previous studies used genotype–phenotype association to identify genes underlying specific morphological traits of Silkie [9,10,11,12, 50, 51]. Although we did not collect phenotye data, our scan of genomic regions under selection, as well as high frequency SV in Silkie, identified both known and new candidate regions and genes that may be linked to unique phenotypes of Silkie. The identified regions under selection harbor 45 candidate genes that are enriched in melanogenesis pathway, which is probably associated with the recessive white plumage. These candidate regions also overlap with previously reported QTLs associated Silkie-specific phenotypes, such as polydactyly, color of skin and comb, feather pigmentation,as well as general poultry domestication phenotypes, including egg number and yolk weight [60]. Several candidate genes we identified have been reported to be under selection in previous studies [3, 32, 50]. One study also reported different regions under selection [61], which is at least partially due to a different experimental design and dataset used.
In addition to the morphological diversity among chicken breeds, there are also notable behavioral differences, although these have been given less attention in research. For instance, Silkie chickens are renowned for their docility and considered as one of the gentlest breeds. In our genome-wide scan of selective signals, we identified several candidate regions that contain genes associated with neurological and psychological diseases in human (Table 2). One candidate region with strong signatures of selection contains both PDSS2 and SOBP, as well as their 5' upstream regions (Fig. 4b). PDSS2 was previously linked to hookless feathers in Silkie [10]. SOBP was reported to be involved in brain activity and cognition ability, and a disruptive mutation could cause autosomal recessive mental retardation and intellectual disability [43]. Recent studies also showed a higher level of SOBP expression in Silkie’s heart and brain relative to other organs [10, 62]. It is worth future investigation whether the selected haplotype in Silkie leads to both hookless feathers and mild temper, and whether genetic hitchhiking in selection of one trait leads to changes in the other. Additionally, both CTBP1 and SNRPN are related to the Prader-Willi syndrome (PWS) [44, 47, 48]. The psychiatric and behavioral symptoms of the PWS include unstable mood bursts, aggressiveness, impulsive behavior, and a limited ability to cope with changes [63]. CTBP1 might also be related to the Wolf-Hirschhorn syndrome (WHS), of which patients usually exhibited mental retardation, seizures, and developmental defects [45, 64]. Future functional validation or genotype–phenotype association studies are needed to establish a direct link between these variants to distinct behavioral phenotype in Silkie.
We discovered several new SVs with high frequency in the Silkie breed, including an inversion on Chr2 (67,174,958 – 67,785,951), which could potentially influence the expression of the GMDS gene. GMDS encodes GDP-mannose 4,6 dehydratase in the protein fucosylation pathway. A missense mutation in GMDS had been shown to cause neural development defects in zebrafish [65]. Disruption of GMDS expression, along with alteration in other genes due to chromosomal abnormality, can lead to immunodeficiencies, congenital malformations in various organs and mental retardation in human patients [66,67,68]. Another deletion (Chr1: 178,824,081 – 178,824,670) intersects with an exon of TNFRSF19 (TNF Receptor Superfamily Member 19). TNFRSF19 is widely expressed during human embryogenesis, as well as in the adult brain, hair and follicles [69], and has been shown to regulate tumor cells proliferation and migration [70, 71] and melanoma growth [72]. In addition, we found a 17.7 kb deletion on Chr13 that was associated with the feathered shank phenotype [51]. Another independent dominant locus that contributes to the feathered shank phenotype is a SNP 25 kb upstream of TBX5 on Chr15 (12,573,054) [51]. This variant is present at a frequency of 71.7% (33/46) in our Silkie samples, but not found in any other chicken population.
We selected relatively high-quality resequencing samples using criteria such as sequencing depth and the number of available samples per population, thus the total breed variety is limited, and this sampling strategy may influence some of our analysis. Moreover, inferences on introgression may be inconclusive due to potential hybridization with unsampled chicken populations, other RJF subspecies or Gallus species. Furthermore, many of the selected regions do not cover annotated genes or known epigenetic markers, which limits our understanding of these regions. Additionally, the power to detect large SVs is restricted when only short reads are available. Future studies should incorporate long-read sequencing technology to examine the SVs in the genome of Silkie and other chicken populations.
