
Abstract
Background
Polycystic ovary syndrome (PCOS) is an endocrinopathy in childbearing-age females which can cause many complications, such as diabetes, obesity, and dyslipidemia. The metabolic disorders in patients with PCOS were linked to gut microbial dysbiosis. However, the correlation between the gut microbial community and dyslipidemia in PCOS remains unillustrated. Our study elucidated the different gut microbiota in patients with PCOS and dyslipidemia (PCOS.D) compared to those with only PCOS and healthy women.
Results
In total, 18 patients with PCOS, 16 healthy females, and 18 patients with PCOS.D were enrolled. The 16 S rRNA sequencing in V3-V4 region was utilized for identifying the gut microbiota, which analyzes species annotation, community diversity, and community functions. Our results showed that the β diversity of gut microbiota did not differ significantly among the three groups. Regarding gut microbiota dysbiosis, patients with PCOS showed a decreased abundance of Proteobacteria, and patients with PCOS.D showed an increased abundance of Bacteroidota compared to other groups. With respect to the gut microbial imbalance at genus level, the PCOS.D group showed a higher abundance of Clostridium_sensu_stricto_1 compared to other two groups. Furthermore, the abundances of Faecalibacterium and Holdemanella were lower in the PCOS.D than those in the PCOS group. Several genera, including Faecalibacterium and Holdemanella, were negatively correlated with the lipid profiles. Pseudomonas was negatively correlated with luteinizing hormone levels. Using PICRUSt analysis, the gut microbiota community functions suggested that certain metabolic pathways (e.g., amino acids, glycolysis, and lipid) were altered in PCOS.D patients as compared to those in PCOS patients.
Conclusions
The gut microbiota characterizations in patients with PCOS.D differ from those in patients with PCOS and controls, and those might also be related to clinical parameters. This may have the potential to become an alternative therapy to regulate the clinical lipid levels of patients with PCOS in the future.
Similar content being viewed by others

Background
PCOS is an endocrine disease with a high prevalence (15%) according to the Rotterdam Consensus criteria [1]. Despite the heterogeneous symptoms and signs of PCOS, its imperative clinical manifestations include oligo-ovulation, polycystic ovaries, and androgen excess (biochemical or clinical) [2]. PCOS can exert a wide range of effects on long-term health and metabolic morbidities, such as diabetes, dyslipidemia, obesity, and coronary heart disease [3, 4]. Notably, patients with PCOS commonly have dyslipidemia, a risk factor for reproductive outcomes. Some forms of dyslipidemia, such as total cholesterol (TC), are negatively correlated with live birth in patients with PCOS [5,6,7]. The serum level of triglyceride (TG) is also negatively correlated with the rate of oocyte maturation [8]. Meanwhile, lipids can modulate glucose metabolism and exert side effects on early reproductive outcomes in patients with PCOS undergoing in-vitro fertilization (IVF), such as the number of retrieved oocytes [9, 10].
The gut microbiota, considered as the second genome, is an essential symbiotic partner of human cells and engages in extensive interactions with them [11]. It plays critical roles in the human host, including steroid hormone biosynthesis, immune system regulation, and metabolic health [12]. Furthermore, certain metabolites produced by the gut microbiota, such as short chain fatty acids (SCFAs), can also regulate multiple metabolic pathways [13]. Gut microbial dysbiosis might be an influencing factor in human diseases, such as obesity, PCOS, and diabetes [14, 15]. Previous studies have demonstrated a correlation between PCOS and an imbalance in the abundance in Bacteroidetes and Firmicutes [16, 17]. A recent study reported that germ-free mice transplanted with stool from patients with PCOS had elevated testosterone levels, and impaired ovarian function [18]. In addition, the gut microbial composition plays a non-negligible role in the variations in blood-lipid levels [19, 20]. The dysbiosis of gut microbiota, specifically the phyla Bacillota and Bacteroidetes, has been implicated in the regulation of lipid levels among patients with dyslipidemia [21, 22]. Therefore, aberrations in the gut microbial composition may be related to lipid metabolic disorders in patients with PCOS.
However, limited evidence has indicated a correlation between dyslipidemia in women with PCOS and gut microbial communities. In our study, we aimed to identify several important gut microbial compositions in PCOS, particularly in PCOS with dyslipidemia, and determine their association with lipid parameters and sex hormones. These findings have the potential to recommend precision therapies for PCOS patients in the future.
