
Abstract
Background
Acinetobacter baumannii is an opportunistic human pathogen that causes a variety of infections in immunosuppressed individuals and patients in intensive care units. The success of this pathogen in nosocomial settings can be directly attributed to its persistent nature and its ability to rapidly acquire multidrug resistance. It is now considered to be one of the top priority pathogens for development of novel therapeutic approaches. Several high-throughput techniques have been utilised to identify the genetic determinants contributing to the success of A. baumannii as a global pathogen. However, targeted gene-function studies remain challenging due to the lack of appropriate genetic tools.
Results
Here, we have constructed a series of all-synthetic allelic exchange vectors – pALFI1, pALFI2 and pALFI3 – with suitable selection markers for targeted genetic studies in highly drug resistant A. baumannii isolates. The vectors follow the Standard European Vector Architecture (SEVA) framework for easy replacement of components. This method allows for rapid plasmid construction with the mutant allele, efficient conjugational transfer using a diaminopimelic acid-dependent Escherichia coli donor strain, efficient positive selection using the suitable selection markers and finally, sucrose-dependent counter-selection to obtain double-crossovers.
Conclusions
We have used this method to create scar-less deletion mutants in three different strains of A. baumannii, which resulted in up to 75% deletion frequency of the targeted gene. We believe this method can be effectively used to perform genetic manipulation studies in multidrug resistant Gram-negative bacterial strains.
Similar content being viewed by others


Background
Acinetobacter baumannii is one of the most prevalent multidrug-resistant hospital pathogens globally. A. baumannii is now considered a global threat in clinical settings due to its propensity to rapidly acquire antimicrobial resistance determinants, and its ability to persist on various abiotic surfaces facilitating nosocomial transmission [1,2,3]. Due to widespread multidrug resistance (MDR) in this species, the WHO and CDC have listed carbapenem-resistant A. baumannii as a top priority pathogen for research and development of new antimicrobial treatments [4, 5].
In order to combat this problematic pathogen, it is important to identify the genetic determinants contributing to its success. However, studies of gene knockouts have been limited due to the paucity of advanced genetic manipulation tools. While there are many well-established gene modification techniques that work broadly in Gram-negative bacteria, gene manipulation in A. baumannii is still challenging. This is especially true for extensively drug resistant clinical isolates, which are resistant to most of the antibiotics that are commonly used in laboratory for selection [6,7,8,9, 13, 14]. Given the rise of highly drug resistant isolates of A. baumannii, studies involving MDR and virulent isolates will best reflect the molecular dynamics behind A. baumannii pathogenesis. Therefore, it is of utmost importance to develop genetic manipulation tools that are suitable for use in highly drug resistant strains of A. baumannii.
Allelic exchange mutagenesis is an efficient method used in bacterial genome editing. This method enables construction of gene knockouts, knock-ins and other site-directed mutations in a wide range of bacterial species [10, 11]. To facilitate allelic exchange, a suicide vector is constructed with a copy of the mutant allele flanked by regions adjacent to the gene of interest that are identical to the recipient chromosome. The suicide vectors for allelic exchange usually contain an origin of transfer (OriT) and therefore conjugation is most often used to introduce the plasmid carrying the mutant allele to the recipient strain. Allelic exchange mutagenesis is a two-step procedure, where a site-specific chromosomal integration event mediated by homologous recombination occurs in the first step, followed by the plasmid backbone excision from the chromosome via a second crossover event, which results in allelic exchange. The first homologous recombination event, also known as a single crossover, results in the recipient acquiring resistance to the antibiotic marker encoded on the suicide vector. The antibiotic-resistant merodiploids are selected and then subject to a counter-selection method to isolate the double-crossovers [10, 11]. The majority of suicide vectors used in Gram-negative bacteria include a sacB gene from Bacillus subtilis encoding levansucrase for counter-selection. Levansucrase is an enzyme that confers sucrose sensitivity in Gram-negative bacteria by catalysing the hydrolysis of sucrose into glucose and the toxic product levan in the periplasm [6, 11, 12]. Double crossover mutants are isolated by growing the single crossover mutants in the presence of sucrose, as only those bacteria that have lost the sacB gene can grow under this condition. Although allelic exchange mutagenesis is one of the most common techniques to create marker-less mutants in Gram-negative bacteria [10, 11], and has been previously used in A. baumannii [13, 14], many widely used allelic exchange vectors lack suitable selection markers for MDR strains and are also difficult to modify or edit due to their lack of suitable restriction sites and complex architectures. Therefore, this method is still not easily applied to many clinical A. baumannii isolates.