Extreme artificial selection and long-term inbreeding could lead to inbreeding depression and an excess of genetic burden on the genome of a species. Selective breeding, especially of fancy breeds, inevitably involves inbreeding to consolidate preferred phenotypes. Many studies have shown that domesticated populations often bear more genetic diseases than natural populations [73, 74]. An elevated genetic load was observed in domesticated chicken compared with RJF [24]. Investigations on genomic signatures of domesticated breeds, including our study, could provide useful information for establishing protocols for sustainability and genetic conservation of the Silkie breed.
Conclusion
This study conducted a comprehensive analysis of the Silkie genome using comparative methods with seven other IDGBs (YVC,YNLC, TBC, DULO, WHYC, LX and TLF), a commercial line (WLH) and the wild ancestor of the domesticated chicken (RJF). Our results showed that Silkie exhibits lower genetic diversity and a relatively higher inbreeding level and is substantially differentiated from other domestic chicken. Demographic history analysis indicated a prolong decline in effective population size, and the observed genomic characteristics in the Silkie genome are likely attributed to the enduring bottlenecks and strong artificial selection processes. Furthermore, this study identified genomic regions under selection and high-frequency structural variants in Silkie. In addition to several previously reported genes that have been associated with Silkie-specific phenotypes, several new candidate genes were uncovered that may be associated with artificial selection during the domestication of Silkie. These findings enhance our comprehension of the influence of domestication and selective breeding on the genome of Silkie, offering valuable insights to inform future conservation and breeding efforts.
Materials and methods
Sample collection, DNA extraction and genomic data
Ten male Silkie chicken were sampled from a poultry farm in Taihe, Jiangxi Province, China (Table S1). Blood samples were obtained from the brachial vein underneath the wing by venipuncture and stored in EDTA tubes at -80 °C before DNA extraction. DNA was extracted from each sample using DNeasy Blood & Tissue Kit (QIAGEN, Germany), and a sequencing library with an insertion size of 350 bp was prepared using NEB Next® Ultra™ Kit (NEB, USA). Whole genome resequencing (WGS) was performed on an Illumina NovaSeq 6000 platform using a pair-end library and read length of 150 bp. The average sequencing depth is 19.2× (16.06× ~ 21.06× ; Table S1).
We retrieved publicly available WGS data of 13 Silkie individuals from three previous studies, with the mean sequencing depth at 9.58× (Table 1; Table S2) [16, 19, 20]. WGS data from RJF [16, 17] and other domestic chicken breeds were selected based on breed variety, individual sequencing depth (minimum of 8×), and population sample size (minimum of 8 individuals per population, except for TLF with six samples available) (Table S2). In total, our dataset consisted of WGS data from 108 samples (Table 1).
SNP discovery
WGS data were processed following the GATK (v4.0.2.0) Best Practices for calling germline SNPs and small insertions/deletions (INDEL) [75]. Prior to analysis, the quality of the raw reads for each individual was assessed using FastQC (v0.11.8) [76]. Adaptor sequences and low-quality reads were trimmed with Trimmomatic (0.39) using default parameters except for MINLEN: 51 [77]. Filtered reads were aligned to the chicken reference genome Galgal6 (GCA_000002315.5, Ensembl [78]) using Burrows-Wheeler-Aligner (bwa 0.7.16a) [79], and duplicated reads were marked using Picard “MarkDuplicates” (v2.19.0) [80]. Base recalibration and variant calling were performed using “BaseRecalibrator” and GATK joint-calling pipeline “HaplotypeCaller” and “GenotypeGVCFs” [75].
The output VCF file was filtered with Read Depth (DP) < 4, QualByDepth (QD) < 4, Quality (QUAL) < 30, Symmetric Odds Ratio of 2 × 2 contingency table to detect strand bias (SOR) > 3, Fisher Strand (FS) > 60, MappingQuality (MQ) < 40, MQRankSum < -12.5 and ReadPosRankSum < -8. Biallelic autosomal (Chr1-Chr28) variants with > 90% genotyping rate across all 108 samples were retained. SNPs in repetitive regions were removed based on UCSC annotation (https://genome.ucsc.edu/). One RJF sample was removed because of its unusual homozygosity for ancestral alleles, as it had only 1/10 the number of variants compared to other samples, despite having high-quality data. Consequently, 107 individuals were included in the downstream analysis.