Results
Study participant characteristics
Table 1, depicts the clinical features of 52 participants. There were no significant differences in body mass index (BMI) and age between the groups. PCOS groups showed markedly higher levels of luteinizing hormone (LH), LH: follicle-stimulating hormone (LH: FSH) ratio, androstenedione, and dehydroepiandrosterone sulfate (DHEA-S) than controls. Among the three groups, no differences in parameters were observed, including progesterone, estradiol, FSH, testosterone, prolactin, homeostatic model assessment for insulin resistance (HOMA-IR), and sex hormone binding globulin (SHBG). Regarding plasma lipid parameters, patients with PCOS.D showed significantly increased levels, including TC and TG, compared with healthy controls and patients with PCOS. Furthermore, patients with PCOS.D exhibited a higher level of low-density lipoprotein cholesterol (LDL-C) than PCOS group.
Gut microbial diversity among three groups
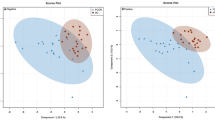
The α diversity of communities in the three groups was analyzed, including the number of observed operational taxonomic units (OTUs), Chao1, Simpson, as well as Shannon indices. The rarefaction curve indicated that OTU richness reached saturation in all three groups as sample sized increased (Fig. 1A). Furthermore, the α diversity of patients with PCOS was increased than that of controls. However, no significant differences between the Control and PCOS.D or between the PCOS and PCOS.D groups were observed (Fig. 1B-D). We identified common and unique genes among the three groups by comparing their gene sequences, as shown in Fig. 1E using a Venn diagram. The β diversity of the gut microbial communities was assessed according to the principal coordinate analysis (PCoA). As shown in Fig. 1F, samples from the three groups could not be completely discriminated.

Gut microbial diversity in all participants. (A) The rarefaction curve based on the number of observed OTUs in the three groups. The boxplots of α diversity among the three groups in Chao1 (B), Simpson (C), and Shannon indices (D). (E) Venn diagram displaying the common and distinct OTUs among the three groups. (F) PCoA analysis through weighted UniFrac metric on gut microbial communities among the three groups
Gut microbial dysbiosis
Ten bacterial phyla were identified to be different between the groups. Patients with PCOS and PCOS.D showed a notable decrease in Proteobacteria when compared to healthy women. Meanwhile, patients with PCOS showed more abundance of Firmicutes than controls. Compared to both the PCOS and Control groups, PCOS.D group exhibited a higher abundance of Bacteroidota (Fig. 2A). At the family level, the abundance of Pseudomonadaceae was decreased in the PCOS and PCOS.D groups compared to that in healthy women. In addition, PCOS.D group showed a higher abundance of Prevotellaceae than the other two groups, and PCOS group exhibited a higher level of Lachnospiraceae than the controls. The levels of Ruminococcaceae and Bifidobacteriaceae were decreased markedly in the PCOS.D than those in the PCOS group (Fig. 2B and C). In genus level, we found the abundance of Pseudomonas in Control group is higher compared with other groups. Patients with PCOS.D have an increased level of Prevotella_9, while a decreased level of Faecalibacterium than other two groups (Fig. 2D).
Next, we further analyzed the significantly different gut bacterial genera between two groups using linear discriminant analysis effect size (LEfSe) and the default of linear discriminant analysis (LDA) was three. Patients with PCOS have lower levels of Pseudomonas compared to healthy women, but higher levels of Bacteroides, Prevotella_9, Blautia, and Faecalibacterium(Fig. 3A). In the PCOS.D. group, the abundance of Prevotella_9, Romboutsia, and Clostridium_sensu_stricto_1 was markedly increased when comparing with the Control group (Fig. 3B). Faecalibacterium, Comamonas, and Holdemanella were at significantly lower levels in the PCOS.D group than those in the PCOS group. Additionally, the abundance of Clostridium_sensu_stricto_1 is higher in the PCOS.D group compared to the PCOS group (Fig. 3C).

