
Abstract
Disease can act as a driving force in shaping genetic makeup across populations, even species, if the impacts influence a particularly sensitive part of their life cycles. White-nose disease is caused by a fungal pathogen infecting bats during hibernation. The mycosis has caused massive population declines of susceptible species in North America, particularly in the genus Myotis. However, Myotis bats appear to tolerate infection in Eurasia, where the fungal pathogen has co-evolved with its bat hosts for an extended period of time. Therefore, with susceptible and tolerant populations, the fungal disease provides a unique opportunity to tease apart factors contributing to tolerance at a genomic level to and gain an understanding of the evolution of non-harmful in host-parasite interactions. To investigate if the fungal disease has caused adaptation on a genomic level in Eurasian bat species, we adopted both whole-genome sequencing approaches and a literature search to compile a set of 300 genes from which to investigate signals of positive selection in genomes of 11 Eurasian bats at the codon-level. Our results indicate significant positive selection in 38 genes, many of which have a marked role in responses to infection. Our findings suggest that white-nose syndrome may have applied a significant selective pressure on Eurasian Myotis-bats in the past, which can contribute their survival in co-existence with the pathogen. Our findings provide an insight on the selective pressure pathogens afflict on their hosts using methodology that can be adapted to other host-pathogen study systems.
Similar content being viewed by others

Introduction
Infectious diseases are natural processes affecting wildlife and contribute to ecosystem stability [1]. However, over the past decades, these processes have been intensified due to anthropogenic activities, such as urbanisation, and environmental and climate change [1,2,3]. Anthropogenic impacts on the incidence of epizootic diseases have been identified as an increasing threat to wildlife conservation [1, 3]. The role of infectious diseases in population declines and/or extinction has recently received growing recognition, but there is still a critical need to identify and understand the impacts and projections of diseases on populations [4].
White-nose disease, or white-nose syndrome as it is often referred to, (WND/WNS, please see Whiting-Fawcett et al. 2024 [5] for details on nomenclature) is an epizootic caused by a fungal infection affecting hibernating bats in Eurasia and North America [6]. In North America, it is considered one of the most detrimental wildlife diseases of recent decades [7], with mass mortality of affected species causing unprecedented population collapses in many of the affected areas [6, 8] since it was first discovered in the winter of 2006–2007 [6]. The causative agent, the cold-loving fungus Pseudogymnoascus destructans (= Geomyces destructans) [6], is endemic to the Palearctic, where it does not cause significant mortality in bat hosts [9, 10] due to a potentially extended coevolution between the pathogen and the hosts [11].
Host mortality caused occurs via the disruption of normal patterns of hibernation [12, 13]. Hibernation, characterised by bouts of torpor and arousals, allows bats to survive periods of food scarcity by reducing their energy expenditure via decreased metabolism. Infected North American Myotis lucifugus arouse three times more frequently from torpor in the final third of the hibernation period [13], expending large amounts of the fat reserves that are expected to last until insect food is available again in the spring. The psychrophilic fungus invades host tissue during the extended bouts of torpor when the host immune system is downregulated [14]. Pseudogymnoascus destructans infection induces the production of inflammatory cytokines during the arousals that take place during hibernation [15,16,17]. The presumed irritation, such as pain and itchiness, associated with inflammation and immunopathology, in addition to compounds produced by the fungus [18] and evaporative water loss from open sores [19] have been suggested to increase the frequency of arousals. This leads to premature consumption of fat reserves essential for hibernation and death in the more susceptible bat species [12, 13].
In the Palearctic, P. destructans infects a number of different bat species, mostly in the genus Myotis [9, 20,21,22]. The coevolution between the fungus and the bat hosts is potentially extensive [11, 23], and although no direct evidence exists, it has been suggested that Myotis species may have experienced similar population declines as a result of disease in the past [24]. The most extensively studied Palearctic species, Myotis myotis, elicits neither an immediate immune response or more frequent arousals to the infection which prevents the bats from emaciation [25,26,27], suggesting that these bats utilise tolerance as a response to infection [28, 29]. Tolerance allows the host to evade harmful immunopathology which appears to be a major factor contributing to mortality of bats in the Nearctic [30].
Diseases are a strong driving force of natural selection [31]. Selection pressures result in the modification of the hosts’ genetic diversity and leave behind distinctive signatures in the genome. The nature of these signatures depends on the evolutionary timescale of interest. Among these identifiable footprints of selection are selective sweeps, whereby an advantageous mutation eliminates or reduces variation in the population at linked neutral sites as the mutation increases in frequency. Additionally, alterations in host genetic makeup can be detected in protein coding regions as signals of natural selection; the ratio amino acid changing (non-synonymous or dN) to amino acid preserving (synonymous or dS) substitutions, also known as the Omega (ω) ratio, is used to infer selection. Under neutral evolution the rates are equal (ω = 1). Whereas, an excess of non-synonymous changes is a sign of positive selection (ω > 1), the converse is associated with a predominant negative selection pressure (ω < 1) [32]. Based on the evidence of a long term association between Palearctic bats and P. destructans [23], it is likely that bats have evolved inheritable mechanisms leading to infection tolerance [29]. Signatures of such mechanisms could be detected within protein coding genes associated with wound healing, immune responses and hibernation physiology [15, 26, 27, 33].
Because infected Palearctic Myotis bats, overlapping in range with the pathogen, do not experience significant mortality [9, 10] or elicit a transcriptional response to infection [27], we hypothesise that the genomes of these bat species show signs of positive selection in genes that are associated with immune system signalling and function in response to fungal antigens. To investigate the potential impact of extended coexistence with P. destructans on the genetic makeup of Palearctic Myotis hosts, we use a variety of approaches to detect selection. First, we constructed a whole-genome dataset of 12 Myotis species (1 Nearctic, 11 Palearctic) to investigate the general overall patterns of selection in Palearctic Myotis and conduct a branch test to detect any lineage-specific selection in either the Palearctic M. myotis, which is considered the tolerant main host of P. destructans [9], and the susceptible Nearctic M. lucifugus sampled prior to the arrival of the pathogen. Second, we also assessed the potential of more recent positive selection with selective sweep analysis using single nucleotide polymorphisms (SNPs) in in the same two species, the Nearctic M. lucifugus and Palearctic M. myotis. Additionally, we conducted a systematic literature search to list genes already found to be associated with host responses during infection with P. destructans. Finally, the combined gene set curated from a literature search, sweep analysis and the phylogenetic branch tests were used to test for the presence of inheritable variation within Myotis. The identification of positive selection among these genes may highlight variations that contribute to tolerance among Palearctic lineages of Myotis. The identified gene variants and mechanisms contributing to survival in the Palearctic can be applied to conservation genetic approaches to predict the fate of affected populations.
Methods
Sample collection, DNA extraction and sequencing
Samples for DNA extraction were obtained from existing collections of the authors. No bats were sampled as a part of the present study.
Eleven out of the twelve species sampled represent the main lineages of Myotis from the Palearctic clade (M. bechsteinii, M. daubentonii, M. brandtii, M. mystacinus, M. dasycneme, M. frater, M. pequinus, M. petax, M. myotis, M. nattereri, M. ikonnikovi) with distributions ranging from the Iberian Peninsula to Japan (See Fig. 1). All species reside in the temperate or boreal zone and utilise extended periods of torpor, hibernation, over the winter months [34]. These species overlap in range with the predicted range of P. destructans [35], and all but M. ikonnakovii and M. frater have been described with the fungal infection, but with no signs of mortality [21]. As a whole, these Palearctic Myotis represent taxa that potentially have an extended infection history with the pathogen and are considered tolerant. Of the Palearctic species, the responses to infection by P. destructans have been thoroughly documented M. myotis, a species considered as the primary host for the fungus [9, 26, 27, 36]. The final species is M. lucifugus, the only representative of the Nearctic clade of Myotis in our study. The species has been extensively studied with regards to WND [12, 13, 37, 38] and its responses to infection have been documented [15,16,17, 27, 39,40,41,42]. The purpose of this species in our dataset is to act as an outlier against which the Palearctic species are compared. The estimated divergence times for the species within our dataset range from between 19 and 29.5 mya for the split between Nearctic (M. lucifugus) and Palearctic (all other species) Myotis and c. 5–22 mya within the Palearctic Myotis [43]. Full details for each sample are given in Supplementary Table S1.