Genetic diversity and population structure
We estimated several summary statistics of population genetic diversity, including pairwise nucleotide diversity (π) and Watterson’s theta (\(\theta\)w) [81] for each population using pixy (v1.1.1beta) [82] and custom scripts. The inbreeding level for each sample was estimated by inbreeding coefficient (F) and runs of homozygosity (ROH) with PLINK (v1.9) [83]. The parameters used for calling ROH are --homozyg-density 50, --homozyg-kb 100, --homozyg-snp 33. Linkage disequilibrium (LD) for each population was estimated by calculating genotype correlation coefficient (r2), using PopLDdecay with a setting of -MAF 0.05 [84].
Principal component analysis (PCA) and ADMIXTURE (v1.3.0) were used to infer population structure, and pairwise fixation indices (FST) were calculated to quantify population genomic differentiation [25, 26]. For all three analyses, variant sites with > 2% missing data were removed. R packages gdsfmt and SNPrelate were used for both PCA and FST calculation [25]. For ADMIXTURE, SNPs were further pruned using PLINK v1.9 with parameters “--indep-pairwise 50 10 0.2” to generate the required input BED file [83]. The analysis was performed for K = 2 to 8 and the best K was determined by comparing cross-validation errors.
Demographic history inference
SMC++ (ver 1.15.2) [28] was used to characterize the historical effective population size (Ne) of each chicken population with the standard pipeline and additional “--spline cubic” in “estimate” step for a smoother graphic presentation.
Population splits and gene flows were inferred using the maximum likelihood method in TreeMix (v1.13) [29]. The phylogenetic tree was rooted with RJF as the outgroup, and WLH was excluded from the model. Up to 9 migration events were modeled without sample size correction (-noss) and with blocks of 500 SNPs for bootstrapping. The resulting trees were plotted using the plotting_func.R in the TreeMix program.
For both analyses, the mutation rate was set as 1.8 × 10–9 per site per year, and the generation time was assumed to be one year [27].
Genome-wide scan for selective signals
Three methods were employed to detect positively selected regions in Silkie, i.e. cross population extended haplotype homozygosity (XP-EHH) [85], ratio of nucleotide diversity (πratio) and pairwise FST. In all three methods, Silkie was set as the focal population and all other domesticated chickens (excluding RJF) were combined as the reference population. All three methods were performed using a sliding-window approach with non-overlapping 20 kb windows across autosomal regions of the chicken genome. After filtering windows with less than 5 SNPs or more than 50% of the sites missing, 45,553 windows (911.06 Mb, 96.5% of autosomal region) remained for the selective signal analyses.
XP-EHH detects loci with reduced haplotype diversity caused by recent positive selection, which drives the favored haplotype towards fixation faster than the background recombination [85]. VCF files of Silkie and other domesticated chickens were first phased with Beagle (ver. 25Nov19.28d) [86]. XP-EHH was then calculated for each locus using selscan (ver 1.2.0a) [87], and averaged across loci in each 20 kb window.
FST and π statistics were calculated using pixy v1.1.1beta, which provides an unbiased estimation of population diversity and divergence while accounting for missing data [82]. Raw GVCF files were processed with VCFtools and filtered with “--max-maf 0 --minQ 30 --remove-indels --max-missing 0.5 --min-meanDP 4” for invariant sites. Repetitive regions were also excluded based on the UCSC annotation. Invariant and variant sites were combined using “bcftools concat --allow-overlaps” [88]. πratio equals πSilkie/πnon-Silkie, where non-Silkie includes rest of the IDGBs and WLH individuals.
Raw statistics were normalized by Zx = (x-μ)/σ, where x is the raw statistic value, and μ and σ are the mean and standard deviation of that statistic, respectively. The distributions of ZFST, Zπratio and ZXP-EHH were shown in Figure S3. We used cut-offs of the top 1% for FST and XP-EHH, and the bottom 1% for πratio. Windows captured by all three methods were considered positively selected windows.
Structural variants discovery
Three software, i.e. LUMPY [52], Manta [53] and GRIDSS [54], were used to detect structural variants (SV) using default parameters. These software were chosen based on their overall performance in detecting SVs form short reads sequencing data [55]. BAM files from the above GATK BaseRecalibrator step were used as input files. The output VCF files from each method of all samples were merged using SURVIVOR v1.0.3 [89]. Intra-autosomal SVs longer than 50 bp, identified by at least two of three methods, were investigated with additional filters including frequency \(\ge\) 70% in Silkie individuals and \(\le\) 50% in all other domesticated populations. For each deletion or breakends of SV, genes that overlapped or were closest to the SV were identified.