The results of gut microbiota in participants. (A) A phylum-level heatmap of the gut microbiota. (B) Gut microbiota of three groups at phylum level. (C) Abundance comparison of three groups at family level. (D) Gut microbiota of three groups at genus level

The comparison of gut bacteria at genus level among the groups. LEfSe and Cladogram analyses of significantly different classification units between Control and PCOS (A), Control and PCOS.D (B), PCOS and PCOS.D (C)
PICRUSt analysis
We explored the functional characterization of the gut bacteria by PICRUSt analysis. In Fig. 4A, carbohydrate metabolism was increased in patients with PCOS, but amino acid and lipid metabolism were increased in the Control group. Compared with healthy women, patients with PCOS and PCOS.D showed increased synthesis in certain amino acids (such as valine, leucine, and isoleucine) but lipid metabolism (such as lipid protein biosynthesis and fatty acid biosynthesis) was decreased (Fig. 4B and C). In addition, patients with PCOS showed a significant increase in amino acid, glycolysis and lipid metabolism compared to those patients with PCOS.D (Fig. 4D).

Pathway features related to gut microbial communities based on PICRUSt analysis. (A) The heatmap of functions in gut microbiota among the three groups. PICRUSt analysis between Control and PCOS (B), Control and PCOS.D (C), and PCOS and PCOS.D (D). The data was statistically significant (P < 0.05)
Correlation analysis
Finally, we analyzed the correlation between clinical characteristics of the three groups and the gut microbiota. The abundance of Prevotella_9 and Blautia exhibited a positive correlation with sex-hormones, including LH and DHEA levels. Holdemanella was negatively correlated with the level of TC and LDL-C. In addition, Achromobacter was positively correlated with TC and high-density lipoprotein cholesterol (HDL-C). (Fig. 5A). Pseudomonas displayed a negative correlation with LH and LDL-C levels (Fig. 5B and C). Faecalibacterium was positively correlated with SHBG, while negatively correlated with the TG and TC levels (Fig. 5D and F). Additionally, Clostridium_sensu_stricto_1 had a significantly negative correlation with the FSH level (Fig. 5G).

Correlation between clinical parameters in patients with PCOS and gut microbial communities. (A) Correlations of gut microbial genera and clinical parameters. Spot colors indicate the R value of Spearman correlation between clinical parameters and gut microbial compositions. *P < 0.05 and **P < 0.01. (B) The correlation between the abundance of Pseudomonas and the serum level of LH. (C) The correlation between the abundance of Pseudomonas and the serum level of LDL-C. (D) The correlation between the abundance of Faecalibacterium and the level of TC. (E) The correlation between the abundance of Faecalibacterium and the level of TG. (F) The correlation between the abundance of Faecalibacterium and the serum level of SHBG. (G) The correlation between the abundance of Clostridium_sensu_strictio_1 and the serum level of FSH
Discussion
PCOS, a heterogeneous endocrine disease in reproductive-aged females, with an estimated prevalence ranging approximately between 6 and 20%; however, this estimated may vary depending on the diagnostic criteria used [23]. Although the etiology of this disease remains unknown, PCOS is regarded as a polygenic hereditary disease that is also influenced by the environment, such as diet and lifestyle [24, 25]. In the past decade, research has established a correlation between gut microbiota and the clinical manifestations of PCOS as well as its metabolic complications [17, 26, 27]. However, the relationship of the gut microbiota and PCOS with dyslipidemia has not been investigated.
In a previous research, Torres et al. illustrated that compared with healthy women, patients with PCOS had lower α diversity and no significant clustering of unweighted UniFrac distances. Meanwhile the higher abundance of Bacteroides and Porphyromonas in PCOS patients elucidated the association between hyperandrogenism and alterations in gut microbiota [26]. However, a separate study indicated that Akkermansia and Ruminococcaceae were lower in women with obesity and PCOS, while their β diversity was significantly clustered compared with that in healthy women without obesity [16]. Numerous factors modulate the effects on the gut microbiota and host, including socioeconomic status, genes, BMI, lifestyle, and drugs [28,29,30,31,32]. PCOS is an intricate metabolic disease, correlated with susceptibility genes, environmental factors, and lifestyle [33, 34]. Due to the various effects of different factors on the study, we made efforts to minimize their impact by excluding women with a high BMI, taking various medicines, and having frequent diarrhea.
