
Abstract
Background
Reproductive performance plays an important role in animal welfare, health and profitability in animal husbandry and breeding. It is well established that there is a negative correlation between performance and reproduction in dairy cattle. This relationship is being increasingly considered in breeding programs. By elucidating the genetic architecture of underlying reproduction traits, it will be possible to make a more detailed contribution to this. Our study followed two approaches to elucidate this area; in a first part, variance components were estimated for 14 different calving and fertility traits, and then genome-wide association studies were performed for 13 reproduction traits on imputed sequence-level genotypes with subsequent enrichment analyses.
Results
Variance components analyses showed a low to moderate heritability (h2) for the traits analysed, ranging from 0.014 for endometritis up to 0.271 for stillbirth, indicating variable degrees of variation within the reproduction traits. For genome-wide association studies, we were able to detect genome-wide significant association signals for nine out of 13 analysed traits after Bonferroni correction on chromosome 6, 18 and the X chromosome. In total, we detected over 2700 associated SNPs encircling more than 90 different genes using the imputed whole-genome sequence data. Functional associations were reviewed so far known and potential candidate regions in the proximity of reproduction events were hypothesised.
Conclusion
Our results confirm previous findings of other authors in a comprehensive cohort including 13 different traits at the same time. Additionally, we identified new candidate genes involved in dairy cattle reproduction and made initial suggestions regarding their potential impact, with special regard to the X chromosome as a putative information source for further research. This work can make a contribution to reveal the genetic architecture of reproduction traits in context of trait specific interactions.
Similar content being viewed by others
Background
Breeding programs in dairy cattle have in the past decades focused on performance, even accelerated by genomic selection. This has resulted in a significant increase in milk production, but has had a negative impact on reproduction traits due to antagonistic genetic relationships [1,2,3]. Boundary conditions for breeding for higher reproductive performance are the low heritability of reproduction traits, in contrast to the moderate heritability of production traits [4,5,6]. Genomic selection, however, has been shown to be a useful tool in breeding for low heritability traits, as demonstrated in the German Holstein population [7]. In this context, an increasing consideration and reallocation of the weighted traits included in the total merit index is being applied in practice [7, 8]. Furthermore, decreased fertility has an impact on both, animal welfare and the economics of dairy production [2]. Impaired fertility remains one of the major factors for culling animals, accounting for up to 20% of all culling reasons in dairy cattle [9, 10]. The additional costs of poor fertility are also clearly shown in the estimation of the total merit index (RZ€) for German Holsteins, where, for example, stillbirth is calculated with a cost of €137.50 per case [11]. This highlights the close relationship between animal welfare and economically successful dairy farming, particularly in German Holstein breeding, with regards to reproductive performance. Improving reproductive efficiency can increase animal welfare by reducing the proportion of involuntary culling in herds, lowering individual animal diseases, and simultaneously reducing variable costs for dairy farmers while enhancing their merit.
To gain insights into the regulatory pathways affecting fertility and identify associated genomic regions, genomic studies and the use of molecular genetic markers have been established as effective methods [12, 13]. Several studies have investigated genetic associations for reproduction traits in Holstein dairy cattle using genome-wide association studies (GWAS) on autosomes [14,15,16]. Capturing the causal region or quantitative trait loci (QTL) of interest depends on both the density of markers used and the structure of linkage disequilibrium (LD) [17]. Therefore, the accuracy is limited due to the distribution of marker arrays used and the LD structure behind them [18]. Beneficial, the usage of whole-genome sequence (WGS) data is decoupled from LD structure dependency since the causative mutation itself is likely included [17]. Moreover, using WGS data in GWAS allows fine mapping for complex traits [19] and offers high potential for revealing the underlying genetic architecture, including quantitative trait nucleotides (QTN) [17, 20, 21]. In addition to sequencing animals, genotype imputation has the potential to predict genotypes that have not been directly genotyped in a study, based on a reference panel of haplotypes [22]. Imputation combines the advantages of high marker density in WGS data with the ability to predict large-scale cohort data in a cost-effective manner, to be used for GWAS or fine-mapping [23].
The aim of this study is to improve the genetic characterisation of reproduction traits in German Holstein cattle and to identify putative influential genomic regions using chip and imputed WGS data. For pedigree-based analyses based on first lactation phenotypic records, we initially included a group of 34,497 primiparous German Holstein cows. In addition, we utilised imputed WGS data for GWAS of 13 reproduction traits using a mixed linear model association (MLMA) [24]. Our approach identified more than 2700 distinct genome-wide significantly associated SNPs and promising genomic regions on autosomes and the X chromosome. Finally, we conducted enrichment analyses to contextualise our findings.
Material & methods
Animals and phenotypes
Phenotypic and SNP chip data were provided from the “KuhVision” project, a cow reference population including 252,285 German Holstein cows representing the genomic pattern of German Holstein Friesian [7]. The national computing centre VIT (Vereinigte Informationssysteme Tierhaltung w.V., Verden, Germany) provided all data for animals included [7]. This large dataset was filtered according to the steps described below in order to ensure a sufficient number of observations in each effect class. The intention is thereby a potentially strong environmental impact, that needs to be accounted for in the analyses. At this, having a sufficient number of observations in each class would make sure that the environmental and genetic effects can precisely be estimated and differentiated from each other. After filtering, a subset of 34,497 primiparous Holstein cows born between 2013 and 2018 with observations for reproductive performance and 13 disease traits, including the three reproduction diseases of interest in this study, was available. In general, the disease traits were recorded on farms, mainly by the farmers themselves, claw trimmers, and veterinarians. They were binary coded, which implies that 0 means “no disease during the lactation” and 1 “at least one event of disease during the lactation”. At this, multiple disease events over the course of the lactation were not considered. In order to generate the final dataset of 34,497 cows, we applied the following filter steps. First, all cows with less than 270 and more than 305 days in milk (DIM) were excluded, resulting in a mean of 302.43 DIM in the final dataset. Then, all cows whose age at first calving (AFC), recorded in months, was below 21 and above 36 months were excluded. Thus, the mean AFC in the final dataset was 24.83 months and each class of AFC consisted of at least 20 individuals. Next, our intention was to avoid biased results because of farms with an incomplete recording of disease cases. Hence, farms with less than 10 cows having an event of diseases during their lactations were excluded, resulting in a total of 103 farms having on average 345 cows per farm. In the last step, the multicode herd-year-season (hys) was generated by combining the cow’s farm, year and season of calving, whereby the partitioning of seasons followed the calendric partitioning. Here, the filtering implied that each class of hys had to consist of at least 20 individuals. Finally, the pedigree consisted 90,407 animals covering two complete generations in the final dataset. The workflow and filtering criteria were first described in Schneider et al. [25].
Phenotypic trait data
A total of 13 fertility and calving traits were considered for which breeding values are routinely estimated (Table 1). The deregressed proofs (DRPs) were based on the official breeding value estimation in 2021. Basic principle for the calculations are based on the deregression method firstly presented by Jairath et al. [26]. The adaption of this method on estimated breeding values in German Holstein cattle is described by Liu and Masuda [27]. For the pedigree-based analyses, we included 34,497 animals for each trait. In contrast, the number of available DRPs per trait ranged from 24,559 (calving ease direct, CEd) up to 34,494 (stillbirth maternal, SBm) available for genome-wide association analysis (Table 1).