Availability of data and materials
Raw sequencing reads are deposited in NCBI SRA database (BioProject PRJNA896380). Scripts used are available on GitHub (https://github.com/rosehuang24/SK_genome_paper).
References
Wang MS, Thakur M, Peng MS, Jiang Y, Frantz LAF, Li M, et al. 863 genomes reveal the origin and domestication of chicken. Cell Res. 2020;30(8):693–701.
Peters J, Lebrasseur O, Deng H, Larson G. Holocene cultural history of Red jungle fowl (Gallus gallus) and its domestic descendant in East Asia. Quatern Sci Rev. 2016;142:102–19.
Li D, Li Y, Li M, Che T, Tian S, Chen B, et al. Population genomics identifies patterns of genetic diversity and selection in chicken. BMC Genomics. 2019;20(1):263.
Zhang J, Nie C, Li X, Ning Z, Chen Y, Jia Y, et al. Genome-wide population genetic analysis of commercial, indigenous, game, and wild chickens using 600K SNP microarray data. Front Genet. 2020;11:543294.
Rubin CJ, Zody MC, Eriksson J, Meadows JR, Sherwood E, Webster MT, et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature. 2010;464(7288):587–91.
Ministry of Agriculture of the People’s Republic of China. Announcement 1346: AGI2010-02-00245; 2010. [cited 2022 Sept 02]. Available from: http://www.moa.gov.cn/nybgb/2010/dsiq/201805/t20180530_6148371.htm.
Haw SG. Marco Polo's China: a Venetian in the realm of Khubilai Khan. Routledge; 2006. p. 130.
Ka-Shu Wong G, Liu B, Wang J, Zhang Y, Yang X, Zhang Z, et al. A genetic variation map for chicken with 2.8 million single-nucleotide polymorphisms. Nature. 2004;432(7018):717–22.
Dorshorst B, Molin AM, Rubin CJ, Johansson AM, Stromstedt L, Pham MH, et al. A complex genomic rearrangement involving the endothelin 3 locus causes dermal hyperpigmentation in the chicken. PLoS Genet. 2011;7(12):e1002412.
Feng C, Gao Y, Dorshorst B, Song C, Gu X, Li Q, et al. A cis-regulatory mutation of PDSS2 causes silky-feather in chickens. PLoS Genet. 2014;10(8):e1004576.
Imsland F, Feng C, Boije H, Bed’hom B, Fillon V, Dorshorst B, et al. The Rose-comb mutation in chickens constitutes a structural rearrangement causing both altered comb morphology and defective sperm motility. PLoS Genet. 2012;8(6):e1002775.
Guo Y, Gu X, Sheng Z, Wang Y, Luo C, Liu R, et al. A Complex Structural Variation on Chromosome 27 Leads to the Ectopic Expression of HOXB8 and the Muffs and Beard Phenotype in Chickens. PLoS Genet. 2016;12(6):e1006071.
Chen L, Wang X, Cheng D, Chen K, Fan Y, Wu G, et al. Population genetic analyses of seven Chinese indigenous chicken breeds in a context of global breeds. Anim Genet. 2019;50(1):82–6.
Zhang C, Lin D, Wang Y, Peng D, Li H, Fei J, et al. Widespread introgression in Chinese indigenous chicken breeds from commercial broiler. Evol Appl. 2019;12(3):610–21.
Nie C, Almeida P, Jia Y, Bao H, Ning Z, Qu L. Genome-wide single-nucleotide polymorphism data unveil admixture of Chinese indigenous chicken breeds with commercial breeds. Genome Biol Evol. 2019;11(7):1847–56.
Luo W, Luo C, Wang M, Guo L, Chen X, Li Z, et al. Genome diversity of Chinese indigenous chicken and the selective signatures in Chinese gamecock chicken. Sci Rep. 2020;10(1):14532.
Wang MS, Li Y, Peng MS, Zhong L, Wang ZJ, Li QY, et al. Genomic Analyses Reveal Potential Independent Adaptation to High Altitude in Tibetan Chickens. Mol Biol Evol. 2015;32(7):1880–9.