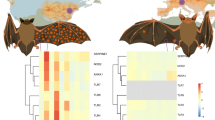
Phylogenetic relationships among 12 Myotis species based on species tree reconstruction using 2,515 genes. Support values represent Astral Support/bootstrap values from the two runs of ASTRAL-III, unless otherwise shown all nodes had 1.00/1.00 support. Distribution maps for each species are colour coded according to the taxa on the phylogeny, with the P. destructans distribution being shown in grey. The distribution maps are based on IUCN (International Union for Conservation of Nature, https://www.iucnredlist.org Accessed on 03 October 2023). Map of distribution range of P. destructans based on Blomberg et al. (2023) [35]. Figure adapted from Harazim et al. 2018 [33]
DNA extraction was carried out using one of the following methods: (1) QIAmp DNA Mini Kit (Qiagen, Germany); (2) DNeasy Blood and Tissue Kit (Qiagen, Germany); (3) SDS Extraction method (full details can be found in Supplementary Methods 1), with extractions being stored at -80 ºC. Library preparation and sequencing was carried out by a variety of providers (see Supplementary Table S1) as follows. For samples processed at: (1) Novogene, United Kingdom: Library preparation was carried out with the Novogene NGS DNA Library Preparation Set (Cat No. PT004) prior to sequencing on a NovaSeq6000 platform to generate 2 × 150 bp reads. (2) Novogene, China: 1.5 µg of DNA was used as input for the TruSeq Nano DNA HT Sample preparation kit (Illumina), with a 350 bp insert. Samples were sequenced on a NovaSeq6000 to generate 2 × 150 bp reads. (3) DNA Sequencing and Genomics Laboratory (BIDGEN): Libraries were prepared using the Nextera™ DNA Flex Library Preparation Kit (Illumina) and sequenced on a NovaSeq6000 to generate 2 × 150 bp reads. (4) University of Liverpool Centre for Genomics Research (CGR): Libraries were prepared using the NEBNext Ultra II FS Kit (~ 300 bp inserts) on the Mosquito platform using a 1/10 reduced volume protocol. Samples were sequenced on a NovaSeq6000 to generate 2 × 150 bp reads. For those samples sequenced at CGR, the provider also carried out read trimming as follows: Adapter removal with Cutadapt (version 1.2.1, [44], followed by trimming with Sickle (version 1.200 [45]), with a minimum window quality score of 20, and removal of reads less than 15 bp.
All raw sequencing reads have been submitted to the NCBI SRA under bioproject PRJNA1051501. In addition to the 18 samples sequenced here, sequencing data from a further six M. lucifugus, originally sampled among the first bats in North America to die of the mycosis, at the onset of the white nose-epizootic in North America [46], were downloaded from the NCBI SRA (see Supplementary Table S2).
Data processing and read mapping
The study is built on four datasets curated in parallel; (i) phylogenetic dataset; (ii) SNP dataset; (iii) literature search dataset and (iv) curated gene set. An overview of how the four datasets are related and which analyses they were used for is summarised in Fig. 2.
Raw reads were quality checked with FASTQC (version 0.11.8, [47], followed by ambiguous base removal with Prinseq (version 0.20.4, [48]. Adapter removal and quality filtering was undertaken with Trimmomatic (version 0.39, [49], using library appropriate adapter sequences and the following settings: ILLUMINACLIP:< adapter>:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:25 MINLEN:50.
All samples were mapped against the M. myotis genome (GCF_014108235.1 mMyoMyo1.p) using bowtie2 (version 2.4.1 [50]) utilising the sensitive-local option. The resulting sam file was converted to a sorted bam file with samtools (version 1.10 [51]). Duplicate removal was carried out with Picard (version 2.27.5 [52]) MarkDuplicates option, with a lenient validation stringency, coordinate sort order and the removal of duplicates. Appropriate reads groups were added to each sample using the AddorReplaceReadGroups function of Picard, with the resulting bam file was indexed with samtools. A consensus assembly was created for each sample with ANGSD (version 0.939, [53] using the following settings: -minQ 20 -doCounts 1 -minMapQ 20 -setMinDepth 3 -dofasta 2.

Phylogenetic dataset construction and analysis
Orthologue identification
In order to identify single copy orthologues, the BUSCO Mammalia_odb10 reference dataset [54] was used. At the time of analysis, the current version of BUSCO did not output nucleotide sequences, therefore Metaeuk (Release 5-34c21f2, [55]) was used to identify gene sequences (settings: easy-predict –cov 0.6 –filter-mas 1 –metaeuk-eval 0.0001 -metaeuk-tcov 0.6 –min-length 40). Result files were filtered to remove any duplicated genes and genes identified in less than 8 individuals. Both the protein and nucleotide sequences produced by Metaeuk were taken forward.
Initial protein alignments were generated using MAFFT (version 7.429, [56]), using the ‘auto’ option and manually checked to ensure accuracy, screened for the presence of pseudogenes, reading frame errors and alignment errors using Geneious 11.0.3 (https://www.geneious.com). After screening, any alignments with less than 8 sequences, < 50% of the ORF or those lacking either M. myotis or M. lucifugus sequences were discarded. This resulted in a final dataset of 2,515 genes (Supplementary Table S3). Amino acid alignments were converted into codon alignments utilising the nucleotide sequences extracted by Metaeuk with Pal2Nal (Version 14, [57]). Alignments were cleaned with gblocks (0.91b, [58]) with the following settings: -t = c, -b3 = 8, -b5 = 15 -b5 = a, b1 = 50% of the number of sequences + 2 and b2 = 50% of the number of sequences + 4. The final gene alignments can be accessed at Zenodo DOI: https://doi.org/10.5281/zenodo.11353518. Annotations for each BUSCO gene code were retrieved from OrthoDB v10 [59].
Phylogenetic analysis
To reconstruct the evolutionary relationships a multispecies coalescent analysis was used. Individual gene trees were reconstructed with IQ-TREE2 (2.1.3, [60]), with ModelFinder [61] being run to determine the optimal model and 1000 ultrafast bootstraps (UFBoot2) approximations [62]. For species tree inference two approaches were undertaken with ATSTRAL-III (5.7.8, [63]), first default parameters were used, while the second analysis incorporated the bootstrap trees from IQ-TREE2 with 500 replicates.
Inferring selection associated with M. myotis and M. lucifugus
Phylogenetic branch tests were implemented to detect selection acting on particular lineages. Two independent analyses were carried out, in the first analysis the branch leading to M. myotis was labelled as the foreground branch, while in the second the branch leading to M. lucifugus was considered the foreground. For each analysis individual gene trees were used. In order for the labelled branches to be read correctly, all of the extra information associated with each branch, such as the branch length and bootstrap scores, were removed with PareTree (version 1.02, Available from: http://emmahodcroft.com/PareTree.html), followed by the branch of interest being labelled. Branch models allow for the labelled (or foreground) branch to have an ω independent of the rest of the phylogeny and were carried out with Paml 4.9j [64]. P-values were generated from the LRT tests using the R software (version 4.1.2, [65]) function pchisq, with correction for multiple tests being carried out with the holm-bonferroni correction using the p.adjust function.
SNP dataset construction and analysis
SNP calling
In parallel to the phylogenetic dataset and branch tests (see Fig. 2.), SNPs were called from M. myotis and M. lucifugus, seven individuals from each species, for selective sweep detection. Because of low numbers of individuals and low sequencing coverage in many of the individuals, we used two SNP callers after which overlapping SNPs were extracted. For M. myotis we used the aligned deduplicated files with read groups (see Data processing and read mapping) for both callers. We also aligned M. lucifugus samples to a draft chromosome-level M. lucifugus assembly (mMyoLuc1, NCBI BioProject PRJNA973719) using the same settings as the M. myotis samples (see Data processing and read mapping), with deduplication and read group addition. The first caller was ANGSD version 0.940 [53, 66] with settings -uniqueOnly 1 -remove_bads 1 -only_proper_pairs 1 -C 50 -baq 1 -minMapQ 20 -minQ 20 -minInd 7 -doCounts 1 -setMinDepth 10 -setMaxDepth 100 -GL 2 -doMajorMinor 1 -doMaf 1 -SNP_pval 1e-6 -doPost 2 -doGeno 3 -doBcf 1 -doGlf 2. The second SNP caller was GATK HaplotypeCaller version 4.3.0.0 [53] with -ERC GVCF and by scaffold (-L). The scaffolds were combined with picard’s GatherVcfs function. All the individual vcfs were combined with GATK’s CombineGVCFs tool. The variants were genotyped with GATK GenotypeGVCFs with settings --heterozygosity 0.001 and -stand-call-conf 20 and SNPs selected with GATK SelectVariants with settings --select-type-to-include SNP --restrict-alleles-to BIALLELIC. SNPs were filtered GATK’s VariantFiltration with --filter-expression “QD < 2.0 || FS > 60.0 || MQ < 40.0 || MappingQualityRankSum < -12.5 || SOR > 3.00 || ReadPosRankSum > -8.00” --filter-name “snp_filter” and selected from the files with vcftools --remove-filtered-all, version 0.1.16 [67].