In our study, we found an imbalance in the gut microbiota in the patients with PCOS and those with PCOS.D. We observed a decreased abundance of Pseudomonas in patients with PCOS and PCOS.D compared to the controls. Pseudomonas, a member of the Proteobacteria phylum, is one of the most common bacteria involved in steroid degradation [35, 36]. Multiple studies have demonstrated that Proteobacteria use different pathways in steroid decomposition (such as sex hormones and cholesterol) [36]. Pseudomonas plays a role in degrading bile salts via candidate genes that can oxidize the corresponding aldehydes [37, 38]. In addition to investigating the mechanism of bile salt degradation in Pseudomonas in vitro, one study elucidated that lipase from Pseudomonas may hydrolyze goat blood triglycerides and cholesterol [39]. Altogether, these in vitro and in vivo results illustrate that Pseudomonas might be associated with lipid metabolism in patients with PCOS. Contrary to our results, a recent study on PCOS reported an increased abundance of Pseudomonas compared with that in healthy women, which was attributed to the sulfatase from Pseudomonas that could hydrolyze DHEA [40]. DHEA is then bioconverted to androstenediol [41]. Therefore, the abundance of Pseudomonas increased significantly in patients with PCOS. However, Pseudomonas also hydrolyzes testosterone to regulate steroid metabolism [42]. In the present study, we found that healthy females had an increased abundance of Pseudomonas compared with patients with PCOS. Meanwhile, Pseudomonas showed a negative correlation with serum LH and LDL-C levels. The findings suggest a potential involvement of Pseudomonas in patients with PCOS presenting with imbalances in sex hormone and lipid metabolism However, the mechanism between Pseudomonas and lipid metabolism disorders in PCOS patients should be further studied.
Furthermore, the patients with PCOS.D had a higher abundance of Clostridium_sensu_stricto_1 than that in the other two groups. The function of this gut microbiota genus in host health is not particularly clear yet. An increased level of Clostridium_sensu_stricto_1 is associated with metabolic disorders, such as diabetes [43], obesity [44, 45], and non-alcoholic fatty liver disease (NAFLD) according to previous studies [46].Clostridium sensu stricto is positively related to TG and TC levels in the patients with NAFLD [46]. Clostridium spp. could develop bowel inflammation [47, 48], which results in intestinal barrier dysfunction that can increase dyslipidemia [49]. These findings suggest that Clostridium_sensu_stricto_1 has a strong association with dyslipidemia. However, interestingly, a recent study found the abundance of Clostridium_sensu_stricto_1 had a positive correlation with HDL and a negative correlation with very-low density lipoprotein [43]. More studies are needed to reveal the role of Clostridium_sensu_stricto_1 in PCOS.
In our study, Faecalibacterium and Holdemanella played critical roles in PCOS as compared to those in PCOS.D. Faecalibacterium could produce butyrate that influences carbohydrate and lipid metabolism [50, 51]. An enriched abundance of Faecalibacterium can ameliorate dyslipidemia and obesity [45, 52]. Similarly, Holdemanella genus was negatively associated with gynoid fat ratios in women [53]. Our study found that Faecalibacterium and Holdemanella were negatively correlated with TG and TC levels. Taken together, our results indicate the increasing abundance of Faecalibacterium and Holdemanella, especially Faecalibacterium, which might be a potential target drug development and treatment in patients with PCOS, especially those with PCOS.D.
Metabolic pathways of gut microbiota, such as amino acid and lipid metabolism, are significantly down-regulated in PCOS and PCOS.D groups identified in our results. Conversely, the gut microbiota of PCOS patients showed an upregulation in carbohydrate metabolism pathway. An imbalance in metabolites, including lipid proteins and fatty acids, may aggravate lipid metabolism dysfunction in patients with PCOS. Fatty acids can promote a pro-inflammatory response, which facilitates embryo implantation [54]. Indeed, Pseudomonas spp. is associated with the activation of fatty acid biosynthesis [55], which is consistent with our research. In the PCOS group, metabolic pathways of amino acids, lipid and glycolysis are increased compared with those in patients with PCOS.D. Reportedly, amino acid metabolism is able to produce SCFAs [56]. SCFAs, particularly butyrate, are important substrates for maintaining gut integrity and regulating host metabolism (e.g., glucose homeostasis and lipid metabolism) [57, 58]. Faecalibacterium, a key gut bacterium produces butyrate, which is negatively correlated with lipid parameters according to our results. Altogether, the reduced abundance of Faecalibacterium in patients with PCOS.D may be associated with lipid metabolism.
Conclusions
In summary, our study showed that the compositions of gut microbiota were significantly different among the Control, PCOS, and PCOS.D groups. We have identified imbalanced gut compositions in patients with PCOS.D, characterized by increased levels of Clostridium_sensu_stricto_1 and decreased abundance of Faecalibacterium and Holdemanella in PCOS.D, when compared to those in individuals with PCOS. Meanwhile, the abundance of Faecalibacterium was found to be significantly associated with serum lipid levels, such as TG and TC, as well as SHBG, suggesting a potential pivotal role in the pathogenesis of PCOS.D. Besides, the metabolic pathways of gut microbiota function were significantly decreased in patients with PCOS.D. However, given that multiple factors affect the host gut microbiota, the number of clinical samples should be expanded to validate our results. Further studies are warranted to reveal whether gut microbiota dysbiosis affects dyslipidemia pathogenesis in patients with PCOS. These findings could help us fully understand the gut microbial pathogenesis of PCOS and promote its personalized medicine.