Jeong H, Kim K, Caetano-Anolles K, Kim H, Kim BK, Yi JK, et al. Whole genome sequencing of Gyeongbuk Araucana, a newly developed blue-egg laying chicken breed, reveals its origin and genetic characteristics. Sci Rep. 2016;6:26484.
Yi G, Qu L, Liu J, Yan Y, Xu G, Yang N. Genome-wide patterns of copy number variation in the diversified chicken genomes using next-generation sequencing. BMC Genomics. 2014;15(1):1–16.
Fan WL, Ng CS, Chen CF, Lu MY, Chen YH, Liu CJ, et al. Genome-wide patterns of genetic variation in two domestic chickens. Genome Biol Evol. 2013;5(7):1376–92.
Ulfah M, Kawahara-Miki R, Farajalllah A, Muladno M, Dorshorst B, Martin A, et al. Genetic features of red and green junglefowls and relationship with Indonesian native chickens Sumatera and Kedu Hitam. BMC Genomics. 2016;17:320.
Wang Q, Li D, Guo A, Li M, Li L, Zhou J, et al. Whole-genome resequencing of Dulong Chicken reveal signatures of selection. Br Poult Sci. 2020;61(6):624–31.
Weng Z, Xu Y, Li W, Chen J, Zhong M, Zhong F, et al. Genomic variations and signatures of selection in Wuhua yellow chicken. PLoS One. 2020;15(10):e0241137.
Wang MS, Zhang JJ, Guo X, Li M, Meyer R, Ashari H, et al. Large-scale genomic analysis reveals the genetic cost of chicken domestication. BMC Biol. 2021;19(1):118.
Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28(24):3326–8.
Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–64.
Axelsson E, Smith NG, Sundström H, Berlin S, Ellegren H. Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol Biol Evol. 2004;21(8):1538–47.
Terhorst J, Kamm JA, Song YS. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat Genet. 2017;49(2):303–9.
Pickrell JK, Pritchard JK. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLOS GENET. 2012;8(11):e1002967.
Dunn IC, Paton IR, Clelland AK, Sebastian S, Johnson EJ, McTeir L, et al. The chicken polydactyly (Po) locus causes allelic imbalance and ectopic expression of Shh during limb development. Dev Dyn. 2011;240(5):1163–72.
Chu Q, Yan Z, Zhang J, Usman T, Zhang Y, Liu H, et al. Association of SNP rs80659072 in the ZRS with polydactyly in Beijing you chickens. PLoS One. 2017;12(10):e0185953.
Chang C-M, Coville J-L, Coquerelle G, Gourichon D, Oulmouden A, Tixier-Boichard M. Complete association between a retroviral insertion in the tyrosinase gene and the recessive white mutation in chickens. BMC Genomics. 2006;7(1):1–15.
Miller CT, Beleza S, Pollen AA, Schluter D, Kittles RA, Shriver MD, et al. cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007;131(6):1179–89.
Sulem P, Gudbjartsson DF, Stacey SN, Helgason A, Rafnar T, Magnusson KP, et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat Genet. 2007;39(12):1443–52.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391–406.
Seco CZ, de Castro LS, Van Nierop JW, Morín M, Jhangiani S, Verver EJ, et al. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am J Hum Genet. 2015;97(5):647–60.
Ogawa Y, Kono M, Akiyama M. Pigmented macules in Waardenburg syndrome type 2 due to KITLG mutation. Exp Dermatol. 2017;40:860–4.
Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, et al. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann Neurol. 2005;58(6):829–39.
Schaffer BA, Bertram L, Miller BL, Mullin K, Weintraub S, Johnson N, et al. Association of GSK3B with Alzheimer disease and frontotemporal dementia. Arch Neurol. 2008;65(10):1368–74.
Zhang K, Yang C, Xu Y, Sun N, Yang H, Liu J, et al. Genetic association of the interaction between the BDNF and GSK3B genes and major depressive disorder in a Chinese population. J Neural Transm. 2010;117(3):393–401.
Huang YQ, Deng XM, Du ZQ, Qiu X, Du X, Chen W, et al. Single nucleotide polymorphisms in the chicken Lmbr1 gene are associated with chicken polydactyly. Gene. 2006;374:10–8.