Both SNP caller files from both species were filtered with vcftools using settings --min-alleles 2 --max-alleles 2 --minDP 5 --maxDP 100 --max-missing 1. In order to select the overlapping SNPs from the files, we used Bedtools intersect version 2.30.0 [68] (bedtools intersect -u -a GATK.vcf -b ANGSD.vcf) and filtered the overlapping SNPs with vcftools --positions. Finally, the SNPs with differing genotypes within an individual from the overlap set was first detected with vcftools --diff-site-discordance and removed with vcftools --exclude-positions.
Selective sweep detection
For the selective sweep detection we used RAiSD version 2.9 [69] excluding known genome gaps. RAiSD was run for each species and the resulting outlier windows (highest µ values 0.05%) were filtered using a conservative threshold (α = 0.0005) in R version 4.3.0. The genes from these windows that were above the threshold were extracted with bedtools intersect using M. myotis NCBI gene annotation release 100 version GCF_014108235.1_mMyoMyo1 in M. myotis. In M. lucifugus samples a draft annotation was used. The mMyoLuc1 draft assembly was annotated by porting over gene annotations from mMyoMyo1 using Liftoff (v1.6.3, [70] and the settings “-exclude_partial -cds -polish;” these annotations were further cleaned using AGAT (v0.9.2, [71]).
Nucleotide diversity
Nucleotide diversity, Watterson Theta, and Tajima’s D were estimated for both species separately using ANGSD. First, the dosaf 1 function was used to calculate the site allele frequency spectrum likelihood (saf) for each species based on individual genotype likelihoods using the same quality specification as in genotype calling. Then, the realSFS function was used to optimize the saf and estimate the unfolded site frequency spectrum (SFS; [72]). Nucleotide diversity, Watterson Theta, and Tajima’s D were calculated for each site with the commands saf2theta and thetaStat in ANGSD.
Literature search
We carried out a literature search to identify genes previously determined to be linked to White-Nose tolerance, using Web of Science (Clarivate) (carried out on 17/04/2023). The presence of the following keywords was searched for in the abstract or title or keywords: ((gene OR genome OR genetic OR transcript*) AND (Pseudogymnoascus destructans OR WNS OR white-nose syndrome OR white-nose disease)). The search yielded 134 papers. Upon closer investigation, 11 of these papers looked at bat genetic changes associated with the fungal disease, yielding a total of 172 genes (Supplementary Table S4 A).
Curated gene dataset
Gene identification, extraction and alignment
A curated gene dataset included the genes identified in the literature search, the genes identified in the sweep analysis (see Supplementary Table S4B) and the genes with the top 1% of ω values (from the phylogenetic dataset, see Sect. 3.3 and Supplementary Table S4 C). Gene codes, and aliases were checked against GeneCards [73]. This resulted in a total of 347 genes of interest (Supplementary Table S4D).
To extract the coding (CDS) region of each gene, the M. myotis NCBI gene annotation release 100 version GCF_014108235.1_mMyoMyo1 was searched for corresponding CDS entries. In the case where genes had multiple isoforms, the longest version was extracted. Exon extraction from the mMyoMyo1 genome was carried out using bedtools getfasta (v2.30.0 [68]) with the force strandedness option, followed by concatenation to create a reference CDS sequence. To ensure correct assembly of each gene, a blastp search was carried out against the NCBI nr database. These assembled CDS sequences were used as a reference set, to ensure correct extraction and assembly of sample CDS regions.
Exons were extracted from each consensus assembly using bedtools getfasta using the force strandedness option and assembled against the reference sequence using the ‘assemble to reference’ option in Geneious, with the subsequent CDS assemblies being aligned using MAFFT, using the auto option. Alignments were manually screened for frameshifts and difficult to align regions, those with less than 8 sequences, < 80% of the CDS region and those lacking sequences from M. lucifugus and M. myotis were discarded. This resulted in a final dataset of 300 genes (Supplementary Table S5). Alignments were automatically cleaned with gblocks with the same settings as the phylogenetic dataset (See orthologue identification). The final gene alignments can be accessed at Zenodo DOI: https://doi.org/10.5281/zenodo.11353518.
Curated gene set site selection patterns
To detect patterns of selection within individual genes, site-based likelihood models were used, whereby ω values vary among codons but not branches, therefore the ω for any given codon is averaged across all branches in the phylogeny and does not indicate which species adaptation is occurring in. For each gene, individual gene trees were first constructed under the maximum likelihood framework with IQ-TREE2, with model selection with ModelFinder and 1000 UFBoot2 approximations. For tests of selection, the ω ratio was estimated for each gene using the CODEML package of PAML, with individual gene trees being used. To detect amino acid sites under selection, four-site model comparisons were implemented (M0:M3, M1a: M2a, M7:M8, M8a: M8). These models have been extensively described elsewhere [64]. Briefly, the one ratio model (M0) allows for a single ω for all sites. The nearly neutral (M1a) has two categories of sites, ω = 1 and ω < 1. M2a (positive selection) has the same two categories as the M1a, with the addition of an ω > 1 category. The discrete (M3) model has three categories of sites for which ω can vary. M7 (‘beta’ neutral model) has eight site categories with eight ω taken from a discrete approximation of the beta distribution (range 0–1), meaning that the signal for positive selection cannot be detected. M8 (‘beta’ plus ω) has the same eight categories as the M7 model, plus and additional categories for which ω can vary from 0 to > 1. While the M8a (beta plus ωS = 1) model is similar to the M8 model, with the ω1 being fixed at one. Multiple models and comparisons were used to test the robustness of the patterns observed. The M0:M3 tests whether ω varies between sites. The other comparisons test for positive selection, with the M1a: M2a being a more conservative test, the M7:M8 having more power and the M8a: M8 combining both power and robustness. Likelihood ratio tests (LRTs) between nested models, where ω is allowed to vary above one and the associated null model, allows inference of the selection pressures acting along the protein sequence [64]. P-values were generated from the LRT tests using the R function pchisq, with correction for multiple tests being carried out with the holm-bonferroni correction using the p.adjust function. Codons under positive selection were identified using the BEB (Bayes Empirical Bayes) method under the M2a model.
For visualisation of positively selected sites within the ANXA1, TNFSF4, CXCL16 and ANKRD17 protein structures, Phyre2 [74] was used to predict protein models using a reference bat protein sequence (XP_036186722.1, XP_036199699.1, XP_036196999.1, XP_036191841.1). Where reliable models could be constructed, the resulting structure was used to predict relative solvent accessibility (RSA) with PolyView-2D [75]. Binding sites in the ANXA1 protein were identified by comparison against the Rat (Rattus norvegicus) version of the protein (Uniprot Id: P07150). For transmembrane domain containing proteins, CXCL16 and TNFSF4, the location of transmembrane domains and signal peptides was carried out with Protter [76].
Gene ontology
Functional enrichment was tested using g: Profiler v. e109_eg56_p17_773ec798 [77]. Enrichment tests for both overrepresentation and underrepresentation were performed using the list of 2,515 genes (Phylogenetic Dataset). Overrepresentation enrichment was analysed for the 38 genes identified as under selection in the Curated Gene Set. For all comparisons, human annotations were used with the background list of all annotated genes. GO enrichment was measured using a g: SCS multiple testing correction threshold of 0.05 and the GO: BP, KEGG, Reactome, and WikiPathways databases.
Results
We generated low coverage whole genome data for 18 Myotis specimens, encompassing 12 species (see Fig. 1 for distributions of host species and P. destructans infections). In addition, a further six M. lucifugus samples were downloaded from genbank (Supplementary Table S2). Mapping to the M. myotis reference genome resulted in an average mapping rate of 96% (Range: 85 − 99%) and an average genomic depth of coverage of 18X (Range: 6X − 53X). See Supplementary Table S6 for full sequencing statistics.