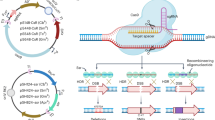
In this study, we have built and validated a set of all-synthetic, Standard European Vector Architecture (SEVA)-based suicide vectors with varied selection markers, with the specific aim of enabling allelic exchange mutagenesis of MDR A. baumannii strains (Fig. 1). Use of these vectors provided an efficient and quick method for genetic manipulations in various drug susceptible and MDR A. baumannii strains with deletion frequencies up to 75%.

Map of pALFI vectors showing how they follow the SEVA architecture. The cargo segment includes the MCS (multi cloning site); AbR segment includes HygR (hygromycin resistance cassette) and AmpR (ampicillin resistance cassette); the replication segment includes R6K (pir-dependent origin of replication); an addition selection segment (which is not standard in SEVA vectors) includes SacB (levansucrase gene) and GFP (green fluorescent protein); The pALFI vectors also include an OriT (origin of conjugational transfer) and T1 (terminator). Each genetic element is flanked with unique restriction sites. The HygR resistance marker in pALFI1 is replaced with TcR in pALFI2 and with Tpm in pALFI3 (Created with BioRender.com).
Results and discussion
We first designed a series of vectors – pALFI1, pALFI2 and pALFI3 – with broadly useful selection markers for MDR A. baumannii strains, as well as properties allowing easy modification in future studies. The pALFI vectors follow the Standard European Vector Architecture (SEVA)[15] for easy replacement of components, and contain a choice of hygromycin, tetracycline or tellurite resistance markers (Fig. 1). The three vectors contain all genetic elements commonly present in suicide vectors: the R6K replication origin for pir-dependent replication, oriT for transfer by conjugation, the sacB gene for selection of double crossovers, and the standard SEVA multiple cloning site for introduction of allelic exchange cassettes [Ortiz-Martín et al., 2006; Biswas, 2015; Hmelo et al., 2015; Cianfanelli et al., 2020].
We created three different vector derivatives to expand the usability of these vectors in susceptible as well as MDR A. baumannii strains. The pALFI1 vector contains the hygR gene encoding an aminoglycoside phosphotransferase that confers resistance to hygromycin, which can be used in strains that are resistant to other aminoglycosides. The tetA gene conferring tetracycline resistance is included in pALFI2 for genetic manipulation in susceptible A. baumannii strains, such as ATCC17978. A non-antibiotic selection marker, Tpm (thiopurine-S-methyltranferase), conferring resistance to tellurite, is included in pALFI3 for use when there are no suitable antibiotic resistance markers. The pALFI- vectors also encode an ampicillin resistance marker, which allows for the selection of transformants in E. coli. We used ampicillin selection for plasmid cloning and propagation, and the other markers only for positive selection of trans-conjugants, thus limiting the use of toxic or expensive selection agents. Vector construction and mutagenesis of target strains was performed using E. coli strain Jke201 – a pir-carrying, diaminopimelic acid (DAP)-auxotrophic donor strain that allows for replication of plasmids containing an R6K origin and the subsequent removal of donor strain on any growth medium not supplemented with DAP [12, 16].
In order to test the efficiency of the pALFI vectors, we deleted the aceI efflux pump gene in two different A. baumannii strains: AB5075_UW [17] using both pALFI1 and pALFI3, and AB0057[18](Table 1) using pALFI1. AceI is a member of the proteobacterial antimicrobial compound efflux (PACE) family of transport proteins and is highly conserved across A. baumannii strains [19, 20]. Similarly, we used pALFI3 to create a deletion mutant lacking sensor histidine kinase gene, BAL062_00580, in another highly MDR strain BAL062 [21, 22]. Initial susceptibility testing showed that the optimal concentrations for selection were 250 µg/ml hygromycin in AB5075_UW and AB0057, and 50 µg/ml potassium tellurite in BAL062 and AB5075_UW.