Genotyping data, quality control
About 96% of the animals were imputed to 50 K level from various versions of the EuroGenomics low-density (LD) chips (Eurogenomics, Amsterdam, NL), while the rest was genotyped with various versions and quality of the Illumina 50 K chips (Illumina Inc., San Diego, CA) and EuroGenomics medium-density (MD) chips (Eurogenomics, Amsterdam, NL). This imputation procedure is described in Segelke et al. [29]. After lifting to reference genome assembly ARS.UCD1.2, 45,613 SNPs of 252,285 cows were imputed to sequence level in two steps. Firstly, animals were imputed to a high-density reference panel (∼ 777 K), and from that to the whole genome sequence level using the Run9 reference panel of the 1000 Bull Genomes consortium [19]. Imputation was performed using Beagle 5.2 [30]. Afterwards, data were quality filtered using the dosage R squared parameter (DR2 > 0.75) [31] and a minor allele frequency cutoff of 1%. A detailed description of the workflow and filtering criteria can be found in Križanac et al. [32]. The final dataset consisted of 17.2 million SNPs. Given the entirely female and large sample size, for subsequent analyses we included the X-chromosomal information to facilitate a more comprehensive examination. This aspect was highlighted in recent studies as the X chromosome may be a potential and important source of information (e.g [33, 34]).
Pedigree based analyses
For the estimation of variance components, Bayesian uni- and bivariate animal models using Markov chain Monte Carlo sampling techniques with a Gibbs sampler as implemented in the R-package MCMCglmm [35] were used. The chain length was 25,000 iterations and the burn-in 5,000 iterations. Traits with a binary coding (namely MET, ZYS, GME, NGV, NR56, NR90, NRh, SBm, and CEm recoded as binary) were analysed with a generalized linear mixed model. The underlying latent variable 𝑙𝑖 with i as the liability for the i-th animal is connected to the binary observation \(\:{y}_{i}\) with a probit link function in the model. Choosing a probit over a logit link function is the more accurate choice for an animal model [36].
The model was defined with \(\:{l}_{i}\) as the vector of liabilities for each individual and 𝜇 as the mean liability, 𝛽 being the vector of fixed effects for age at first calving (EKA), 𝑎 the random effects for the additive genetic effect, ℎ𝑦𝑠 a multicode effect set up of heard, year and season of the individual cow and 𝑒 the residuum with a fixed residual variance of 1. 𝑎 and ℎ𝑦𝑠 are said to be normally distributed with \(\:a\:\sim\:N(0,A{\sigma\:}_{a}^{2})\) and \(\:hys\:\sim\:N(0,I{\sigma\:}_{hys}^{2})\), with A as the pedigree-based relationship matrix and I indicating the identity matrix. The prior for both effects was set to follow a \(\:{\chi\:}^{2}\) distribution with one degree of freedom. Heritabilities for traits with binary coding were calculated with the following formula.
For binary traits, residual variance was fixed to 1. The remaining traits, namely CFc, DOc, FSc, FSh, and CEm as linear, follow a normal distribution and were therefore analysed using the same model, but without the probit link. Prior assumptions are weak with an uninformative prior following \(\:{\sigma\:}_{a}^{2}\:\sim\:inv-gamma\left(\text{0.01,0.01}\right)\) and \(\:{\sigma\:}_{hys}^{2}\:\sim\:inv-gamma\left(\text{0.01,0.01}\right)\). \(\:{y}_{i}\) is the vector of observations. Next to EKA also days in milk (DIM) was chosen as fixed effect.
Trait heritability for these models was calculated in the following manner
Calving ease maternal was analysed in two ways for variance components. In a first run CEm was used as a linear trait and all four factor levels were maintained. In a second approach the levels were merged to achieve a recoded binary trait, where “easy” and “normal” as well as “heavy” and “with vet / caesarean” were taken together, respectively.
Genome-wide association analysis (GWAS)
For genome-wide association analysis, a mixed linear model approach (MLMA) as implemented in the software tool for genome-wide complex trait analysis (GCTA) version 1.93.2 beta was applied [24, 37] using the following model:
where y is the vector of DRPs, a is the mean term, b is the additive affect (fixed effect) of the candidate SNP to be tested for association, x is the SNP genotype indicator variable coded as 0, 1 or 2, g is the polygenic effect captured by the genetic relationship matrix (GRM) and e is the residual. The GRM was calculated between pairs of individuals from the set of SNPs used on chip level before imputation including 44,144 SNP-markers on the autosomes, using the approach of Yang et al. [38]. The majority of GWAS in dairy cattle only relies on autosomes [14, 39]. However, we included the X chromosome as a potential source of information for fertility and reproduction traits due to the previously disclosed relationship between phenotypic expression and sex chromosomes in the context of reproductive performance [33, 40]. In addition, it has been identified that X-linked genes have an significant influence on various complex traits in dairy cattle, including reproduction of Holstein dairy cattle [34]. Threshold for significance of the GWAS statistic was Bonferroni corrected on genome-wide level to account for multiple testing of SNPs included in the MLMA approach with a level of 0.05 ([(0.05/17,222,496), p = 2.903 * 10− 9]). The R package ggplot2 [41] was used in R version 4.2.0 [42] to generate the Manhattan plots for graphical representation.
Variant effect prediction
Ensembl Variant Effect Predictor (VEP) [43] was used to downstream analyse genome-wide significantly associated SNPs. All genes taken into account were known genes confirmed by an approved symbol of the gene nomenclature [44]. A gene was considered significantly associated with a trait if at least one SNP in proximity reached the Bonferroni threshold ([(0.05/17,222,496), p = 2.903 * 10− 9]). For proximity, a window of 10,000 base pairs (bp) downstream and upstream of the variant, according to genome assembly ARS-UCD 1.2, was considered.
Downstream analyses
Enrichment analyses were conducted using the R packages org.Bt.eg.db [45], clusterProfiler [46] and DOSE [47] using default settings. SNPs with MLMA results below p < 1 * 10− 4 were included. To assess the over-representation of genes belonging to particular pathways, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) [48] database for enrichment. The tool g:Profiler [49] was used with default settings to determine the molecular function of gene products and cellular components specific for the findings on the X chromosome only. The R package VennDiagram [50] was used to generate Venn diagrams showing shared candidate genes.
Results
Heritabilities
Observations for events and their incidences are shown in Table 2.
Based on these binary traits and the recoding of CEm, the results for genetic parameter estimations are shown in Table 3. The heritability (h2) for binary traits is based on liability scale with a range of 0.014 (MET) to 0.211 (SBm). For MET the lower 95% quantile of both heritability estimates were close to zero.
GWAS results
We obtained over 2700 genome-wide SNPs significantly associated with 9 out of the 13 traits on various chromosomes as shown in Table 4. The significantly associated SNPs were located on Bos taurus (BTA) chromosome 6 (BTA6), BTA18 and the X chromosome (BTAX). Four different traits were associated with regions on BTA6, whereas the highest number of significantly associated SNPs embracing the broadest regions was found on BTAX.
Chromosome 6
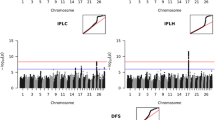
In total, 952 significant SNPs and six associated genes were identified for first to successful insemination heifer (FSh), non-return rate heifer (NRh), calving to first insemination cow (CFc) and days open cow (DOc) on BTA6. The results for these traits are shown in Fig. 1 and the dedicated number of SNPs per trait listed in Table 4. For FSh a significant SNP in 211 bp distance to PTPN13 reached genome-wide significance level. Four tested SNPs appeared significant for NRh between 102,025,927 bp and 102,081,667 bp and directly associated with AFF1 and KLHL8 (two hits for each gene). Within a window from 86,745,798 bp and 87,358,291 bp, a total of 947 SNPs showed up significant for CFc and DOc. Almost a fourth, approximately 220 SNPs, were detected for both traits and were assigned to SLC4A4, GC and NPFFR2.