Laurell T, VanderMeer JE, Wenger AM, Grigelioniene G, Nordenskjöld A, Arner M, et al. A novel 13 base pair insertion in the sonic hedgehog ZRS limb enhancer (ZRS/LMBR1) causes preaxial polydactyly with triphalangeal thumb. Hum Mutat. 2012;33(7):1063–6.
Birk E, Har-Zahav A, Manzini CM, Pasmanik-Chor M, Kornreich L, Walsh CA, et al. SOBP is mutated in syndromic and nonsyndromic intellectual disability and is highly expressed in the brain limbic system. Am J Hum Genet. 2010;87(5):694–700.
Misceo D, Barøy T, Helle J, Braaten Ø, Fannemel M, Frengen E. 1.5 Mb deletion of chromosome 4p16.3 associated with postnatal growth delay, psychomotor impairment, epilepsy, impulsive behavior and asynchronous skeletal development. Gene. 2012;507(1):85–91.
Simon R, Bergemann AD. Mouse models of Wolf–Hirschhorn syndrome. Am J Med Genet C Semin Med Genet. 2008;148(4):275–80.
Mehawej C, Hoischen A, Farah R, Marey I, David M, Stora S, et al. Homozygous mutation in ELMO2 may cause Ramon syndrome. Clin Genet. 2018;93(3):703–6.
Kuslich C, Kobori J, Mohapatra G, Gregorio-King C, Donlon T. Prader-Willi syndrome is caused by disruption of the SNRPN gene. Am J Hum Genet. 1999;64(1):70–6.
Sun Y, Nicholls RD, Butler MG, Saitoh S, Hainline BE, Palmer CG. Breakage in the SNRPN Locus in a Balanced 46, XY, t (,) Prader-Willi Syndrome Patient. Hum Mol Genet. 1996;5(4):517–24.
Li J, Lin X, Wang M, Hu Y, Xue K, Gu S, et al. Potential role of genomic imprinted genes and brain developmental related genes in autism. BMC Med Genomics. 2020;13(1):1–13.
Dorshorst B, Okimoto R, Ashwell C. Genomic regions associated with dermal hyperpigmentation, polydactyly and other morphological traits in the Silkie chicken. J Hered. 2010;101(3):339–50.
Li J, Lee M, Davis BW, Lamichhaney S, Dorshorst BJ, Siegel PB, et al. Mutations Upstream of the TBX5 and PITX1 Transcription Factor Genes Are Associated with Feathered Legs in the Domestic Chicken. Mol Biol Evol. 2020;37(9):2477–86.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15(6):1–19.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–2.
Cameron DL, Schröder J, Penington JS, Do H, Molania R, Dobrovic A, et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 2017;27(12):2050–60.
Cameron DL, Di Stefano L, Papenfuss AT. Comprehensive evaluation and characterisation of short read general-purpose structural variant calling software. Nat Commun. 2019;10(1):3240.
Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic runs of homozygosity record population history and consanguinity. PLoS One. 2010;5(11):e13996.
Vila C, Seddon J, Ellegren H. Genes of domestic mammals augmented by backcrossing with wild ancestors. Trends Genet. 2005;21(4):214–8.
Lin T, Zhu G, Zhang J, Xu X, Yu Q, Zheng Z, et al. Genomic analyses provide insights into the history of tomato breeding. Nat Genet. 2014;46(11):1220–6.
Bosse M, Megens HJ, Frantz LA, Madsen O, Larson G, Paudel Y, et al. Genomic analysis reveals selection for Asian genes in European pigs following human-mediated introgression. Nat Commun. 2014;5:4392.
Hu Z-L, Park CA, Reecy JM. Bringing the animal QTLdb and CorrDB into the future: meeting new challenges and providing updated services. Nucleic Acids Res. 2022;50(D1):D956–61.
Zhang M, Yang L, Su Z, Zhu M, Li W, Wu K, et al. Genome-wide scan and analysis of positive selective signatures in Dwarf Brown-egg Layers and Silky Fowl chickens. Poult Sci. 2017;96(12):4158–71.
Liu W, Li N. Chicken sine oculis binding protein homolog (sobp), a novel gene that may regulate feather development. Poult Sci. 2012;91(8):1950–5.
Cassidy SB, Driscoll DJ. Prader–willi syndrome. Eur J Hum Genet. 2009;17(1):3–13.