Phylogenetic dataset
Screening of single copy orthologues among our 12 Myotis species, resulted in a final dataset of 2,515 genes. Gene completeness ranged from 82 to 100%, with 2,173 of these genes being found in all 12 species (Supplementary Table S3). When this list of 2,515 genes was tested for functional enrichment (Supplementary Fig. S1), we found that some significant enrichment was observed, but mostly for broad functional categories, such as GO:0050794 regulation of cellular process (padj = 2.632 × 10–17) and GO:0140053 mitochondrial gene expression (padj = 4.751 × 10–7). We also tested this gene list for underrepresentation and found that certain categories were significantly underrepresented, such as GO:0000244 spliceosomal tri-snRNP complex assembly (padj = 1.790 × 10–52) and GO:0007186 G protein-coupled receptor signalling pathway (padj = 2.978 × 10–4), but no immune-related categories were found to be underrepresented (Supplementary Fig. S2). We conclude that this list was not strongly biased in favour of any particular subset of functional pathways and that various immune pathways were sufficiently represented.
To ensure the evolutionary relationships between our samples were as expected the 2,515 gene trees were used to reconstruct the species tree (Fig. 1), which resulted in a topology with generally high support. Astral support values of 100% were estimated for all nodes, except for the split between M. daubentonii and M. bechsteinii. Generally speaking, reconstructed relationships agree with previous studies [43, 78,79,80], with the primary difference in our topology being that M. pequinus is recovered as the sister species to M. myotis rather than M. nattereri.
To test for differences in selection in the branches leading to either M. myotis or M. lucifugus, independent branch tests were carried out. In all tests, no genes were found to have a statistically significant difference between the foreground and background ratios (Supplementary Table S7). A comparison of ω values under the neutral model for the phylogeny as a whole revealed strong patterns of purifying selection (ω < 1), with 17 genes having an ω value > 1 (average: 0.20, Range: 0.0001–2.12, Supplementary Fig. 3A, Supplementary Table S7). The gene with the highest ω value was TNFSF4 (BUSCO code:188443at40674, ω of 2.12).
SNP dataset: selective sweeps detection and nucleotide diversity
In addition to the multi species phylogenetic dataset and consecutive branch tests, we also carried out a selective sweep analysis for both M. myotis and M. lucifugus (See Fig. 2.). We called 1,438,586 SNPs for M. lucifugus and 3,915,084 SNPs for M. myotis after filtering with ANGSD, and 4,304,169 and 11,757,539 SNPs with GATK, respectively (Supplementary Table S8). Of these, the final set of overlapping SNPs was for M. lucifugus 927,923 SNPs, and for M. myotis 3,005,290 SNPs (Supplementary Table 8). In M. lucifugus, there were 464 outlier selective sweep windows which covered 17 genes (Supplementary Fig. S4, Supplementary Table S9). In M. myotis, there were 1,499 outlier windows and 174 genes (Supplementary Fig. S5, Supplementary Table S10). Three outlier genes were shared between the two species: ARHGEF4, PLSCR1 and PLOD2. Nucleotide diversity, Watterson Theta, and Tajima’s D were higher in M. myotis than in M. lucifugus (Supplementary Fig. S6A). Furthermore, some of the sweep regions matched with the low diversity regions in the whole genome level diversity plots (Supplementary Fig. S6A and B). When unidentified LOC-genes were removed, total of 154 sweep genes were added to the curated gene list (see below).
Curated gene set: codon test results
A final gene set of 300 protein coding genes were identified and extracted from our dataset. These 300 genes consisted of those genes identified in: (I) Literature search, (II) Selective Sweep analysis and (III) overall ω values from the phylogenetic dataset. An overview of how these datasets are related is shown in Fig. 2, and the overlap between each of these categories is shown in Fig. 3.

Venn Diagram of the curated gene set (300 genes in total) highlighting which dataset(s) they originate from and which analysis, if any, where they are significant. M0 Omega refers to the top 1% of omega values from the branch test null model (Phylogenetic Dataset); Sweep refers to the genes linked to the SNPs identified in our selective sweep analysis; Literature refers to all the genes identified from our literature search; and Codon Test refers to the 38 genes identified as under positive selection from our curated gene dataset
Of the 300 genes, 270 were identified in all 12 species (Supplementary Table S5). To determine the selective pressures acting on these genes, an ω value was calculated for each orthologous gene set using site-based likelihood models. We found the majority of genes to be evolving under purifying selection when a single ω value is calculated across the entire coding region using the one-ratio (M0) model, mean = 0.32, range 0.0001–2.12 (Supplementary Fig. S3B, Supplementary Table S11). Twenty-one genes have an ω > 1, indicating the protein as a whole is under positive selection (Supplementary Table S12 A). As seen in the phylogenetic dataset, TNFSF4 had the highest ω of 2.12. Rate heterogeneity among sites was tested using the M0:M3 comparison; significant among site variation was observed in 107 of the 300 genes tested. This, however, is not a test of positive selection. The three different model comparisons allow for the identification of positive selection among codons, each varying in their power and robustness, with ω values for each codon being averaged across the phylogeny. The genes identified in each comparison are given in Supplementary Table S12B. A total of 46 genes were identified in one or more comparisons, with 38 genes being identified in all three tests. In these 38 genes showing the strongest evidence for positive selection, we identified specific amino acids sites under significant selection. The number of sites identified as statistically significant under the robust M2a model ranges from 0 to 46 for each gene (Supplementary Table S13). Of the 38 genes showing the strongest signal for selection, six were from the selective sweep genes, 11 from the top 1% of omega values and 22 from the literature search (Fig. 3).
The 38 genes that were identified with the strongest evidence for selection were further examined for functional enrichment using a statistical enrichment analysis and human gene ontology databases. We found that this gene list was highly enriched for genes with functional annotations related to many immune-related categories (Supplementary Table S14) and categories related to inflammatory immune responses were particularly common (Fig. 4). For example, in the GO: Biological Process database, we found enrichment of GO:0045087 innate immune response (padj = 5.515 × 10− 6), GO:0006954 inflammatory response (padj = 5.836 × 10− 6), GO:0032637 interleukin-8 production (padj = 9.751 × 10− 6) GO:0002250 adaptive immune response (padj = 3.393 × 10− 4), and GO:0030097 hemopoiesis (padj = 3.735 × 10− 3). In the KEGG database, we found enrichment for category KEGG:04613 Neutrophil extracellular trap formation (padj = 3.185 × 10− 6); in the Reactome database, we found enrichment for categories involved in the TLR2/4 pathway, such as REAC: R-HSA-5,602,498 MyD88 deficiency (TLR2/4) (padj = 3.612 × 10− 5); and in the WikiPathways database, we found enrichment of category WP: WP4493 Cells and molecules involved in local acute inflammatory response (padj = 2.953 × 10− 5). Together, these functional enrichment results demonstrate that the majority of the genes identified under the strongest selection by our screens are related to inflammatory immune responses to the fungal pathogen mediated by both innate and adaptive mechanisms.

An overview of some of the key categories enriched in our 38 genes under positive selection. The four genes discussed in further detail are highlighted in red
The gene, TNFSF4 had the highest overall ω in the Phylogenetic and Curated Gene datasets, with an overall ω of 2.12. In addition, 9 sites were identified in our codon tests as being under positive selection, with ω values of ~ 9 (Supplementary Table S13, Fig. 5A). All nine sites are located in the extracellular portion of the protein (Supplementary Fig. S7). Modelling of the three dimensional protein structure was successful for ~ 60% of the protein sequence (Supplementary Model 1, Fig. S5B, a 360o view is shown in Supplementary File S1); assessing the solvent accessibility of the resulting model showed that residue 121 is fully exposed to the solvent, with an additional 3 sites (22, 64, 66) at least partially exposed, with RSA values > 5, and the remaining 5 being mostly buried within the protein structure (RSA < 5, Supplementary Fig. S8).
Among the 38 genes with strong evidence of positive selection, the gene CXCL16 was the only gene to have originated from multiple datasets (being identified in the literature screen [27] and among the genes with an ω > 1 in the Phylogenetic dataset). The protein as a whole has an ω of 1.4, with six codons being identified as being under positive selection (ω range: 4.6–4.8, Supplementary Table S13, Fig. 5A). We were unable to model the structure of CXCL16 with high confidence, however all six sites occur within the extracellular region of the protein (Supplementary Fig. S9).