Manhattan plots of QTL mapping results for significant traits on chromosome 6. Calving to first insemination (CFc), days open (DOc), first to successful insemination (FSh) and non-return rate heifers (NRh). Negative decadic logarithm of p-value of each SNP is shown on the y-axes, on x-axes the 29 autosomes and X chromosome is shown. The red line represents the significance threshold on genome-wide level p = 2.903 * 10− 9
Chromosome 18
Analyses of CEd and SBd led to significant peaks on BTA18 for both traits, shown in Fig. 2. For CEd eight SNPs reached Bonferroni threshold. The SNPs found were between 57,055,186 bp and 57,062,793 bp and displayed an association with CTU1. Regarding SBd, the same CTU1 associated SNPs could be found. In addition, significant associated SNPs were identified in a distal region on BTA18 between 59,506,758 bp and 60,085,291 bp.

Manhattan plots of QTL mapping results for significant traits on chromosome 18. Calving ease direct (CEd) and stillbirth direct (SBd). Negative decadic logarithm of p-value of each SNP is shown on the y-axes, on x-axes the 29 autosomes and X chromosome is shown. The red line represents the significance threshold on genome-wide level p = 2.903 * 10− 9
X Chromosome
For three traits (CEm, SBm, NGV) several genome-wide significant SNPs were detected on BTAX, including a various number of associated genes (Fig. 3 and Additional file 1). For SBm, a region between 30,670,566 and 57,379,944 bp was found to be significantly affected by 1233 SNPs. A set of 35 different genes was found to be associated with the SNPs. For CEm, a region between 32,856,421 bp and 50,341,339 bp, including 328 SNPs with a total of 26 announced genes, were identified. An overlap of 22 genes between 32,856,421 bp and 45,950,746 bp shared a number of common significant SNPs for CEm and SBm. Between 85,905,388 bp and 102,397,780 bp, a total of 838 significant SNPs were identified for NGV, along with 45 different genes described within this region.

Manhattan plots of QTL mapping results for significant traits on chromosome X. Calving ease maternal (CEm), retained placenta (NGV), and stillbirth maternal (SBm). (A) Genome-wide results, negative decadic logarithm of p-value of each SNP is shown on the y-axes, on x-axes the 29 autosomes and X chromosome is shown. The red line represents the significance threshold on genome-wide level p = 2.903 * 10− 9. (B) The detailed presentation of BTAX per trait, including the significantly associated region, Megabase pairs (Mb) are shown on the x-axes. The SNPs that exceed the significance threshold are highlighted in dark blue
Gene enrichment
The functional enrichment analysis was performed for all trait related genes identified in GWAS. To evaluate the list of reasonable genes, we related the genes identified by the genome-wide significant associated SNPs in GWAS to those identified with the SNPs showing a p-value p < 1 * 10− 4. No significant results were obtained for DOc, SBd, and SBm. For the other six traits, we identified over-represented genes in 26 different pathways using the KEGG database. The results were specific to each trait, with no overlap between them. Figure 4 shows a common dot plot used for visualising the functional enrichment results. For BTAX, an additional enrichment approach was conducted using the g:Profiler tool [49]. The results of this approach are presented directly in the discussion and summarized in context. In this context, 33 distinct genes out of 83 genome-wide significant identified on BTAX (detailed summary in Additional file 1) could finally be evaluated in the context of the in-depth enrichment analysis.

Functional enrichment dot plot. Calving ease direct (CEd), calving ease maternal (CEm), calving to first insemination (CFc), first to successful insemination heifer (FSh), retained placenta (NGV) and non-return rate heifers (NRh). The result for over-represented genes in 26 different pathways (y-axis) and the corresponding traits (x-axis). The thickness of dots represents GeneRatio and the color represents the associated adjusted p-value
Discussion
This study analysed a sample of 34,497 primiparous German Holstein cows to estimate genetic parameters for reproduction traits based on first lactation records using pedigree-based analyses. Additionally, DRPs were used to perform GWAS on imputed WGS data and downstream analyses. This enabled the estimation of variance components for these functional traits and identification of genome-wide significant SNPs, indicating potential candidate genes. Genetic parameters from variance component estimation showed results in line with previous studies for cattle [4, 9, 11], for example h² = 0.041 for CFc [9], and therefore the suitability of our subset for the conducted analyses. Due to the comprehensive number of animals included in this analysis, even for the low heritable functional traits (e.g. MET), reliable results escorted by tight HPD intervals were obtained. Like outlined by Berry et al. [4] for reproductive performance, various factors can affect the variance component estimation, also resulting in a population specific estimation. Nevertheless, there are limitations arising from the available data here, as only reproductive performance of the first lactation was taken into account, regardless of for how long each animal stayed in the population, although this is highly linked to reproductive performance and vice versa. DRPs provide an alternative method for handling traits with raw phenotypes, which are binary coded, while also allowing for the consideration of multiple observations within and between lactations. This increased the variance within traits to more accurately identify associated SNPs or regions [51]. The primary advantage of DRPs is their greater informativeness compared to raw observations of own performance, as they are estimated using the full reference population. An additional weighting could have been added, such as effective daughter contributions [52], but due to the selection of the cohort and therefore pre-correction of similar information quality origin from the same reference population, additional weighting was not applied. Striking signals were found on two autosomes (BTA6, BTA18) and BTAX. Testing more than 17.2 million SNPs in this large cohort led to an increased capability to identify associations for the tested functional traits. Furthermore, the inclusion of BTAX allowed us to identify two major regions including several genes in context of reproductive performance and revealed a new potential information source.
Chromosome 6
A significantly associated SNP was found on BTA6 at 101,529,627 bp located with PTPN13 (protein tyrosine phosphatase non-receptor type 13) for FSh in a 2118 bp distance. Kolbehdari et al. [53] detected this gene as chromosome-wide significant, affecting direct calving ease. The PTPN13 encoded protein belongs to the protein tyrosine phosphatase (PTP) family, which are signalling molecules involved in various cellular processes such as cell growth, differentiation, and mitotic cycle processes [54]. In contrast, no peak or genome-wide significant associated SNP was detected for FSc. It could be hypothesized that PTPN13 influences heifers and cows to different extents. Heifers undergo their own growth and development, in addition to potential embryonic development after successful insemination [55]. This could lead to bias according to the heifer’s own demands for development and an overall increase in cell growth and differentiation, which could explain the difference between FSh compared to FSc in terms of association results. In humans, PTPN13 is discussed as a potential tumour suppressor that regulates cell growth in various tissues, resulting in better outcomes for those affected [56, 57]. This suggests that PTPN13 may have two potential points of interaction. Firstly, the development of heifers requires resources for growth and tissue differentiation, unlike adult cows where requirements are no longer divided into growth and maintenance. During placentation and gestation, there is a physiological process of tissue development and remodelling [49].
The significantly associated SNPs for NRh were identified between 102,025,927 and 102,081,667 bp and were found to be locally close to AFF1 (ALF transcription elongation factor 1) and for KLHL8 (Kelch like family member 8) SNPs were within and close to the gene. AFF1 has already been suggested in the literature to be associated with conception rate in dairy cows [58] and as important transcription factor in the molecular regulation of puberty in beef cattle [59]. Furthermore, AFF1 was reported to influence daughter pregnancy rate, cow and heifer conception rate including a large dominance effect for heifer conception rate [39]. Specific gonadal studies identified AFF1 expression in the ovary, epididymis, and testis of mice [60]. Our results with an association between NRh and KLHL8 are in agreement with the literature. KLHL8 was previously proposed as a potential candidate gene for NRh and involved in oogenesis [61]. In a study by Koh et al. [62], KLHL8 was detected in plasma of low-fertility heifers functionally assigned to cellular and metabolic processes. Exosomes of high and low fertility heifers were isolated from plasma, processed and afterwards analysed by mass spectrometry. The KLHL8 product, Kelch-like protein 8, was one of two unique proteins only present in plasma of low fertility heifers. Beside this, Koh et al. [62] further showed a katalytic activity of both unique proteins.