Battaglia A, Carey JC, South ST. Wolf–Hirschhorn syndrome: A review and update. Am J Med Genet C Semin Med Genet. 2015;169(3):216–23.
Song Y, Willer JR, Scherer PC, Panzer JA, Kugath A, Skordalakes E, et al. Neural and synaptic defects in slytherin, a zebrafish model for human congenital disorders of glycosylation. PLoS One. 2010;5(10):e13743.
Andrieux J, Devisme L, Valat AS, Robert Y, Frnka C, Savary JB. Prenatal diagnosis of ring chromosome 6 in a fetus with cerebellar hypoplasia and partial agenesis of corpus callosum: case report and review of the literature. Eur J Med Genet. 2005;48(2):199–206.
Pace NP, Maggouta F, Twigden M, Borg I. Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome. Mol Cytogenet. 2017;10:9
Haliburton GD, McKinsey GL, Pollard KS. Disruptions in a cluster of computationally identified enhancers near FOXC1 and GMDS may influence brain development. Neurogenetics. 2016;17(1):1–9.
Kojima T, Morikawa Y, Copeland NG, Gilbert DJ, Jenkins NA, Senba E, et al. TROY, a newly identified member of the tumor necrosis factor receptor superfamily, exhibits a homology with Edar and is expressed in embryonic skin and hair follicles. J Biol Chem. 2000;275(27):20742–7.
Loftus JC, Dhruv H, Tuncali S, Kloss J, Yang Z, Schumacher CA, et al. TROY (TNFRSF19) promotes glioblastoma survival signaling and therapeutic resistance. Mol Cancer Res. 2013;11(8):865–74.
Paulino VM, Yang Z, Kloss J, Ennis MJ, Armstrong BA, Loftus JC, et al. TROY (TNFRSF19) is overexpressed in advanced glial tumors and promotes glioblastoma cell invasion via Pyk2-Rac1 signaling. Mol Cancer Res. 2010;8(11):1558–67.
Spanjaard RA, Whren KM, Graves C, Bhawan J. Tumor necrosis factor receptor superfamily member TROY is a novel melanoma biomarker and potential therapeutic target. Int J Cancer. 2007;120(6):1304–10.
Mooney JA, Yohannes A, Lohmueller KE. The impact of identity by descent on fitness and disease in dogs. Proc Natl Acad Sci. 2021;118(16):e2019116118.
Marsden CD, Ortega-Del Vecchyo D, O’Brien DP, Taylor JF, Ramirez O, Vilà C, et al. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci. 2016;113(1):152–7.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Bioinformatics B. FastQC: a quality control tool for high throughput sequence data. Cambridge, UK: Babraham Institute; 2011.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, et al. Ensembl 2021. Nucleic Acids Res. 2021;49(D1):D884–91.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Picard toolkit: Broad institute; 2019. Available from: https://broadinstitute.github.io/picard/.
Watterson G. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7(2):256–76.
Korunes KL, Samuk K. pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol Ecol Resour. 2021;21(4):1359–68.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4(1):s13742-015-0047–8.
Zhang C, Dong S-S, Xu J-Y, He W-M, Yang T-L. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics. 2019;35(10):1786–8.
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913–8.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97.
Szpiech ZA, Hernandez RD. selscan: an efficient multithreaded program to perform EHH-based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10(2):giab008.
Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 2017;8(1):1–11.
Acknowledgements
We thank Westlake Supercomputer Center for HPC and computation assistance.
Funding
This work was supported by Zhejiang Provincial Natural Science Foundation of China grants LR21C030001, National Natural Science Foundation of China grant 32370662, "Pioneer" and "Leading Goose" R&D Program of Zhejiang 2024SSYS0032, and Research Center for Industries of the Future (RCIF) at Westlake University.
Author information
Authors and Affiliations
Contributions
YZ designed and supervised research; RH and CZ collected samples; RH analyzed data; YZ and RH interpreted the data. RH drafted the paper; YZ revised and edited the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal handling process were evaluated and approved by the Institutional Animal Care and Use Committee (IACUC, approval No. AP#19–045-ZY) at Westlake University. The study is reported in accordance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, R., Zhu, C. & Zhen, Y. Genetic diversity, demographic history, and selective signatures of Silkie chicken. BMC Genomics 25, 754 (2024). https://doi.org/10.1186/s12864-024-10671-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10671-x