Methods
Participants
64 reproductive-aged women were recruited at the First Affiliated Hospital of Anhui Medical University from February 1st to December 30th 2022. However,52 participants were screened out from 64 volunteers and provided fresh stool samples. Three participants were excluded because one of them declined to participate, and the others had used traditional Chinese medicine within a month. Another two healthy participants were excluded because they failed to take part in follow-ups. One patient with PCOS was lost contact, and three patients were excluded due to insulin resistance. Additionally, another patient with PCOS.D was exclude due to relocation, and two patients were excluded for lost contact. The control group consisted of sixteen healthy females with regular menstrual periods, normal levels of sex hormones, and normal follicle morphology and count. These females underwent assisted reproduction due to “male factors.” The remaining 36 participants were stratified into two groups, PCOS (n = 18) and PCOS.D groups (n = 18), based on their lipid levels (Fig. 6). PCOS was diagnosed by the revised 2003 Rotterdam criteria; briefly, if two of the following three conditions were present: oligomenorrhea (menstrual interval > 35 d) or amenorrhea (absence of menstruation > 90 d), together with hyperandrogenism, or polycystic ovaries in ultrasound (≥ 12 antral follicles in diameter of 2–9 mm or ovary volume ≥ 10 cm3). Participants those had thyroid disease, elevated prolactin levels, or Cushing disease were excluded [59]. Patients with IR (HOMA-IR index ≥ 2.69) [60], as well as smoking, hypertension, diabetes, pregnancy, obesity, liver, and other serious diseases were excluded. The participants were treated without any drugs, such as antibiotics, probiotics, or metformin, for < 3 months, as evaluated by two professional physicians. All the protocols in this study were approved by the Ethics Committee of the First Affiliated Hospital of Anhui Medical University (PJ 2023-08-42).

The flowchart of study participants
Clinical examination
The clinical parameters, including age, BMI (= weight[kg]/height[m]2) and the number of days in the menstrual cycle of all individuals were evaluated by a research assistant. Parameters (including progesterone, estradiol, LH, FSH, testosterone, prolactin, androstenedione, LDL-C, TC, TG and HDL-C) were analyzed by electrochemiluminescence immunoassay (Roche Diagnostics). The levels of TC ≥ 5.2 mmol/L, TG ≥ 1.7 mmol/L, HDL-C ≤ 1.0 mmol/L, and LDL-C ≥ 3.4 mmol/L, was defined as dyslipidemia according to the Chinese guidelines for lipid management (2023) [61].DHEA-S and SHBG were measured by chemiluminescence immunoassay (Beckman Coulter Unicel Dxl 800) Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were analyzed by a biochemical analyzer (Liaison XL, Diasorin, Saluggia, Italy).
Sequencing
The DNA was extracted using CTAB/SDS method, followed by purity on 1% agarose gels. 16 S rRNA genes were amplified with V3-V4 primers. The PCR products underwent a denaturation and extension, then were purified by a Qiagen Gel Extraction Kit (Qiagen, Germany). After quality evaluation by a Qubit@2.0 Fluorometer (Thermo Scientific), the library was sequenced on an Illumina platform. Finally, 250 bp paired-end reads were generated.
Bioinformatics analysis
The quality of the tags was determined by the Fastp software. The sequences were removed to obtain effective tags by Vsearch (Version 2.15.0) [62]. The QIIME2 software was used to perform species annotation. The Mothur software was used to assess α diversity (including observed OTUs, Chao 1, Simpson and Shannon index) [63]. To calculate β diversity, PCoA analysis was performed. Different community structures between groups were analyzed by ANOSIM and QIIME2 software. In addition, t-test and LEfSe analysis (LDA score threshold: three) were performed to identify the significantly different species or biomarkers [64]. Finally, functional annotation analysis was performed using PICRUSt2 software (Version 2.1.2-b).