A region with overlapping association signals was found on BTA6 for CFc and DOc, as DOc is dependent on CFc and FSc. Therefore, it is reasonable to assume that a shared genomic region affects both traits. The region containing SNPs that are significantly associated with both traits is situated between 86,745,798 bp and 87,358,291 bp. It has been previously described that SLC4A4 (solute carrier family 4 member 4) is associated with milk production and clinical mastitis [63, 64], as well as with somatic cell score [39]. SLC4A4 is involved in the regulation of bicarbonate secretion and absorption, encoding a sodium-bicarbonat-cotransporter [54]. For neonatal Holstein calves, the role of SLC4A4 inside metabolic pathways and intracellular pH control in ruminal epithelium tissue was assessed using gene expression analysis [65]. It is possible that particular characteristics of the endometrium affect the implantation process.
GC (GC vitamin D binding protein) is involved in vitamin D metabolism together with transport and was already described in context of milk fever [66], mastitis resistance [63, 64], body condition score and calving interval [63] in dairy cattle. In humans, several associations between vitamin D-binding Protein and reproductive health were described [67]. It is known that pregnancy increases the demand of vitamin D throughout the time of pregnancy and lactation and in case of deficiency, the metabolic system is incapable to fulfil the requirement neither of the mother, nor the developing foetus [68]. Vitamin D is involved in different processes according to reproduction and production traits, underlined through the several traits found in literature. This indicates a possible direct effect on reproductive performance due to a limited amount of vitamin D for the developing foetus. On the other hand, there may be an indirect effect through an increased demand for milk yield during early lactation, which could lead to decreased reproductive performance, resulting in extended time for CFc and DOc.
NPFFR2 (neuropeptide FF receptor 2) is a G protein-coupled receptor for neuropeptide FF, which is involved in modulating the opioid system and regulating cardiovascular and neuroendocrinological function [69]. Bonini et al. [70] described an upregulated mRNA expression of NPFFR2 in the human placenta. In addition, there is a known physiological interaction between endogenous opioids and gonadotropin secretion in various mammalian species [71, 72]. It is therefore reasonable to assume that any deviation from the hormonal control cycle may result in a disruption that makes it more difficult to achieve pregnancy, thereby increasing the time between calving and successful insemination.
Chromosome 18
On BTA18, SNPs significantly associated with CEd and SBd were located in a region between 57,005,186–60,085,251 bp, which encompasses the CTU1 (cytosolic thiouridylase subunit 1) gene. CTU1 is involved in the sulphur relay system in humans [73] along with tRNA modifications of the uridine at position 34. This modification occurs in an interplay with the estrogen receptor α [74]. Abo-Ismail et al. [14] were able to detect a region on BTA18 between 56.9 and 59.9 Mb, including CTU1, associated with calving performance and rump traits. The potential interplay with the estrogen receptor appears to be a plausible physiological reason for the association between CEd and SBd and restrictions in light calving. The association of the rump trait may also be linked to the ability of light calving or even an increased likelihood of heavy births, as shown by Cue et al. [75].
X Chromosome
To gain a better understanding of the significant SNPs found for CEm, NGV and SBm on BTAX, we utilised the g:Profiler tool [49]. By using the g:GOSt option, we conducted an enrichment analysis for the identified genes. Since there is a lack of evidence-based biological processes that are known to be associated with the gene products, we outline the molecular functions of the gene products. In addition, we outline the cellular components to describe the physical location of a gene product in the cell. Default settings were used. Additionally, we considered the dependency of multiple testing due to the overlap of functional terms, as described in Reimand et al. [76]. Figure 5 presents the results for molecular function and cellular components as Venn diagrams.

Venn diagram of BTAX associated genes. Gene associations according to the g:profiler results. Overlapping shapes represent interception between different parts for the same gene. (A) Associated cellular components for identified genes. (B) Associated molecular functions for identified genes
A total of 33 distinct genes were identified, 11 of which were identified for molecular function, 30 for cellular component, and seven were shared between both categories (FMR1, MTM1, GABRA3, PABPC5, GLRA4, CLCN5, DDX3X). The cellular component category had the highest variation of genes, ranging from 11 different genes for ‘supramolecular complex’ to 18 genes for ‘cell junction’. To refine our results, we focused on genes that were most frequently affected in function or components for each trait tested that was associated with BTAX. The identified gene was then considered as the most promising gene.
Starting with the cellular components (as shown in Fig. 5A), the gene FMR1 (Fragile X messenger ribonucleoprotein 1) was found to interact with all five available components. FMR1 is located in the region between 30,624,825 and 30,664,682 bp harbouring several SNPs significantly associated with the trait SBm. Additionally, FMR1 is involved in two molecular functions: ‘RNA strand annealing’ and ‘poly-purine tract binding’. The FMR1 gene is well-described in humans in the context of disturbed fertility in women and the associated Fragile X Syndrome (FXS) [77]. This effect is associated with a dynamic mutation that increases the number of CGG triplet repeats across generations. A variation in methylation and the number of CGG repeats occurs within the untranslated region of the first exon [78, 79]. This circumstance leads to an ovarian dysfunction and is physiologically associated with an increased level of FSH, premature ovarian failure or an earlier menopause before the age of 40 years [80, 81]. Mihm et al. [82] identified a decline in FSH levels as a crucial factor in the selection process of the dominant follicle in cattle. These physiological interactions are consistent with the characteristics of CEm, NGV and SBm. The changes in FSH levels and CGG triplet numbers may affect birth-related observations. The extent of this variation seems to be logically derived from the stages that are influenced, both in terms of time and intensity. These stages cover severe births, stillbirths and postnatal behaviour.
The gene GABRA3 (Gamma-aminobutyric acid type A receptor subunit alpha 3), which encodes for the gamma-aminobutyric acid type A receptor subunit alpha 3, showed a cluster between four of the five components, except for ‘supramolecular complex’, including significant associated SNPs for CEm and SBm. This gene is located at 34.602 Mb up to 34.842 Mb on BTAX, bordered by a region including further genes matching gamma-aminobutyric acid type A receptor subunits (GABRE, GABRQ). Both of these genes also showed a significant association of SNPs with CEm and SBm. GABRA3 encodes the alpha 3 subunit in GABAA receptors, which is part of a receptor complex that exhibits functional diversity depending on subunit composition [83]. The alpha 3 subunit is associated with both anxiogenesis and anxiolysis, as described by Atack et al. [84] and Dias et al. [85]. Additionally, Rudolph et al. [86] stated that the muscle relaxant activity of diazepam is mediated by this subunit in mice. In humans, GABRA3 is associated with fetal brain development and is considered a candidate gene for Rett syndrome, a neurodevelopmental disorder that primarily affects females [87]. GABAA receptors are critical binding structures for allopregnanolone, which acts as a potent allosteric modulator for these receptors. Allopregnanolone concentrations vary during pregnancy and play an important role in protecting pregnancy and birth outcomes in various mammalian species, including sheep and humans [88]. Sheep and cattle, both ruminants, have similar placentation ratios. Therefore, it is reasonable to compare the effect observed in the foetal brain of sheep and lambs with that of cows and calves in terms of hormonal circulation. In sheep, the level of allopregnanolone increases during late gestation, reaching a maximum near term, and then decreases after birth [88]. An imbalance in this regulation, due to a lack of GABRA3, could explain the problems around birth, compared to cattle and both traits CEm and SBm.