The counter-selection plates containing 5% sucrose were found to contain some sucrose-resistant merodiploids, which may have occurred due to inactivating mutations in the sacB gene [11]. An additional selection step, where colonies from sucrose plates were transferred into fresh sucrose as well as selective agar plates (containing hygromycin or tellurite) helped in the identification of colonies in which plasmid excision had not occurred. A total of 20 colonies that grew on sucrose plates but not on selection plates were screened by colony PCR. Deletion frequencies of 75% and 50% was observed with pALFI1 in AB5075_UW and AB0057, respectively. The use of pALFI3 in AB5075_UW and BAL062 resulted in deletion frequencies of 40% and 50% respectively (Supplementary Fig. 2). The deletion frequencies were calculated by dividing the number of positive colonies per strain by the total number of colonies screened.
In theory, the frequency of allelic exchange using suicide vectors would be expected to be around 50%, however this is often not the case. The deletion frequencies using the pALFI vectors was observed to be between 40 and 75%, demonstrating the reasonable efficiency of this method. Taken together, the whole method from cloning the mutant allele in suicide vector to obtaining a knockout mutant took a total of 8–10 days (Fig. 2).

An overview of allelic exchange mutagenesis protocol using pALFI vectors
Mutant allele is first cloned into an all elic exchange vector (pALFI) and transformed into E. coli Jke201 or another suitable donor strain for biparental mating. This step can ideally be completed in two working days. In the second step, the knockout vector is introduced into the Acinetobacter recipient strain by conjugation and screened for single-crossover mutants (positive selection). This step can be completed in 2–3 days. In the final step, single crossovers are transferred onto sucrose plates (negative selection) for mutant screening, which can be completed in 4 working days. Typically, the whole process can be completed in 8–10 working days. A detailed step by step protocol is included as the supplementary material S1 (Created with BioRender.com).
Conclusions
Genetic manipulation enables understanding of gene-phenotype relationships and allows for the identification of novel therapeutic targets to combat problematic pathogens, such as A. baumannii. The allelic exchange vectors generated in this study were found to be efficient for obtaining scar-less mutants in MDR A. baumannii isolates. We believe that this method and the allelic exchange vectors can also be effectively used for gene-function studies in other highly multidrug resistant bacterial species, in which genetic manipulation is troublesome.
Methods
Bacterial strains, growth media and conditions
Bacterial strains were cultured in Luria-Bertani (LB) media at 37 °C unless otherwise stated. E. coli Jke201 was used for plasmid propagation and as a donor strain for conjugation [12], and was cultured in the presence of 100µM of diaminopimelic acid (DAP) (Sigma Aldrich). A. baumannii strains AB5075_UW, AB0057, BAL062 were included in this study as recipient strains (Tables 1 and 2).
Chemically competent cells for E. coli Jke201 were prepared using the calcium chloride/MES (2-(N-morpholino) ethanesulfonic acid) method. Briefly, cells were grown to mid exponential phase (OD600nm 0.4–0.6) from overnight cultures in the presence of 15mM MgCl2. Bacterial cultures were chilled on ice for 40 min before harvesting. The cell pellets were washed twice in 10ml ice-cold solution A (50 mm CaCl2, 10mM MnCl2, 10mM MES at pH 6.3). The cells were then resuspended in 1.5ml solution A containing 15%(v/v) glycerol and 100 µl aliquots were either used directly or frozen and stored at -80 °C.
Transformation was performed using the heat shock method [23]. E. coli transformants were selected in the presence of 100 µg/ml ampicillin (Sigma Aldrich). For positive selection of trans-conjugants, a final concentration of 250 µg/ml hygromycin (Invitrogen) or 10 µg/ml tetracycline (Amresco) or 50 µg/ml potassium tellurite (Sigma Aldrich) was used. For counter-selection, fresh low-salt LB plates (10 g/L tryptone, 5 g/L NaCl, 5 g/L yeast extract, 15 g/L agar) with 5% sucrose (Sigma Aldrich) were prepared.