Statistical analysis
SPSS 24.0 (IBM SPSS, Inc., Chicago, IL, USA) was used for statistical analysis. Data was expressed as means ± standard deviations. Differences between the groups were compared by one-way ANOVA or nonparametric Mann–Whitney U test. P < 0.05 was considered as significantly different.
Data availability
The raw data used in the study are available from the corresponding author on reasonable request. The sequencing data have been submitted to China National GeneBank DataBase (accession number, CNP0004508).
Abbreviations
- PCOS:
-
Polycystic ovary syndrome
- PCOS.D:
-
PCOS and dyslipidemia
- NAFLD:
-
Nonalcoholic fatty liver disease
- BMI:
-
Body mass index
- LH:
-
Luteinizing hormone
- SHBG:
-
Sex hormone binding globulin
- HOMA-IR:
-
Homeostatic model assessment for insulin resistance
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- OTUs:
-
Operational taxonomic units
- PCoA:
-
Principal coordinate analysis
- ANOSIM:
-
Analysis of similarity
- LDA:
-
Linear discriminant analysis
- Lefse:
-
Linear discriminant analysis effect size
- HDL-C:
-
High-density lipoprotein cholesterol
- LDL-C:
-
Low-density lipoprotein cholesterol SCFAs, short-chain fatty acids
- DHEA-S:
-
Dehydroepiandrosterone sulfate
- IVF:
-
In-vitro fertilization
- FSH:
-
Follicle-stimulating hormone
- FPG:
-
Fasting plasma glucose
- FINS:
-
Fasting plasma insulin
References
Fauser BC, Tarlatzis BC, Rebar RW, Legro RS, Balen AH, Lobo R, Carmina E, Chang J, Yildiz BO, Laven JS, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97(1):28–38.e25.
Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7(4):219–31.
Lizneva D, Suturina L, Walker W, Brakta S, Gavrilova-Jordan L, Azziz R. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril. 2016;106(1):6–15.
Cooney LG, Dokras A. Beyond fertility: polycystic ovary syndrome and long-term health. Fertil Steril. 2018;110(5):794–809.
Gao L, Li M, Wang Y, Zeng Z, Xie Y, Liu G, Li J, Zhang B, Liang X, Wei L, et al. Overweight and high serum total cholesterol were risk factors for the outcome of IVF/ICSI cycles in PCOS patients and a PCOS-specific predictive model of live birth rate was established. J Endocrinol Invest. 2020;43(9):1221–8.
Chan JL, Kar S, Vanky E, Morin-Papunen L, Piltonen T, Puurunen J, Tapanainen JS, Maciel GAR, Hayashida SAY, Soares JM, editors. Jr. : Racial and ethnic differences in the prevalence of metabolic syndrome and its components of metabolic syndrome in women with polycystic ovary syndrome: a regional cross-sectional study. Am J Obstet Gynecol. 2017;217(2):189 e181-189 e188.
Jiang X, Lu X, Cai M, Liu Y, Guo Y. Impact of dyslipidemia on the cumulative pregnancy outcomes after first ovarian stimulation. Front Endocrinol (Lausanne). 2022;13:915424.
Liu T, Liu D, Song X, Qu J, Zheng X, Li J, Yang R, Yang S, Zhang X, Wang H, et al. Lipid metabolism was Associated with oocyte in vitro maturation in women with polycystic ovarian syndrome undergoing unstimulated natural cycle. Front Cell Dev Biol. 2021;9:719173.
Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta-cell dysfunction. Diabetes. 2007;56(9):2328–38.
Jiang H, Si M, Tian T, Shi H, Huang N, Chi H, Yang R, Long X, Qiao J. Adiposity and lipid metabolism indicators mediate the adverse effect of glucose metabolism indicators on oogenesis and embryogenesis in PCOS women undergoing IVF/ICSI cycles. Eur J Med Res 2023:28(1).
Cani PD. Human gut microbiome: hopes, threats and promises. Gut. 2018;67(9):1716–25.
Lynch SV, Pedersen O. The human intestinal microbiome in Health and Disease. N Engl J Med. 2016;375(24):2369–79.
de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–32.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19(1):55–71.
Thackray VG. Sex, microbes, and polycystic ovary syndrome. Trends Endocrinol Metab. 2019;30(1):54–65.
Liu R, Zhang C, Shi Y, Zhang F, Li L, Wang X, Ling Y, Fu H, Dong W, Shen J, et al. Dysbiosis of Gut Microbiota Associated with Clinical parameters in Polycystic Ovary Syndrome. Front Microbiol. 2017;8:324.