SYN1 (Synapsin I) was identified for NGV and shared the same four cellular components as GABRA3 described above. SYN1 is located more distally on BTAX between 85.929 Mb and 85.992 Mb and is a member of a gene family that encodes for neuronal phosphoproteins [54]. Synapsins play an essential role in regulating vesicles, especially in accelerating synaptic vesicle traffic through repetitive stimulation [89]. In the context of neurological disorders, many diseases are associated with synapsins. Their expression patterns are described in the literature [90]. Synapsin 1, in particular, has been highlighted as an important mediator for glucocorticoids [91] and has been detected as a member of hormonal adjustment in GnRH and LH release [92] in the field of reproductive physiology. In dairy cattle, GnRH treatment has been shown to have a positive effect on reducing the number of services per conception and shortening the days open in cases of NGV [93]. However, GnRH is generally proposed as a mediator to improve fertility in low or moderate fertility cows, especially heifers [94]. It may be difficult to separate the general hormonal effect from the clinical persistence of NGV. A decreased neutrophil function and recruitment has been linked to causing NGV in dairy cattle [95]. Neutrophils are involved in the immune response to treatments such as infections or injuries. It seems reasonable to expect an effect in the case of NGV when the decreased function in neutrophils is challenged by an infection reaction.
For molecular functions (refer to Fig. 5B), fewer genes are related compared to the results for cellular components, and there is a lower proportion of shared elements. The previously discussed GABRE3 accounts for the majority of functions, including ‘ligand-gated anion channel activity’, ‘GABA-receptor activity’ and ‘poly-purine tract binding’. Based on the intersection between molecular function and cellular component, the PABPC5 (Poly(A) binding protein cytoplasmic 5) gene, appears to be the most likely candidate to affect reproductive performance issues such as SBm and CEm in cattle.
PABPC5 was identified for SBm as well as CEm. It belongs to the cytosolic poly(A) binding protein family and is involved in protein binding at the 3’ end of the poly(A) tail [96]. Its location on BTAX is between 39.337 Mb and 39.339 Mb. An involvement in mitochondrial metabolism and apoptosis is described [97], and an association between premature ovarian failure in ovarian diseases as well as ovarian cancer is linked to PABPC5 in humans [98]. Furthermore, this is underlined functionally by PABPC5 expression in testis and ovarian tissue [98] and the known involvement of poly(A)-binding proteins in germ cell development [99].
The enrichment of the identified genes on BTAX indicates the contribution of this gonosome to reproductive performance. Two regions have shown significant associations with different calving and fertility traits. The relationship between hormones and tissues of the dam and offspring during gestation is a complex and physiologically well balanced interplay. Therefore, the number of accounting genes seems reasonable. Several studies have been conducted to investigate the multifactorial problems affecting reproductive performance in dairy cattle [28, 100, 101]. The genes presented in this section, as determined by enrichment analysis, have a direct influence on reproductive performance (e.g. FMR1) or are linked to pregnancy-dependent physiological processes (e.g. SYN1). However, the use of BTAX for GWAS is still increasing. Methods to improve the joint inclusion of autosomes and gonosomes are currently under development [34, 102]. KEGG pathway enrichment analyses also revealed a high proportion of signalling pathways (7 out of 26), distributed across almost all traits. This is consistent with the previously presented genes, such as PTPN13 [54] or SLC4A4 [65], which were also identified in the context of signalling pathways. It is important to note that the associated adjusted p-values should be viewed with caution, as they may make it more difficult to draw strong conclusions from the results of the KEGG enrichment analysis. Only three traits achieved an adjusted p-value below 0.05. The difficulty in quantification may be attributed to the low proportion of previously identified genome-wide significantly associated genes. However, the corresponding lambda values of the GWAS statistics suggest that the detected number of SNPs and their associated genes are unlikely to have been overestimated. It is known that the detection of associated markers in functional traits is challenging [9], which may limit potential downstream analyses. However, the results from g:Profiler suggested a promising approach for narrowing down and enriching downstream analyses as a follow-up to the obtained GWAS results.
Conclusion
We identified candidate genes for fertility in dairy cattle. More than 2700 genome-wide significantly associated SNPs were detected representing more than 90 different genes. Major association signals on BTA6 and BTA18 are in line with previous research, while our WGS based approach, in conjunction with downstream analyses, allowed for the identification of putative candidate genes on BTAX. Some of the genes, such FMR1 and PABPC5 have been directly related with reproductive disturbances in humans, mice, or sheep. Considering the interplay between reproduction and performance in dairy cattle, the relevance of BTAX appears to be evident. Thus, the analyses can help to better explain the genomic architecture for reproduction traits.
Data availability
The SNP chip genotype data, deregressed proofs, and phenotypes cannot be shared publicly, as they are the property of the national computing center VIT (Vereinigte Informationssysteme Tierhaltung w.V., Verden) in Germany. Summary statistics can be provided by the senior author JT (jens.tetens@uni-goettingen.de) upon reasonable request.
References
Walsh SW, Williams EJ, Evans ACO. A review of the causes of poor fertility in high milk producing dairy cows. Anim Reprod Sci. 2011;123:127–38. https://doi.org/10.1016/j.anireprosci.2010.12.001
Pryce JE, Royal MD, Garnsworthy PC, Mao IL. Fertility in the high-producing dairy cow. Livest Prod Sci. 2004;86:125–35. https://doi.org/10.1016/S0301-6226(03)00145-3
Hoekstra J, van der Lugt A, van der Werf J, Ouweltjes W. Genetic and phenotypic parameters for milk production and fertility traits in upgraded dairy cattle. Livest Prod Sci. 1994;40:225–32. https://doi.org/10.1016/0301-6226(94)90090-6
Berry DP, Wall E, Pryce JE. Genetics and genomics of reproductive performance in dairy and beef cattle. Animal. 2014;8(Suppl 1):105–21. https://doi.org/10.1017/S1751731114000743
Hansen LB, Freeman AE, Berger PJ. Yield and Fertility relationships in dairy cattle. J Dairy Sci. 1983;66:293–305. https://doi.org/10.3168/jds.S0022-0302(83)81789-5
Simianer H, Solbu H, Schaeffer LR. Estimated genetic correlations between disease and yield traits in dairy cattle. J Dairy Sci. 1991;74:4358–65. https://doi.org/10.3168/jds.S0022-0302(91)78632-3
VIT. Jahresbericht 2020: Vereinigte Informationssysteme Tierhaltung w. V. 2021. https://www.vit.de/fileadmin/Wir-sind-vit/Jahresberichte/vit-JB2020-gesamt.pdf. Accessed 14 Sep 2023.
Hardie LC, Haagen IW, Heins BJ, Dechow CD. Genetic parameters and association of national evaluations with breeding values for health traits in US organic holstein cows. J Dairy Sci. 2022;105:495–508. https://doi.org/10.3168/jds.2021-20588
Egger-Danner C, Cole JB, Pryce JE, Gengler N, Heringstad B, Bradley A, Stock KF. Invited review: overview of new traits and phenotyping strategies in dairy cattle with a focus on functional traits. Animal. 2015;9:191–207. https://doi.org/10.1017/S1751731114002614
Wangler A, Blum E, Böttcher I, Sanftleben P. Lebensleistung und Nutzungsdauer von Milchkühen aus der Sicht einer effizienten Milchproduktion. Züchtungskunde. 09.2009;2009:341–60.