Construction of suicide vectors
Suicide vector pALFI was constructed by combining the origin of conjugational transfer (OriT), origin of replication (R6K), and multiple cloning site (MCS) from the SEVA database [15]. The sacB gene sequence with its promoter region was obtained from pKNG101_Tc [24]. pALFI1 was generated by combining the hygromycin (HygR) and ampicillin (AmpR) resistance cassettes and a reporter gene (green fluorescent protein, GFP). The GFP gene, preceded by the amvR promoter [25] was intended for use as a second selection marker for single crossovers, however the signal was not high enough under normal growth conditions to reliably indicate colonies containing the plasmid. Each segment of the pALFI suicide vector is flanked with unique restriction sites, adopting the SEVA standard [15] (Fig. 1). The plasmid sequence of pALFI1 was submitted for plasmid synthesis (GENEWIZ). Once the pALFI1 plasmid was synthesized, pALFI2 was generated by replacing hygR with tetA with its native regulator tetR from A. baumannii AB0057. pALFI3 was generated by replacing hygR with tpm (thiopurine-S-methyltranferase) conferring resistance to tellurite from pFOKT [12](Supplementary Fig. 1). The TcR and Tpm markers were cloned using the SwaI/ScaI restriction sites. Plasmids used in this work are listed in Table 2. Primers used to amplify the tetracycline and tellurite resistance cassettes are listed in Table S1.
Cloning the mutant allele into the suicide vector
In order to amplify the knockout fragment containing approximately 700 bp upstream and downstream region of targeted gene, flanking primers with 15-18 bp overlap (Table S1) were designed. The upstream and downstream regions were first amplified using the high-fidelity Platinum SuperFi PCR master mix (Invitrogen) and purified from a 1% agarose gel (QiAquick gel extraction kit, Qiagen). The fragments were then spliced together by overlap extension PCR and gel purified. Plasmids were isolated from overnight cultures using Wizard Plus SV miniprep kit (Promega). Plasmid and the knockout fragment were digested using SpeI-HF or BamHI-HF (New England Biolabs) for 2 h at 37 °C and gel purified. The restriction digested knockout fragment and plasmid were ligated together using T4 DNA ligase (New England Biolabs) and transformed into E. coli Jke201 using the heat-shock method. After a heat shock, the cells were resuspended in pre-warmed LB broth supplemented with 100µM DAP and incubated at 37 °C for 2 h before plating on an ampicillin (100 µg/ml) selection plate. Transformants were screened by colony PCR using the primers flanking the upstream and downstream regions of the targeted gene (Table S1).
Conjugation, positive selection and counterselection
Recipient A. baumannii strains AB5075_UW, AB0057, and BAL062 were streaked on fresh LB agar plates containing no antibiotics and incubated overnight at 37°C. The donor strain, E. coli JKe201 was streaked on a fresh LB agar plate supplemented with 100µM DAP and 100 µg/ml ampicillin and incubated overnight at 37°C. Cells from each plate were resuspended in sterile phosphate buffered saline (PBS) and adjusted to an optical density (OD600nm) of 40 for the donor strain and 20 for the recipient strains. Equal volumes of recipient and donor strains were mixed and spotted on a very dry LB agar plate supplemented with 100µM DAP. The conjugation patch was left to dry and incubated at 37°C for 2 h.
For positive selection, a loopful of cells from the conjugation patch was streaked on LB agar plate containing 250 µg/ml hygromycin (pALFI1) or 20–100 µg/ml potassium tellurite (pALFI3) and incubated at 37°C overnight. The conjugation plate was incubated at 28°C overnight and a loopful of cells was again transferred onto the selective LB agar plates the next day (16-18 h). The selection conditions were optimised for each A. baumannii strains to avoid background growth. The single crossover mutants were screened by colony PCR to confirm the presence of the resistance cassette.