(A) An overview of the sites under positive selection and their amino acid changes relative to their phylogenetic position for our four proteins (ANKRD17, ANXA1, CXCL16, TNFSF4) of interest. The phylogenetic tree shown is the same as that shown in Fig. 1. (B) Three-dimensional model of TNFSF4, with the residues under selection being highlighted in pink. (C) Plot of omega value along the length of the ANKRD17 protein, positions with an * are those identified as being under positive selection. (D) Three-dimensional model of ANXA1, with the calcium binding sites being shown in yellow, sites under positive selection have been coloured as follows: Pink: Codons under selection in our study; Orange: Codons under selection in both this study and Harazim et al. 2018 [33]; Aqua: Codons under selection in Harazim et al. 2018 [33]
Of the 11 sweep genes also identified in the codon analysis, ANKRD17 had the largest proportion of codons under positive selection. Overall, ANKRD17 had an ω of 0.3. A total of 16 codons were identified as being under positive selection all with an ω value of ~ 8 (Supplementary Table S13, Fig. 5A). All 16 codons occur towards the end of the coding region (Fig. 5C), with very little variation seen in approximately the first half of the coding sequence (Supplementary Fig. S10). It was not possible to model the structure of this protein with high confidence, so no inference on where the 16 codons are in the three-dimensional structure is possible.
Annexin A1 (ANXA1) was among the genes with positively selected sites, that had previously been identified in other studies [27, 33], with some evidence for codon specific selection occurring. Overall, ANXA1 had an ω of 0.5, with 10 codons being identified as under positive selection with ω values of ~ 7.8 (Supplementary Tables 12 and 13, Fig. 5A). Half of the sites identified are located in the N-terminal region of the protein (5 out of 10 sites identified are within the range 22–27). Relative solvent accessibility calculations based on the modelled structure (Supplementary Model 2, Fig. S5D, a 360o view is shown in Supplementary File S2), show residue 27 is fully exposed to the solvent, while residues 22, 23 and 219 are partially exposed (RSA ≥ 5) and the remaining residues are either completely or mostly buried within the protein (Supplementary Fig. S11).
Discussion
Our comprehensive genomic study, utilising candidate genes sourced from a variety of datasets, detected strong positive selection in a suite of genes in Palearctic species of Myotis. Although a myriad of different factors may contribute to selection pressures across the genome in our focal species, a considerable proportion of the genes have been associated with responses to fungal infection.
The species tree generated in this study using 2,515 genes generally agrees with the more recent studies on Myotis relationships [43]. One should bear in mind, that the tree produced for this study is limited due to the scope of the study and number of species used, and no novel results on evolutionary relationships can be inferred. Investigation into patterns of selection presents a difficult statistical challenge, which can be influenced by tree topology [81]. We acknowledge that the use of gene trees, as opposed to the species tree, may influence our results. However, we believe the use of gene trees to be appropriate for a number of reasons. Evolutionary relationships among Myotis species have been shown to be discordant [82,83,84]. Secondly, constraining the topology for a given gene can overestimate the substitution rate on constrained branches, which in turn overestimates the ω ratio, leading to an increase in false positives [85] Indeed, within our dataset despite the high branch support for relationships, the conflict between gene trees and species trees is high. The level of conflict can be assessed with quartet scores, which show an overall score of 0.79 for the species tree topology, however some branches have scores as low as 0.34 (Supplementary Fig. S12), emphasising the high level of discordance among Myotis species relationships. We believe by not constraining our phylogenetic branch tests to the species tree topology we provide a more conservative estimation of selection patterns.
The patterns of selection on the proteins in the phylogenetic dataset highlight overall functional constraint (ω < 1). This is a common occurrence with proteins generally being under purifying selection as a whole, with adaptation occurring within proteins at selected sites/regions [86, 87]. Of the 2,515 genes tested, 17 had overall ω values indicative of positive selection. Of these genes showing positive selection, the strongest signal was detected in TNFSF4. However, we found no evidence for lineage specific selection associated with either M. myotis, the species most associated with infection in the Palearctic [9, 21] or M. lucifugus, an affected species in the Nearctic [8], when these were contrasted against the other Myotis species in a branch test. From this analysis, the top 1% of highest ω values were then included in the subsequent curated gene set.
We also looked at signals of selection using selective sweep analysis in European M. myotis, using naïve M. lucifugus sampled at the onset of the epizootic in North America as a comparison. Over a hundred genes were outliers in the M. myotis population (Fig. 2, Supplementary Fig. S5) that has coevolved with the fungal pathogen, in contrast to the M. lucifugus, where only seventeen genes were in the sweep regions (Supplementary Fig. S4). Three genes were shared between the species, most notably phospholipid scramblase 1 (PLSCR1), an important gene in the antiviral responses [88]. It is important to note that while selective sweep analyses can provide valuable insights into the genetic basis of adaptation, they have limitations, and the conclusions drawn should be interpreted with caution. In our case, sample size and sequencing coverage were the most important factors limiting the analyses. Other evolutionary processes and demographic factors as well can also influence patterns of genetic diversity, and it can be challenging to distinguish the causality between these. Therefore, multiple lines of evidence and additional studies are often necessary to confirm the effects of selection on specific loci.
We constructed a curated gene set of 300 proteins from the literature search, sweep analysis and overall ω values to investigate selection patterns within genes potentially associated with tolerance to infection by P. destructans. As with our phylogenetic dataset, most genes had an ω < 1, indicative of overall purifying selection. Twenty-one genes including TNFSF4 showed signals for positive selection over the whole gene. Among our 300 genes, we found evidence for selection acting on at least one amino acid site in 38 genes.
We found that TNFSF4 showed the most positive selection amongst the genes analysed in our study. Interestingly, this gene did not originate from the candidate set of genes gleaned from literature on WND, but rather from the phylogenomic analysis that took an unbiased approach to identifying genes for our downstream analyses. This protein, tumor necrosis factor (ligand) superfamily, member 4, also known as OX40L or CD252, is a potent activator of inflammatory signalling when it is expressed on antigen-presenting cells where it can activate OX40 on T lymphocytes, NK cells, neutrophils, and others [89]. TNFR/TNF superfamily members can control diverse aspects of immune function. The mostly extracellular protein that is encoded by TNFSF4, in conjunction with its partner TNFRSF4, is associated with strong regulation of conventional CD4 and CD8 T cells, modulation of NKT cell and NK cell function, and mediation of cross-talk with professional antigen-presenting cells and diverse cell types such as mast cells, smooth muscle cells and endothelial cells. Additionally, TNFSF4-TNFSRF4 interactions alter the differentiation and activity of regulatory T cells [89] and TNFSF4 mediates adhesion of activated T cells to endothelial cells during infection, inducing secretion of proinflammatory cytokines [90]. This gene clearly has a diverse function in the immune system, including that of viral immunity, but has also been specifically linked to responses to fungal infections. Several investigations have demonstrated that TNFSF4 expression is upregulated in mice sensitized to Aspergillus fumigatus [91] and that TNFSF4 polymorphisms are associated with the risk of developing invasive aspergillosis infection [92]. In addition, blocking TNFSF4, either transcription or translation, has produced strong therapeutic effects in multiple animal models of autoimmune and inflammatory disease [89]. Because of the diverse function of the TNFR/TNF superfamily, we cannot associate positive selection in the gene to just extended exposure to P. destructans, as it may also be associated with coevolution with viruses [93].
CXCL16 is another notable gene, filtering through to the final dataset from both the top 1% of genes in the phylogenetic dataset and the literature search, being previously observed as an upregulated gene in naïve infected M. lucifugus [27]. The protein, expressed as either a soluble or a transmembrane form, is a marker of inflammation in humans and is produced by monocytes/macrophages, B cells, dendritic cells, keratinocytes, and endothelial cells [94]. As the sole ligand for the receptor CXCR6, soluble CXCL16 promotes the directional migration of CXCR6+ cells, such as CD4+ effector memory T cells and natural killer T-cells [95], with expression being induced by inflammatory cytokines [96]. All sites under selection in our study are in the extracellular section of the transmembrane polypeptide, indicating possible functional changes in the interactions between CXCL16 and its receptor, CXCR6.