VIT. Estimation of Breeding Values for Milk Production Traits, Somatic Cell Score, Conformation, Productive Life and Reproduction Traits in German Dairy Cattle. 2022. https://www.vit.de/fileadmin/DE/Zuchtwertschaetzung/Zws_Bes_eng.pdf. Accessed 1 Feb 2024.
Ashwell MS, Heyen DW, Sonstegard TS, van Tassell CP, Da Y, VanRaden PM, et al. Detection of Quantitative Trait Loci Affecting Milk Production, Health, and Reproductive traits in Holstein cattle. J Dairy Sci. 2004;87:468–75. https://doi.org/10.3168/jds.S0022-0302(04)73186-0
Goddard ME, Hayes BJ, Meuwissen THE. Genomic selection in livestock populations. Genet Res (Camb). 2010;92:413–21. https://doi.org/10.1017/S0016672310000613
Abo-Ismail MK, Brito LF, Miller SP, Sargolzaei M, Grossi DA, Moore SS, et al. Genome-wide association studies and genomic prediction of breeding values for calving performance and body conformation traits in Holstein cattle. Genet Sel Evol. 2017;49:82. https://doi.org/10.1186/s12711-017-0356-8
Seidenspinner T, Bennewitz J, Reinhardt F, Thaller G. Need for sharp phenotypes in QTL detection for calving traits in dairy cattle. J Anim Breed Genet. 2009;126:455–62. https://doi.org/10.1111/j.1439-0388.2009.00804.x
Müller M-P, Rothammer S, Seichter D, Russ I, Hinrichs D, Tetens J, et al. Genome-wide mapping of 10 calving and fertility traits in Holstein dairy cattle with special regard to chromosome 18. J Dairy Sci. 2017;100:1987–2006. https://doi.org/10.3168/jds.2016-11506
Druet T, Macleod IM, Hayes BJ. Toward genomic prediction from whole-genome sequence data: impact of sequencing design on genotype imputation and accuracy of predictions. Heredity. 2014;112:39–47. https://doi.org/10.1038/hdy.2013.13
Uffelmann E, Huang QQ, Munung NS, de Vries J, Okada Y, Martin AR, et al. Genome-wide association studies. Nat Rev Methods Primers. 2021;1:1–21. https://doi.org/10.1038/s43586-021-00056-9
Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46:858–65. https://doi.org/10.1038/ng.3034
Wientjes YCJ, Bijma P, Calus MPL, Zwaan BJ, Vitezica ZG, van den Heuvel J. The long-term effects of genomic selection: 1. Response to selection, additive genetic variance, and genetic architecture. Genet Sel Evol. 2022;54:19. https://doi.org/10.1186/s12711-022-00709-7
Xiang R, MacLeod IM, Daetwyler HD, Jong G, de, O’Connor E, Schrooten C, et al. Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat Commun. 2021;12:860. https://doi.org/10.1038/s41467-021-21001-0
Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. https://doi.org/10.1038/nrg2796
van Binsbergen R, Bink MC, Calus MP, van Eeuwijk FA, Hayes BJ, Hulsegge I, Veerkamp RF. Accuracy of imputation to whole-genome sequence data in Holstein Friesian cattle. Genet Sel Evol. 2014;46:41. https://doi.org/10.1186/1297-9686-46-41
Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nat Genet. 2014;46:100–6. https://doi.org/10.1038/ng.2876
Schneider H, Segelke D, Tetens J, Thaller G, Bennewitz J. A genomic assessment of the correlation between milk production traits and claw and udder health traits in Holstein dairy cattle. J Dairy Sci. 2023;106:1190–205. https://doi.org/10.3168/jds.2022-22312
Jairath L, Dekkers J, Schaeffer LR, Liu Z, Burnside EB, Kolstad B. Genetic evaluation for Herd Life in Canada. J Dairy Sci. 1998;81:550–62. https://doi.org/10.3168/jds.S0022-0302(98)75607-3
Zengting Liu Y, Masuda. A deregression method for single-step genomic model using all genotype data. IB. 2021:41–51.
Westwood CT, Lean IJ, Garvin JK. Factors influencing fertility of holstein dairy cows: a Multivariate description. J Dairy Sci. 2002;85:3225–37. https://doi.org/10.3168/jds.S0022-0302(02)74411-1
Segelke D, Chen J, Liu Z, Reinhardt F, Thaller G, Reents R. Reliability of genomic prediction for German holsteins using imputed genotypes from low-density chips. J Dairy Sci. 2012;95:5403–11. https://doi.org/10.3168/jds.2012-5466
Browning BL, Zhou Y, Browning SR. A one-penny Imputed Genome from Next-Generation reference panels. Am J Hum Genet. 2018;103:338–48. https://doi.org/10.1016/j.ajhg.2018.07.015
Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–23. https://doi.org/10.1016/j.ajhg.2009.01.005
Križanac A-M, Reimer C, Heise J, Liu Z, Pryce J, Bennewitz J et al. Sequence-based GWAS in 180 000 German Holstein cattle reveals new candidate genes for milk production traits. Preprint at https://doi.org/10.1101/2023.12.06.570350.
Pacheco HA, Rezende FM, Peñagaricano F. Gene mapping and genomic prediction of bull fertility using sex chromosome markers. J Dairy Sci. 2020;103:3304–11. https://doi.org/10.3168/jds.2019-17767
Sanchez MP, Escouflaire C, Baur A, Hozé C, Capitan A. Sequence-based association analyses on X chromosome in six dairy cattle breeds. Rotterdam, Netherlands; 2022.
Hadfield JD. MCMC methods for Multi-response Generalized Linear mixed models: the MCMCglmmR Package. J Stat Soft. 2010. https://doi.org/10.18637/jss.v033.i02
Villemereuil Pde. Tutorial - Estimation of a biological trait heritability using the animal model: How to use the MCMCglmm R package; 2012.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9. https://doi.org/10.1038/ng.608
Jiang J, Ma L, Prakapenka D, VanRaden PM, Cole JB, Da Y. A large-scale genome-wide Association study in U.S. Holstein cattle. Front Genet. 2019;10:412. https://doi.org/10.3389/fgene.2019.00412
Arishima T, Sasaki S, Isobe T, Ikebata Y, Shimbara S, Ikeda S, et al. Maternal variant in the upstream of FOXP3 gene on the X chromosome is associated with recurrent infertility in Japanese black cattle. BMC Genet. 2017;18:103. https://doi.org/10.1186/s12863-017-0573-8
Wickham H. Ggplot2: elegant graphics for data analysis. Cham: Springer international publishing; 2016.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2022.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, et al. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. https://doi.org/10.1186/s13059-016-0974-4
Tweedie S, Braschi B, Gray K, Jones TEM, Seal RL, Yates B, Bruford EA. Genenames.org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. 2021;49:D939–46. https://doi.org/10.1093/nar/gkaa980
Marc Carlson. org.Bt.eg.db: genome wide annotation for bovine. Bioconductor; 2017.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. https://doi.org/10.1089/omi.2011.0118
Yu G, Wang L-G, Yan G-R, He Q-Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinf (Oxford England). 2015;31:608–9. https://doi.org/10.1093/bioinformatics/btu684
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. https://doi.org/10.1093/nar/28.1.27
Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47:W191–8. https://doi.org/10.1093/nar/gkz369
Hanbo Chen. VennDiagram: Generate High-Resolution Venn and Euler Plots: R package version 1.7.3. 2022. https://CRAN.R-project.org/package=VennDiagram. Accessed 4 Mar 2024.