For counter-selection, at least three trans-conjugant colonies were selected and streaked on freshly prepared low-salt LB agar plates containing 5% sucrose. The plates were incubated at room temperature (RT) for 24-48 h until well defined colonies were obtained. The colonies from sucrose plates were patched onto a fresh selective LB agar plate as well as a fresh LB agar plate supplemented with sucrose to confirm the excision of the plasmid backbone. The selection plates were incubated at 37°C and the sucrose plates were incubated at RT. Colonies that grew on the LB sucrose plate but not on the selective LB plate were screened by PCR to identify the knockout mutants. Genomic DNA was extracted from the deletion mutants using the DNeasy ultraclean microbial kit (Qiagen) and mutants were confirmed by genome sequencing. A detailed step by step protocol is included as the supplementary material S1.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Harding CM, Hennon SW, Feldman MF. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol. 2018;16(2):91–102.
Tucker AT, Nowicki EM, Boll JM, Knauf GA, Burdis NC, Stephen Trent M, et al. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio. 2014;5(4):1–9.
Morris FC, Dexter C, Kostoulias X, Uddin MI, Peleg AY. The mechanisms of disease caused by Acinetobacter baumannii. Front Microbiol. 2019;10:1601.
WHO: Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics [Internet]. 2017; 43:348–365. https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed Accessed 10 Sep 2022
CDC. : Antibiotic resistance threats in the United States [Internet]. Centers for Disease Control and Prevention 2019;1–113. https://www.cdc.gov/drugresistance/biggest-threats.html Accessed 10 Sep 2022
Biswas I. Genetic tools for manipulating Acinetobacter baumannii genome: an overview. J Med Microbiol. 2015;64(7):657–69.
Sykes EME, Deo S, Kumar A. Recent advances in genetic tools for Acinetobacter baumannii. Front Genet. 2020;11:1–19.
Luna BM, Ulhaq A, Yan J, Pantapalangkoor P, Nielsen TB, Davies BW et al. Selectable markers for use in genetic manipulation of extensively drug-resistant (XDR) Acinetobacter baumannii HUMC1 2017; 2: 1–9.
Lucidi M, Visaggio D, Prencipe E, Imperi F, Leoni L. Multidrug-resistant Acinetobacter Species: In vitro and in vivo responses to environmental stressors 2019;1–21: e01334-19
Ortiz-Martín I, Macho AP, Lambersten L, Ramos C, Beuzón CR. Suicide vectors for antibiotic marker exchange and rapid generation of multiple knockout mutants by allelic exchange in Gram-negative bacteria. J Microbiol Methods. 2006;67:395–407.
Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, et al. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc. 2015;10:1820–41.
Cianfanelli FR, Cunrath O, Bumann D. Efficient dual-negative selection for bacterial genome editing. BMC Microbiol. 2020;20:1–6.
Amin IM, Richmond GE, Sen P, Koh TH, Piddock LJ, Chua KL. A method for generating marker-less gene deletions in multidrug-resistant Acinetobacter baumannii. BMC Microbiol. 2013;13:1–4.
Oh MH, Lee JC, Kim J, Choi CH, Han K. Simple method for markerless gene deletion in multidrug-resistant Acinetobacter baumannii. Appl Environ Microbiol. 2015;81:3357–68.
Silva-Rocha R, Martínez-García E, Calles B, Chavarría M, Arce-Rodríguez A, de Las Heras A, et al. The standard european Vector Architecture (SEVA): a coherent platform for the analysis and deployment of complex prokaryotic phenotypes. Nucleic Acids Res. 2013;41:666–75.
Harms A, Liesch M, Körner J, Québatte M, Engel P, Dehio C. A bacterial toxin-antitoxin module is the origin of inter-bacterial and inter-kingdom effectors of Bartonella. PLoS Genet. 2017;13:1–22.
Jacobs AC, Mitchell TG, Chad BC, Jennifer KL, Lily CP, McQueary CN, et al. AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio. 2014;5:e01076–14.
Hamidian M, Venepally P, Hall RuthM, Adams MD. Corrected genome sequence of Acinetobacter baumannii strain AB0057, an antibiotic-resistant isolate from lineage 1 of global clone 1. Genome Announc. 2017;5:e00836–17.