Insenser M, Murri M, Del Campo R, Martinez-Garcia MA, Fernandez-Duran E, Escobar-Morreale HF. Gut microbiota and the polycystic ovary syndrome: influence of sex, sex hormones, and obesity. J Clin Endocrinol Metab. 2018;103(7):2552–62.
Qi X, Yun C, Sun L, Xia J, Wu Q, Wang Y, Wang L, Zhang Y, Liang X, Wang L, et al. Gut microbiota-bile acid-interleukin-22 axis orchestrates polycystic ovary syndrome. Nat Med. 2019;25(8):1225–33.
Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JA, Brandsma E, Marczynska J, Imhann F, Weersma RK, et al. The gut Microbiome contributes to a substantial proportion of the variation in blood lipids. Circ Res. 2015;117(9):817–24.
Kalnina I, Gudra D, Silamikelis I, Viksne K, Roga A, Skinderskis E, Fridmanis D, Klovins J. Variations in the relative abundance of gut Bacteria correlate with lipid profiles in healthy adults. Microorganisms 2023;11(11).
Zhou X, Lian P, Liu H, Wang Y, Zhou M, Feng Z. Causal associations between Gut Microbiota and different types of Dyslipidemia: a two-sample mendelian randomization study. Nutrients 2023;15(20).
Miyajima Y, Karashima S, Ogai K, Taniguchi K, Ogura K, Kawakami M, Nambo H, Kometani M, Aono D, Demura M, et al. Impact of gut microbiome on dyslipidemia in Japanese adults: Assessment of the Shika-Machi super preventive health examination results for causal inference. Front Cell Infect Microbiol. 2022;12:908997.
Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–84.
Urbanek M. The genetics of the polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3(2):103–11.
Escobar-Morreale HF, Luque-Ramirez M, San Millan JL. The molecular-genetic basis of functional hyperandrogenism and the polycystic ovary syndrome. Endocr Rev. 2005;26(2):251–82.
Torres PJ, Siakowska M, Banaszewska B, Pawelczyk L, Duleba AJ, Kelley ST, Thackray VG. Gut Microbial Diversity in Women with Polycystic Ovary Syndrome correlates with hyperandrogenism. J Clin Endocrinol Metab. 2018;103(4):1502–11.
He F, Li Y. The gut microbial composition in polycystic ovary syndrome with insulin resistance: findings from a normal-weight population. J Ovarian Res. 2021;14(1):50.
Bowyer RCE, Jackson MA, Le Roy CI, Ni Lochlainn M, Spector TD, Dowd JB, Steves CJ. Socioeconomic status and the gut microbiome: a TwinsUK cohort study. Microorganisms 2019;7(1).
Wang J, Kurilshikov A, Radjabzadeh D, Turpin W, Croitoru K, Bonder MJ, Jackson MA, Medina-Gomez C, Frost F, Homuth G, et al. Meta-analysis of human genome-microbiome association studies: the MiBioGen consortium initiative. Microbiome. 2018;6(1):101.
Dominianni C, Sinha R, Goedert JJ, Pei Z, Yang L, Hayes RB, Ahn J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE. 2015;10(4):e0124599.
Choi JJ, Eum SY, Rampersaud E, Daunert S, Abreu MT, Toborek M. Exercise attenuates PCB-induced changes in the mouse gut microbiome. Environ Health Perspect. 2013;121(6):725–30.
Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Pedersen HK, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262–6.
Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91(6):2100–4.
Parker J, O’Brien C, Hawrelak J, Gersh FL. Polycystic ovary syndrome: an evolutionary adaptation to Lifestyle and the Environment. Int J Environ Res Public Health 2022;19(3).
Holert J, Cardenas E, Bergstrand LH, Zaikova E, Hahn AS, Hallam SJ, Mohn WW, Lindow SE, Tringe S, Garcia J. Metagenomes Reveal Global Distribution of Bacterial Steroid Catabolism in Natural, Engineered, and Host Environments. mBio 2018;9(1).
Bergstrand LH, Cardenas E, Holert J, Van Hamme JD, Mohn WW. Delineation of Steroid-Degrading microorganisms through comparative genomic analysis. mBio. 2016;7(2):e00166.
Holert J, Jagmann N, Philipp B. The essential function of genes for a hydratase and an aldehyde dehydrogenase for growth of Pseudomonas sp. strain Chol1 with the steroid compound cholate indicates an aldolytic reaction step for deacetylation of the side chain. J Bacteriol. 2013;195(15):3371–80.