Thomsen H, Reinsch N, Xu N, Looft C, Grupe S, Kuhn C, et al. Comparison of estimated breeding values, daughter yield deviations and de-regressed proofs within a whole genome scan for QTL. J Anim Breed Genet. 2001;118:357–70. https://doi.org/10.1046/j.1439-0388.2001.00302.x
Liu Z, Reinhardt F, Reents R. The effective daughter contribution concept applied to multiple trait models for approximating reliability of estimated breeding values. IB. 2001:41.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, et al. A whole-genome scan to map quantitative trait loci for conformation and functional traits in Canadian holstein bulls. J Dairy Sci. 2008;91:2844–56. https://doi.org/10.3168/jds.2007-0585
O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–45. https://doi.org/10.1093/nar/gkv1189
Larson RL. Heifer development: reproduction and nutrition. Vet Clin North Am Food Anim Pract. 2007;23:53–68. https://doi.org/10.1016/j.cvfa.2006.11.003
D’Hondt V, Lacroix-Triki M, Jarlier M, Boissiere-Michot F, Puech C, Coopman P, et al. High PTPN13 expression in high grade serous ovarian carcinoma is associated with a better patient outcome. Oncotarget. 2017;8:95662–73. https://doi.org/10.18632/oncotarget.21175
Révillion F, Puech C, Rabenoelina F, Chalbos D, Peyrat J-P, Freiss G. Expression of the putative tumor suppressor gene PTPN13/PTPL1 is an independent prognostic marker for overall survival in breast cancer. Int J Cancer. 2009;124:638–43. https://doi.org/10.1002/ijc.23989
Parker Gaddis KL, Null DJ, Cole JB. Explorations in genome-wide association studies and network analyses with dairy cattle fertility traits. J Dairy Sci. 2016;99:6420–35. https://doi.org/10.3168/jds.2015-10444
Fortes MRS, Reverter A, Nagaraj SH, Zhang Y, Jonsson NN, Barris W, et al. A single nucleotide polymorphism-derived regulatory gene network underlying puberty in 2 tropical breeds of beef cattle. J Anim Sci. 2011;89:1669–83. https://doi.org/10.2527/jas.2010-3681
Alves BCA, Tobo PR, Rodrigues R, Ruiz JC, de Lima VFMH, Moreira-Filho CA. Characterization of bovine transcripts preferentially expressed in testis and with a putative role in spermatogenesis. Theriogenology. 2011;76:991–8. https://doi.org/10.1016/j.theriogenology.2011.04.027
Strucken EM, Bortfeldt RH, Tetens J, Thaller G, Brockmann GA. Genetic effects and correlations between production and fertility traits and their dependency on the lactation-stage in Holstein friesians. BMC Genet. 2012;13:108. https://doi.org/10.1186/1471-2156-13-108
Koh YQ, Peiris HN, Vaswani K, Almughlliq FB, Meier S, Burke CR, et al. Proteome profiling of exosomes derived from plasma of heifers with divergent genetic merit for fertility. J Dairy Sci. 2018;101:6462–73. https://doi.org/10.3168/jds.2017-14190
Lee Y-L, Takeda H, Costa Monteiro Moreira G, Karim L, Mullaart E, Coppieters W, et al. A 12 kb multi-allelic copy number variation encompassing a GC gene enhancer is associated with mastitis resistance in dairy cattle. PLoS Genet. 2021;17:e1009331. https://doi.org/10.1371/journal.pgen.1009331
Olsen HG, Knutsen TM, Lewandowska-Sabat AM, Grove H, Nome T, Svendsen M, et al. Fine mapping of a QTL on bovine chromosome 6 using imputed full sequence data suggests a key role for the group-specific component (GC) gene in clinical mastitis and milk production. Genet Sel Evol. 2016;48:79. https://doi.org/10.1186/s12711-016-0257-2
Naeem A, Drackley JK, Stamey J, Loor JJ. Role of metabolic and cellular proliferation genes in ruminal development in response to enhanced plane of nutrition in neonatal holstein calves. J Dairy Sci. 2012;95:1807–20. https://doi.org/10.3168/jds.2011-4709
Pacheco HA, da Silva S, Sigdel A, Mak CK, Galvão KN, Texeira RA, et al. Gene mapping and gene-set analysis for milk fever incidence in holstein dairy cattle. Front Genet. 2018;9:465. https://doi.org/10.3389/fgene.2018.00465
Grundmann M, von Versen-Höynck F. Vitamin D - roles in women’s reproductive health? Reprod Biol Endocrinol. 2011;9:146. https://doi.org/10.1186/1477-7827-9-146
Jones KS, Assar S, Prentice A, Schoenmakers I. Vitamin D expenditure is not altered in pregnancy and lactation despite changes in vitamin D metabolite concentrations. Sci Rep. 2016;6:26795. https://doi.org/10.1038/srep26795
Ankö M-L, Panula P. Regulation of endogenous human NPFF2 receptor by neuropeptide FF in SK-N-MC neuroblastoma cell line. J Neurochem. 2006;96:573–84. https://doi.org/10.1111/j.1471-4159.2005.03581.x
Bonini JA, Jones KA, Adham N, Forray C, Artymyshyn R, Durkin MM, et al. Identification and characterization of two G protein-coupled receptors for neuropeptide FF. J Biol Chem. 2000;275:39324–31. https://doi.org/10.1074/jbc.M004385200
Genazzani AR, Genazzani AD, Volpogni C, Pianazzi F, Li GA, Surico N, Petraglia F. Opioid control of gonadotrophin secretion in humans. Hum Reprod. 1993;8(Suppl 2):151–3. https://doi.org/10.1093/humrep/8.suppl_2.151
Goodman RL, Coolen LM, Anderson GM, Hardy SL, Valent M, Connors JM, et al. Evidence that dynorphin plays a major role in mediating progesterone negative feedback on gonadotropin-releasing hormone neurons in sheep. Endocrinology. 2004;145:2959–67. https://doi.org/10.1210/en.2003-1305
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–60. https://doi.org/10.1093/nar/gkp896
Kusnadi EP, Timpone C, Topisirovic I, Larsson O, Furic L. Regulation of gene expression via translational buffering. Biochim Biophys Acta Mol Cell Res. 2022;1869:119140. https://doi.org/10.1016/j.bbamcr.2021.119140
Cue RI, Monardes HG, Hayes JF. Relationships of calving ease with type traits. J Dairy Sci. 1990;73:3586–90. https://doi.org/10.3168/jds.S0022-0302(90)79060-1
Reimand J, Kull M, Peterson H, Hansen J, Vilo J. G:Profiler–a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–200. https://doi.org/10.1093/nar/gkm226
Mila M, Alvarez-Mora MI, Madrigal I, Rodriguez-Revenga L. Fragile X syndrome: an overview and update of the FMR1 gene. Clin Genet. 2018;93:197–205. https://doi.org/10.1111/cge.13075
Oberlé I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–102. https://doi.org/10.1126/science.252.5009.1097
Verkerk AJ, Pieretti M, Sutcliffe JS, Fu Y-H, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–14. https://doi.org/10.1016/0092-8674(91)90397-H
Sullivan AK, Marcus M, Epstein MP, Allen EG, Anido AE, Paquin JJ, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20:402–12. https://doi.org/10.1093/humrep/deh635
Sherman SL. Premature ovarian failure in the fragile X syndrome. Am J Med Genet. 2000;97:189–94. https://doi.org/10.1002/1096-8628(200023)97:33C%189::AID-AJMG10363E%3.0.CO;2-J
Mihm M, Good TE, Ireland JL, Ireland JJ, Knight PG, Roche JF. Decline in serum follicle-stimulating hormone concentrations alters key intrafollicular growth factors involved in selection of the dominant follicle in heifers. Biol Reprod. 1997;57:1328–37. https://doi.org/10.1095/biolreprod57.6.1328