From among the genes identified in the selective sweep analysis, ANKRD17 had the highest number of codons under selection. This gene encodes for an ankyrin repeat protein, which has previously been reported to be an important regulator of the cell cycle [97] and may also be involved in innate immune activation by viruses and bacteria [98, 99]. The codons under selection for this gene are clustered in the C-terminus of the protein, outside of the region of ankyrin repeats that has been shown to bind to the NLR-family pattern-recognition receptor, NOD2 [99], and in the region that is putatively responsible for activation of RIG-I-like receptors [98]. This may indicate some cross-talk between antifungal and antiviral innate signalling pathways, or it could be related to the presence of viral coinfections in some bats with WND [100]. Our literature search identified several studies that implicated the involvement of ANXA1, Annexin A1, a protein that is a well-characterized anti-inflammatory inhibitor of cytosolic phospholipase A2, in a variety of human diseases [101]. In addition, ANXA1 is a known target for positive selection in Myotis infected by P. destructans [33], and shows increased transcription in hibernating, infected Nearctic M. lucifugus compared to uninfected conspecifics, or Palearctic M. myotis [27]. Harazim et al. [33]. suggested ANXA1 may act via two different routes in bats infected by P. destructans. First, ANXA1 has the potential to down-regulate the immune responses initiated by bats arousing from torpor, which can lead to immunopathology if left unchecked. ANXA1 appears to regulate the neutrophil response under fungal infection conditions, altering lipid membrane organization and metabolism [102]. ANXA1 has also been found to participate in adaptive immunity against chronic infectious disease [103], by directing the immune response towards a Th1/Th17 response, a response that is associated not only with clearing fungal infection in normothermic mammals, but also the mortality evidenced in susceptible Nearctic populations of M. lucifugus infected by P. destructans [17]. Second, ANXA1 also appears to play a role in wound repair and epithelial recovery, extending its importance beyond the acute phase of inflammation to the equally important healing phase [104, 105]. Our study found five of the seven sites recognized under positive selection in the study by Harazim et al. [33]. , with an addition of four previously undescribed sites. All previously identified sites were confined to the N-terminal region of the protein, where ANXA1 is known to bind to S100A11 and inhibit phospholipase A2 [106, 107]. We found additional evidence for selection at three sites towards the C-terminal end of the protein, whose function is not as well characterized.
We acknowledge that our screen for selection filters out other types of adaptive variation that may play a key role in the development of tolerance. The screen we employ here fails to take into account the role synonymous codon changes have in altering mRNA structure and stability, the rate at which translation occurs and any resulting structural changes in protein conformation [108, 109]. Additionally, by primarily screening protein coding genes any adaptive variation in introns, UTRs and intergenic regions that may potentially alter the expression [110] of key proteins are excluded from our analysis.
Our combined approach to identify genes involved in adaptation to infection by P. destructans revealed a preponderance of genes with known functions involved in defence responses, including both innate and adaptive immune responses. In addition to the specific genes described above, we found strong evidence that pathways that activate inflammatory responses and neutrophil recruitment are under selection. This is highly consistent with the observed pathology associated with infection by P. destructans in naive populations, both during hibernation and after emergence [111]. IL-8 signalling appears to be among the inflammatory pathways that are particularly important, and this cytokine is known to recruit neutrophils to sites of infection and enhance their activation, a process that likely generates significant immunopathology. Other pathways that appear to be involved in adaptation included coagulation and stress responses, although it is not clear how increased or decreased activation of these pathways would enhance disease tolerance. Presumably the driving force of natural selection is to suppress the inflammatory responses that lead to immunopathology, or at least delay them until it is more energetically favourable to mount a resistance response.
Conclusions
A volume of research over the last decade indicates not only the significance of immunopathology in the cascade of physiological events that build up to mortality associated with WND [15,16,17], but also the lack of immunopathology in infected hosts in the Palearctic [25,26,27, 36]. This is suggestive of the evolution of tolerance in Palearctic bats through coevolution over an extended period of time [23, 29], a process that may already have been initiated in Nearctic Myotis also [112, 113]. Selection towards tolerance to the fungal infection in the Palearctic has been suggested [5, 29] and indicated at a genomic level [33], but here, through an exhaustive approach, we can state with higher confidence that genes and pathways associated with fungal infections, and particularly those involved in infection by P. destructans, have been under positive selection in Palearctic species of Myotis. Of course, when investigating patterns of selection across entire genomes of bats, albeit related, across two continents, we acknowledge that there are a plethora environmental factors also driving the observed selection patterns. Therefore, further functional analysis e.g., proteomics and immunological modelling, or experimental validation is needed to elucidate exactly how the observed selection affects response to infection. Nevertheless, attempts to identify these genes, and their variants will allow for the estimation of survival in North American affected species through population genetic approaches to assist in implementing conservation measures [5, 114]. Finally, not only do the results illuminate processes driving tolerance in a bat fungal disease, but bolster our holistic understanding of how novel host-parasite interactions drive selection towards tolerance in host organisms.
Data availability
All raw data generated in this study are accessible at NCBI under bioproject PRJNA1051501. All final gene alignments, and gene trees can be found on Zenodo at https://doi.org/10.5281/zenodo.11353518.
References
Cunningham AA, Daszak P, Wood JLN. One health, emerging infectious diseases and wildlife: two decades of progress? Philosophical Trans Royal Soc B: Biol Sci. 2017;372:20160167.
Baylis M, Risley C. Infectious diseases, climate change effects on. In: Meyers RA, editor. Encyclopedia of sustainability science and technology. New York, NY: Springer; 2012. pp. 5358–78.
Baker RE, Mahmud AS, Miller IF, Rajeev M, Rasambainarivo F, Rice BL, et al. Infectious disease in an era of global change. Nat Rev Microbiol. 2022;20:193–205.
Daszak P, Cunningham AA, Hyatt AD. Emerging infectious diseases of wildlife-threats to biodiversity and human health. Science. 2000;287:443–9.
Whiting-Fawcett F, Blomberg AS, Troitsky T, Meierhofer MB, Field KA, Puechmaille SJ et al. A palearctic view of a bat fungal disease. Conserv Biol. 2024;n/an/a:e14265.
Blehert DS, Hicks AC, Behr M, Meteyer CU, Berlowski-Zier BM, Buckles EL, et al. Bat white-nose syndrome: an emerging fungal pathogen? Science. 2009;323:227–227.
Frick WF, Puechmaille SJ, Willis CKR. White-nose syndrome in bats. In: Voigt CC, Kingston T, editors. Bats in the Anthropocene: conservation of bats in a changing world. Cham: Springer International Publishing; 2016. pp. 245–62.
Frick WF, Puechmaille SJ, Hoyt JR, Nickel BA, Langwig KE, Foster JT, et al. Disease alters macroecological patterns of north American bats. Glob Ecol Biogeogr. 2015;24:741–9.
Puechmaille SJ, Wibbelt G, Korn V, Fuller H, Forget F, Mühldorfer K, et al. Pan-european distribution of white-nose syndrome fungus (Geomyces destructans) not associated with mass mortality. PLoS ONE. 2011;6:e19167.
Fritze M, Puechmaille SJ. Identifying unusual mortality events in bats: a baseline for bat hibernation monitoring and white- nose syndrome. Mammal Rev. 2018;48:224–8.
Fischer NM, Dool SE, Puechmaille SJ. Seasonal patterns of Pseudogymnoascus destructans germination indicate host – pathogen coevolution. Biol Lett. 2020;16:1–5.
Reeder DM, Frank CL, Turner GG, Meteyer CU, Kurta A, Britzke ER et al. Frequent arousal from hibernation linked to severity of infection and mortality in bats with white-nose syndrome. PLoS ONE. 2012;7.
Warnecke L, Turner JM, Bollinger TK, Lorch JM, Misra V, Cryan PM, et al. Inoculation of bats with European Geomyces destructans supports the novel pathogen hypothesis for the origin of white-nose syndrome. Proc Natl Acad Sci U S A. 2012;109:6999–7003.
Meteyer CU, Buckles EL, Blehert DS, Hicks AC, Green DE, Shearn-Bochsler V, et al. Histopathologic criteria to confirm white-nose syndrome in bats. J Vet Diagn Invest. 2009;21:411–4.
Field KA, Johnson JS, Lilley TM, Reeder SM, Rogers EJ, Behr MJ, et al. The white-nose syndrome transcriptome: activation of anti-fungal host responses in wing tissue of hibernating bats. PLoS Pathog. 2015;11:1–29.
Field KA, Sewall BJ, Prokkola JM, Turner GG, Gagnon MF, Lilley TM, et al. Effect of torpor on host transcriptomic responses to a fungal pathogen in hibernating bats. Mol Ecol. 2018;27:3727–43.
Lilley TM, Prokkola JM, Johnson JS, Rogers EJ, Gronsky S, Kurta A, et al. Immune responses in hibernating little brown myotis (Myotis lucifugus) with white-nose syndrome. Proc R Soc B. 2017;284:20162232.
Flieger M, Bandouchova H, Cerny J, Chudíčková M, Kolarik M, Kovacova V, et al. Vitamin B2 as a virulence factor in Pseudogymnoascus destructans skin infection. Sci Rep. 2016;6:33200.