Hassan KA, Liu Q, Henderson PJF, Paulsen IT. Homologs of the Acinetobacter baumannii aceI transporter represent a new family of bacterial multidrug efflux systems. mBio. 2015;6(1):1–5.
Hassan KA, Liu Q, Elbourne LDH, Ahmad I, Sharples D, Naidu V, et al. Pacing across the membrane: the novel PACE family of efflux pumps is widespread in Gram-negative pathogens. Res Microbiol. 2018;169:450–4.
Nhu NTK, Lan NPH, Campbell JI, Parry CM, Thompson C, Tuyen HT, et al. Emergence of carbapenem-resistant Acinetobacter baumannii as the major cause of ventilator associated pneumonia in intensive care unit patients at an infectious disease hospital in southern Vietnam. J Med Microbiol. 2014;63:1386–94.
Nhu NTK, Riordan DW, Nhu TDH, Thanh DP, Thwaites G, Lan NPH, et al. The induction and identification of novel colistin resistance mutations in Acinetobacter baumannii and their implications. Sci Rep. 2016;6:1–8.
Froger A. Hall. Transformation of plasmid DNA into E. coli using the heat shock method. JoVE. 2007;6:e253.
Evans DJ, Evans DG, Sarker MR, Cornelis GR. Suicide cector pKNG101 for gene replacement in Gram-negative bacteria. Mol Microbiol. 1997;23:410–1.
Short FL, Liu Q, Shah B, Clift HE, Naidu V, Li L, et al. The Acinetobacter baumannii disinfectant resistance protein, AmvA, is a spermidine and spermine efflux pump. Commun Biol. 2021;4(1):1–12.
Zurawski DV, Thompson MG, McQueary CN, Matalka Malcolm N, Craft WSJ. Genome sequences of four divergent multidrug-resistant Acinetobacter baumannii strains isolated from patients with sepsis or osteomyelitis. J Bacteriol. 2012;194:1619–20.
Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, et al. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol. 2015;197:2027–35.
Adams MD, Goglin K, Molyneaux N, Hujer KM, Lavender H, Jamison JJ, et al. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol. 2008;190(24):8053–64.
Acknowledgements
We would like to thank Francesca Romana Cianfanelli from University of Basel for providing us with the E. coli Jke201 donor strain and the pFOKT plasmid. We thank members of the Paulsen and Cain lab for helpful discussions.
Funding
This work was supported by the National Health and Medical Research Council grants APP1120298 and APP1127615. Francesca L. Short was supported by Australian Research Council DECRA Fellowship DE200101524. The funders had no role in the study design, data collection and analysis, or the preparation of this paper.
Author information
Authors and Affiliations
Contributions
This work was conceptualized by FS, AP, IP and LL. Experimental work including allelic exchange vector design and construction was conducted by AP and FS. The manuscript was written by AP, with contributions and guidance from FS and IP, and suggestions from LL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material S1
: Step by Step protocol. Supplementary Figure 1: tetA and tpm gene schematics. A. tetA with its native regulator tetR conferring resistance to tetracycline, amplified from A. baumannii AB0057 as incorporated in pALFI2. B. tpm gene with its promoter conferring resistance to tellurite, amplified from pFOKT plasmid as incorporated in pALFI3. Supplementary Figure 2: Verification of gene deletions by colony PCR. Twenty colonies were screened per strain and per vector combination used. Colonies were considered positive for aceI gene deletion if a band of approximately 200bp was detected. The length of full aceI gene amplicon (positive control) is approximately 450 bp. Control samples (both positive and negative) were also included for reference. Deletion frequencies of 75% and 50% was observed with pALFI1 in AB5075_UW and AB0057, respectively for aceI gene deletion. The use of pALFI3 in AB5075_UW for aceI deletion resulted in 40% deletion frequency. Table S1: Primers used in the study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pokhrel, A., Li, L., Short, F.L. et al. A suite of modular, all-synthetic suicide vectors for allelic exchange mutagenesis in multidrug resistant Acinetobacter strains. BMC Microbiol 23, 137 (2023). https://doi.org/10.1186/s12866-023-02844-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-023-02844-7