Goetz T, Arslan A, Wisden W, Wulff P. GABAA receptors: structure and function in the basal ganglia. Prog Brain Res. 2007;160:21–41. https://doi.org/10.1016/S0079-6123(06)60003-4
Atack JR, Hutson PH, Collinson N, Marshall G, Bentley G, Moyes C, et al. Anxiogenic properties of an inverse agonist selective for alpha3 subunit-containing GABA A receptors. Br J Pharmacol. 2005;144:357–66. https://doi.org/10.1038/sj.bjp.0706056
Dias R, Sheppard WFA, Fradley RL, Garrett EM, Stanley JL, Tye SJ, et al. Evidence for a significant role of alpha 3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci. 2005;25:10682–8. https://doi.org/10.1523/JNEUROSCI.1166-05.2005
Rudolph U, Möhler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475–98. https://doi.org/10.1146/annurev.pharmtox.44.101802.121429
Xiang F, Buervenich S, Nicolao P, Bailey ME, Zhang Z, Anvret M. Mutation screening in Rett syndrome patients. J Med Genet. 2000;37:250–5. https://doi.org/10.1136/jmg.37.4.250
Brunton PJ, Russell JA, Hirst JJ. Allopregnanolone in the brain: protecting pregnancy and birth outcomes. Prog Neurobiol. 2014;113:106–36. https://doi.org/10.1016/j.pneurobio.2013.08.005
Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature. 1995;375:488–93. https://doi.org/10.1038/375488a0
Mirza FJ, Zahid S. The role of synapsins in Neurological disorders. Neurosci Bull. 2018;34:349–58. https://doi.org/10.1007/s12264-017-0201-7
Revest J-M, Kaouane N, Mondin M, Le Roux A, Rougé-Pont F, Vallée M, et al. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Mol Psychiatry. 2010;15(1125):1140–51. https://doi.org/10.1038/mp.2010.40
Ayrout M, Simon V, Bernard V, Binart N, Cohen-Tannoudji J, Lombès M, Chauvin S. A novel non genomic glucocorticoid signaling mediated by a membrane palmitoylated glucocorticoid receptor cross talks with GnRH in gonadotrope cells. Sci Rep. 2017;7:1537. https://doi.org/10.1038/s41598-017-01777-2
Leslie KE, Doig PA, Bosu WT, Curtis RA, Martin SW. Effects of gonadotrophin releasing hormone on reproductive performance of dairy cows with retained placenta. Can J Comp Med. 1984;48:354–9.
Besbaci M, Abdelli A, Minviel JJ, Belabdi I, Kaidi R, Raboisson D. Association of pregnancy per artificial insemination with gonadotropin-releasing hormone and human chorionic gonadotropin administered during the luteal phase after artificial insemination in dairy cows: a meta-analysis. J Dairy Sci. 2020;103:2006–18. https://doi.org/10.3168/jds.2019-16439
Kimura K, Goff JP, Kehrli ME, Reinhardt TA. Decreased neutrophil function as a cause of retained placenta in dairy cattle. J Dairy Sci. 2002;85:544–50. https://doi.org/10.3168/jds.S0022-0302(02)74107-6
Jing F, Ruan X, Liu X, Yang C, Di Wang, Zheng J, et al. The PABPC5/HCG15/ZNF331 feedback Loop regulates Vasculogenic Mimicry of Glioma via STAU1-Mediated mRNA decay. Mol Therapy - Oncolytics. 2020;17:216–31. https://doi.org/10.1016/j.omto.2020.03.017
Bhattacharjee RB, Bag J. Depletion of nuclear poly(A) binding protein PABPN1 produces a compensatory response by cytoplasmic PABP4 and PABP5 in cultured human cells. PLoS ONE. 2012;7:e53036. https://doi.org/10.1371/journal.pone.0053036
Blanco P, Sargent CA, Boucher CA, Howell G, Ross M, Affara NA. A novel poly(A)-binding protein gene (PABPC5) maps to an X-specific subinterval in the Xq21.3/Yp11.2 homology block of the human sex chromosomes. Genomics. 2001;74:1–11. https://doi.org/10.1006/geno.2001.6530
Venables JP, Eperon I. The roles of RNA-binding proteins in spermatogenesis and male infertility. Curr Opin Genet Dev. 1999;9:346–54. https://doi.org/10.1016/s0959-437x(99)80052-5
López-Gatius F, Santolaria P, Yániz J, Rutllant J, López-Béjar M. Factors affecting pregnancy loss from gestation day 38 to 90 in lactating dairy cows from a single herd. Theriogenology. 2002;57:1251–61. https://doi.org/10.1016/S0093-691X(01)00715-4
Spencer TE. Early pregnancy: concepts, challenges, and potential solutions. Anim Front. 2013;3:48–55. https://doi.org/10.2527/af.2013-0033
Druet T, Legarra A. Theoretical and empirical comparisons of expected and realized relationships for the X-chromosome. Genet Sel Evol. 2020;52:50. https://doi.org/10.1186/s12711-020-00570-6
Acknowledgements
This work used the Scientific Compute Cluster at GWDG, the joint data centre of Max Planck Society for the Advancement of Science (MPG) and University of Göttingen. We further acknowledge support by the Open Access Publication Funds of the Göttingen University.
Funding
This work is part of the project “QTCC: From Quantitative Trait Correlation to Causation in dairy cattle” and is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer (project number: 448536632). The funders had no role in study design, data collection and analysis, interpretation of data, or writing the manuscript. Publication fee was covered by the Open Access Publication Funds of the Göttingen University. Open Access funding provided by Projekt DEAL.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
LS performed GWAS, downstream analyses and wrote the manuscript. AMK provided imputed WGS data. HS performed pedigree-based analyses. JH provided the 50 K SNP chip dataset. ZL provided the DRPs. CFG participated in imputation of WGS data and downstream analyses. JT supervised the study and participated in the writing of the paper. JT, JB, and GT conceived and supervised the study. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. No animals or animal materials have been used in this study, and ethical approval for the use of animals was not necessary.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12864_2024_10782_MOESM1_ESM.xlsx
Additional file 1: Significant associated genes on BTAX: Result table for identified genes on BTAX and corresponding number of SNPs per trait, as well as position on the chromosome. Included traits are calving ease maternal (CEm), retained placenta (NGV) and stillbirth maternal (SBm)
12864_2024_10782_MOESM2_ESM.xlsx
Additional file 2: Genome-wide significant SNPs per trait: Results of GWAS summary statistics for all genome-wide significant SNPs identified per analysed trait. Results are separate for each trait, namely, calving ease direct and maternal (CEd/CEm), calving to first insemination (CFc), days open (DOc), first to successful insemination heifer (FSh), retained placenta (NGV), non-return rate 56 heifer (NRh) and stillbirth direct and maternal (SBd/SBm)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwarz, L., Križanac, AM., Schneider, H. et al. Genetic and genomic analysis of reproduction traits in holstein cattle using SNP chip data and imputed sequence level genotypes. BMC Genomics 25, 880 (2024). https://doi.org/10.1186/s12864-024-10782-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10782-5