McGuire LP, Mayberry HW, Willis CKR. White-nose syndrome increases torpid metabolic rate and evaporative water loss in hibernating bats. Am J Physiology-Regulatory Integr Comp Physiol. 2017;313:R680–6.
Hoyt JR, Sun K, Parise KL, Lu G, Langwig KE, Jiang T et al. Widespread Bat White-Nose Syndrome Fungus, Northeastern China. Emerg Infect Dis. 2015;22.
Zukal J, Bandouchova H, Brichta J, Cmokova A, Jaron KS, Kolarik M, et al. White-nose syndrome without borders: Pseudogymnoascus destructans infection tolerated in Europe and Palearctic Asia but not in North America. Sci Rep. 2016;6:19829.
Wibbelt G, Kurth A, Hellmann D, Weishaar M, Barlow A, Veith M, et al. White-nose syndrome fungus (Geomyces destructans) in bats, Europe. Emerg Infect Dis. 2010;16:1237–43.
Leopardi S, Blake D, Puechmaille SJ. White-nose syndrome fungus introduced from Europe to North America. Curr Biol. 2015;25:R217–9.
Martinkova N, Backor P, Bartonicka T, Blazkova P, Cerveny J, Falteisek L, et al. Increasing incidence of Geomyces destructans fungus in bats from the Czech Republic and Slovakia. PLoS ONE. 2010;5:e13853.
Fritze M, Costantini D, Fickel J, Wehner D, Czirják GÁ, Voigt CC. Immune response of hibernating European bats to a fungal challenge. Biology Open. 2019;8:1–10.
Fritze M, Puechmaille SJ, Costantini D, Fickel J, Voigt CC, Czirják GÁ. Determinants of defence strategies of a hibernating European bat species towards the fungal pathogen Pseudogymnoascus destructans. Dev Comp Immunol. 2021. 104017.
Lilley TM, Prokkola JM, Blomberg AS, Paterson S, Johnson JS, Turner GG et al. Resistance is futile: RNA-sequencing reveals differing responses to bat fungal pathogen in Nearctic Myotis lucifugus and Palearctic Myotis myotis. Oecologia. 2019;:295–309.
Ayres JS, Schneider DS. Tolerance of infections. Annu Rev Immunol. 2012;30:271–94.
Whiting-Fawcett F, Field KA, Puechmaille SJ, Blomberg AS, Lilley TM. Heterothermy and antifungal responses in bats. Curr Opin Microbiol. 2021;62:61–7.
Mandl JN, Schneider C, Schneider DS, Baker ML. Going to bat(s) for studies of disease tolerance. Front Immunol. 2018;9.
Karlsson EK, Kwiatkowski DP, Sabeti PC. Natural selection and infectious disease in human populations. Nat Rev Genet. 2014;15:379–93.
Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503.
Harazim M, Horáček I, Jakešová L, Luermann K, Moravec JC, Morgan S, et al. Natural selection in bats with historical exposure to white-nose syndrome. BMC Zool. 2018;3:1–13.
Wilson DE, Mittermeier RA, editors. Handbook of the mammals of the World. Vol. 9. Bats. Barcelona: Lynx Edicions; 2019.
Blomberg AS, Lilley TM, Fritze M, Puechmaille SJ. Climatic factors and host species composition at hibernation sites drive the incidence of bat fungal disease. 2023;:2023.02.27.529820.
Hecht-Höger AM, Beate CB, Krause E, Meschede A, Krahe R, Voigt CC, et al. Plasma proteomic profiles differ between European and north American myotid bats colonized by Pseudogymnoascus destructans. Mol Ecol. 2020;29:1745–55.
Cheng TL, Gerson A, Moore MS, Reichard JD, DeSimone J, Willis CKR, et al. Higher fat stores contribute to persistence of little brown bat populations with white-nose syndrome. J Anim Ecol. 2019;88:561–600.
Frick WF, Johnson E, Cheng TL, Lankton JS, Warne R, Dallas J, et al. Experimental inoculation trial to determine the effects of temperature and humidity on white-nose syndrome in hibernating bats. Sci Rep. 2022;12:971.
Johnson JS, Reeder DM, Lilley TM, Czirják GÁ, Voigt CC, McMichael JW et al. Antibodies to Pseudogymnoascus destructans are not sufficient for protection against white-nose syndrome. Ecol Evol. 2015;:2203–14.
Davy CM, Donaldson ME, Willis CKR, Saville BJ, McGuire LP, Mayberry HW, et al. The other white-nose syndrome transcriptome: tolerant and susceptible hosts respond differently to the pathogen pseudogymnoascus destructans. Ecol Evol. 2017;7:7161–70.
Willis CKR, Menzies AK, Boyles JG, Wojciechowski MS. Evaporative water loss is a plausible explanation for mortality of bats from white-nose syndrome. Integr Comp Biol. 2011;51:364–73.
Donaldson ME, Davy CM, Willis CKR, McBurney S, Park A, Kyle CJ. Profiling the immunome of little brown myotis provides a yardstick for measuring the genetic response to white-nose syndrome. Evol Appl. 2017;10:1076–90.
Morales AE, Ruedi M, Field K, Carstens BC. Diversification rates have no effect on the convergent evolution of foraging strategies in the most speciose genus of bats, Myotis*. Evolution. 2019;73:2263–80.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
Joshi N, Fass J, Sickle. A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. https://github.com/najoshi/sickle. 2011.
Lilley TM, Wilson IW, Field KA, Reeder DM, Vodzak ME, Turner GG, et al. Genome-wide changes in genetic diversity in a Population of Myotis lucifugus affected by White-Nose Syndrome. G3: Genes Genomes Genet. 2020;10:2007–20.
Andrews S. FastQC A Quality Control tool for High Throughput Sequence Data. 2015.
Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4.
Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. 2014.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10:giab008.
Picard Tools - By Broad Institute. https://broadinstitute.github.io/picard/. Accessed 28 Aug 2023.
Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:356.
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO Update: Novel and Streamlined Workflows along with broader and deeper phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647–54.
Levy Karin E, Mirdita M, Söding J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome. 2020;8:48.
Katoh K, Standley DM. MAFFT multiple sequence alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34 suppl2:W609–12.
Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–52.
Kriventseva EV, Kuznetsov D, Tegenfeldt F, Manni M, Dias R, Simão FA, et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019;47:D807–11.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the Ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–22.
Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018;19:153.
Yang Z. PAML 4: phylogenetic analysis by Maximum Likelihood. Mol Biol Evol. 2007;24:1586–91.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. 2022.
Kim SY, Lohmueller KE, Albrechtsen A, Li Y, Korneliussen T, Tian G, et al. Estimation of allele frequency and association mapping using next-generation sequencing data. BMC Bioinformatics. 2011;12:231.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Alachiotis N, Pavlidis P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Commun Biol. 2018;1:1–11.
Shumate A, Salzberg SL. Liftoff: accurate mapping of gene annotations. Bioinformatics. 2021;37:1639–43.
Dainat J, Murray K, Hereñú D, Davis E, Crouch K, Sol L et al. AGAT: another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. (Version v0.9.2). 2023. https://doi.org/10.5281/zenodo.8178877
Nielsen R, Korneliussen T, Albrechtsen A, Li Y, Wang J. SNP calling, genotype calling, and sample allele frequency estimation from new-generation sequencing data. PLoS ONE. 2012;7:e37558.
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, et al. The GeneCards suite: from Gene Data Mining to Disease Genome sequence analyses. Curr Protocols Bioinf. 2016;54:1301–13033.
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–58.
Porollo AA, Adamczak R, Meller J. POLYVIEW: a flexible visualization tool for structural and functional annotations of proteins. Bioinformatics. 2004;20:2460–2.
Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30:884–6.
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47:W191–8.
Ruedi M, Mayer F. Molecular Systematics of bats of the Genus Myotis (Vespertilionidae) suggests deterministic ecomorphological convergences. Mol Phylogenet Evol. 2001;21:436–48.
Ruedi M, Stadelmann B, Gager Y, Douzery EJP, Francis CM, Lin L-K, et al. Molecular phylogenetic reconstructions identify East Asia as the cradle for the evolution of the cosmopolitan genus Myotis (Mammalia, Chiroptera). Mol Phylogenet Evol. 2013;69:437–49.
Shi JJ, Rabosky DL. Speciation dynamics during the global radiation of extant bats. Evolution. 2015;69:1528–45.
Diekmann Y, Pereira-Leal JB. Gene Tree affects inference of sites under Selection by the Branch-Site Test of positive selection. Evol Bioinform Online. 2015;11s2:EBO.S30902.
Platt RNII, Faircloth BC, Sullivan KAM, Kieran TJ, Glenn TC, Vandewege MW, et al. Conflicting evolutionary histories of the mitochondrial and nuclear genomes in New World Myotis bats. Syst Biol. 2018;67:236–49.
Foley NM, Harris AJ, Bredemeyer KR, Ruedi M, Puechmaille SJ, Teeling EC et al. Karyotypic stasis and swarming influenced the evolution of viral tolerance in a species-rich bat radiation. Cell Genomics. 2024;4.
Korstian JM, Paulat NS, Platt RN, Stevens RD, Ray DA. SINE-Based phylogenomics reveal extensive introgression and incomplete lineage sorting in Myotis. Genes. 2022;13:399.
Mendes FK, Hahn MW. Gene Tree Discordance causes apparent substitution rate variation. Syst Biol. 2016;65:711–21.
Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and Applications to the HIV-1 envelope gene. Genetics. 1998;148:929–36.
Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–49.
Xu D, Jiang W, Wu L, Gaudet RG, Park E-S, Su M, et al. PLSCR1 is a cell-autonomous defence factor against SARS-CoV-2 infection. Nature. 2023;619:819–27.
Croft M. Control of immunity by the TNFR-Related molecule OX40 (CD134). Annu Rev Immunol. 2010;28:57–78.
Chiang LY, Sheppard DC, Gravelat FN, Patterson TF, Filler SG. Aspergillus Fumigatus stimulates leukocyte adhesion molecules and cytokine production by endothelial cells in Vitro and during Invasive Pulmonary Disease. Infect Immun. 2008;76:3429–38.
Barrios CS, Johnson BD, Henderson D, Fink J, Kelly JN, Kurup KJ. The costimulatory molecules CD80, CD86 and OX40L are up-regulated in aspergillus fumigatus sensitized mice. Clin Exp Immunol. 2005;142:242–50.
Sánchez-Maldonado JM, Moñiz-Díez A, ter Horst R, Campa D, Cabrera-Serrano AJ, Martínez-Bueno M, et al. Polymorphisms within the TNFSF4 and MAPKAPK2 loci influence the risk of developing invasive aspergillosis: a two-stage Case Control Study in the context of the aspBIOmics Consortium. J Fungi. 2021;7:4.
Banerjee A, Baker ML, Kulcsar K, Misra V, Plowright R, Mossman K. Novel insights into Immune systems of bats. Front Immunol. 2020;11.
Hase K, Murakami T, Takatsu H, Shimaoka T, Iimura M, Hamura K, et al. The membrane-bound chemokine CXCL16 expressed on follicle-Associated Epithelium and M cells mediates Lympho-Epithelial Interaction in GALT1. J Immunol. 2006;176:43–51.
Wilbanks A, Zondlo SC, Murphy K, Mak S, Soler D, Langdon P, et al. Expression cloning of the STRL33/BONZO/TYMSTRligand reveals elements of CC, CXC, and CX3C chemokines. J Immunol. 2001;166:5145–54.
Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, et al. The transmembrane CXC-Chemokine Ligand 16 is Induced by IFN-γ and TNF-α and shed by the activity of the disintegrin-like Metalloproteinase ADAM10 1. J Immunol. 2004;172:6362–72.
Deng M, Li F, Ballif BA, Li S, Chen X, Guo L, et al. Identification and functional analysis of a Novel cyclin E/Cdk2 substrate Ankrd17 *. J Biol Chem. 2009;284:7875–88.
Wang Y, Tong X, Li G, Li J, Deng M, Ye X. Ankrd17 positively regulates RIG-I-like receptor (RLR)-mediated immune signaling. Eur J Immunol. 2012;42:1304–15.
Menning M, Kufer TA. A role for the ankyrin repeat containing protein Ankrd17 in Nod1- and Nod2-mediated inflammatory responses. FEBS Lett. 2013;587:2137–42.
Davy CM, Donaldson ME, Subudhi S, Rapin N, Warnecke L, Turner JM, et al. White-nose syndrome is associated with increased replication of a naturally persisting coronaviruses in bats. Sci Rep. 2018;8:15508.
Gerke V, Moss SE, Annexins. From structure to function. Physiol Rev. 2002;82:331–71.
Sanches JM, Rossato L, Lice I, Alves de Piloto Fernandes AM, Bueno Duarte GH, Rosini Silva AA, et al. The role of annexin A1 in Candida albicans and Candida Auris infections in murine neutrophils. Microb Pathog. 2021;150:104689.
Vanessa KHQ, Julia MG, Wenwei L, Michelle ALT, Zarina ZRS, Lina LHK, et al. Absence of Annexin A1 impairs host adaptive immunity against Mycobacterium tuberculosis in vivo. Immunobiology. 2015;220:614–23.
Leoni G, Neumann P-A, Kamaly N, Quiros M, Nishio H, Jones HR, et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest. 2015;125:1215–27.
Fuller NW, McGuire LP, Pannkuk EL, Blute T, Haase CG, Mayberry HW, et al. Disease recovery in bats affected by white-nose syndrome. J Exp Biol. 2020;223:1–12.
Mailliard WS, Haigler HT, Schlaepfer DD. Calcium-dependent binding of S100C to the N-terminal domain of annexin I. J Biol Chem. 1996;271:719–25.
Sakaguchi M, Murata H, Sonegawa H, Sakaguchi Y, Futami J, Kitazoe M, et al. Truncation of Annexin A1 is a Regulatory Lever for Linking Epidermal Growth Factor Signaling with cytosolic phospholipase A2 in normal and malignant squamous epithelial cells *. J Biol Chem. 2007;282:35679–86.
Mitra S, Ray SK, Banerjee R. Synonymous codons influencing gene expression in organisms. RRBC. 2016;6:57–65.
Brule CE, Grayhack EJ. Synonymous codons: choose wisely for expression. Trends Genet. 2017;33:283–97.
Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–34.
Meteyer CU, Barber D, Mandl JN. Pathology in euthermic bats with white nose syndrome suggests a natural manifestation of immune reconstitution inflammatory syndrome. Virulence. 2012;3.
Auteri GG, Knowles LL. Decimated little brown bats show potential for adaptive change. Sci Rep. 2020;10:1–10.
Gignoux-Wolfsohn SA, Pinsky ML, Kerwin K, Herzog C, Hall M, Bennett AB, et al. Genomic signatures of selection in bats surviving white-nose syndrome. Mol Ecol. 2021;30:5643–57.
Cheng TL, Bennett AB, Teague O’Mara M, Auteri GG, Frick WF. Persist or perish: can bats threatened with extinction persist and recover from white-nose syndrome? Integr Comp Biol. 2024;:icae018.
Acknowledgements
We acknowledge Aliaksei Shpak for providing us the M. dasycneme sample.
Funding
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital). This research was supported by the Research Council of Finland (#331515 to TML), the National Natural Science Foundation of China (Grant No. 32371562 to TLJ), the Fundamental Research Funds for the Central Universities (2412023YQ002 to TLJ), the Ministry of Science and Higher Education of the Russian Federation (FMEN-2022-0003 to VVB and EAK, 121051100109-1 to VAI).
Open Access funding provided by University of Helsinki (including Helsinki University Central Hospital).
Author information
Authors and Affiliations
Contributions
TML, VT, VNL, FI and FWF conceived the idea; TML, MF, DMR, TB, TLJ, VB, EK, VI and DF acquired samples; MR, VT and VNL prepared samples for sequencing; VT, KAF VNL completed bioinformatic analyses; VT, VNL, KAF and FI did the graphical figures; TML, VT, VNL, FI, FWF and KAF wrote the first draft of the manuscript; all authors contributed to the final version of the manuscript and have approved the submitted version. All authors have agreed both to be personally accountable for their own contributions ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented in the literature.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No animal experiments took place as a part of the submitted study. All DNA used have been obtained from existing samples in museum collections. TML, as the curator of the mammal collections at the Finnish Museum of Natural History, has permission to utilise the museum samples for the study. No ethics approval is needed.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 15
Supplementary Material 16
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Twort, V.G., Laine, V.N., Field, K.A. et al. Signals of positive selection in genomes of palearctic Myotis-bats coexisting with a fungal pathogen. BMC Genomics 25, 828 (2024). https://doi.org/10.1186/s12864-024-10722-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10722-3