Holert J, Kulic Z, Yucel O, Suvekbala V, Suter MJ, Moller HM, Philipp B. Degradation of the acyl side chain of the steroid compound cholate in Pseudomonas sp. strain Chol1 proceeds via an aldehyde intermediate. J Bacteriol. 2013;195(3):585–95.
Ramani K, Sekaran G. Production of lipase from Pseudomonas gessardii using blood tissue lipid and thereof for the hydrolysis of blood cholesterol and triglycerides and lysis of red blood cells. Bioprocess Biosyst Eng. 2012;35(6):885–96.
Yang Z, Fu H, Su H, Cai X, Wang Y, Hong Y, Hu J, Xie Z, Wang X. Multi-omics analyses reveal the specific changes in gut metagenome and serum metabolome of patients with polycystic ovary syndrome. Front Microbiol. 2022;13:1017147.
Rutkowski K, Sowa P, Rutkowska-Talipska J, Kuryliszyn-Moskal A, Rutkowski R. Dehydroepiandrosterone (DHEA): hypes and hopes. Drugs. 2014;74(11):1195–207.
Iannone M, Botre F, Martinez-Brito D, Matteucci R, de la Torre X. Development and application of analytical procedures for the GC-MS/MS analysis of the sulfates metabolites of anabolic androgenic steroids: the pivotal role of chemical hydrolysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2020;1155:122280.
Vojinovic D, Radjabzadeh D, Kurilshikov A, Amin N, Wijmenga C, Franke L, Ikram MA, Uitterlinden AG, Zhernakova A, Fu J, et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat Commun. 2019;10(1):5813.
Sun Y, Tang Z, Hao T, Qiu Z, Zhang B. Simulated digestion and fermentation in Vitro by obese human gut microbiota of sulforaphane from Broccoli seeds. Foods 2022;11(24).
Burakova I, Smirnova Y, Gryaznova M, Syromyatnikov M, Chizhkov P, Popov E, Popov V. The effect of short-term consumption of lactic acid Bacteria on the gut microbiota in obese people. Nutrients 2022;14(16).
Rom O, Liu YH, Liu ZP, Zhao Y, Wu JF, Ghrayeb A, Villacorta L, Fan YB, Chang L, Wang L et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci Transl Med 2020;12(572).
Fletcher JR, Pike CM, Parsons RJ, Rivera AJ, Foley MH, McLaren MR, Montgomery SA, Theriot CM. Clostridioides difficile exploits toxin-mediated inflammation to alter the host nutritional landscape and exclude competitors from the gut microbiota. Nat Commun. 2021;12(1):462.
Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–34.
Wang W, Zhao J, Gui W, Sun D, Dai H, Xiao L, Chu H, Du F, Zhu Q, Schnabl B, et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br J Pharmacol. 2018;175(3):469–84.
Zhou M, Johnston LJ, Wu C, Ma X. Gut microbiota and its metabolites: Bridge of dietary nutrients and obesity-related diseases. Crit Rev Food Sci Nutr 2021:1–18.
Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A. 2008;105(6):2117–22.
Tong X, Xu J, Lian F, Yu X, Zhao Y, Xu L, Zhang M, Zhao X, Shen J, Wu S et al. Structural Alteration of Gut Microbiota during the Amelioration of Human Type 2 Diabetes with Hyperlipidemia by Metformin and a Traditional Chinese Herbal Formula: a Multicenter, Randomized, Open Label Clinical Trial. mBio 2018;9(3).
Min Y, Ma X, Sankaran K, Ru Y, Chen L, Baiocchi M, Zhu S. Sex-specific association between gut microbiome and fat distribution. Nat Commun. 2019;10(1):2408.
Yang T, Zhao J, Liu F, Li Y. Lipid metabolism and endometrial receptivity. Hum Reprod Update. 2022;28(6):858–89.
McNaught KJ, Kuatsjah E, Zahn M, Prates ET, Shao H, Bentley GJ, Pickford AR, Gruber JN, Hestmark KV, Jacobson DA, et al. Initiation of fatty acid biosynthesis in Pseudomonas putida KT2440. Metab Eng. 2023;76:193–203.
Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. 2019;7(1):91.
Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7(3):189–200.
Bridgeman SC, Northrop W, Melton PE, Ellison GC, Newsholme P, Mamotte CDS. Butyrate generated by gut microbiota and its therapeutic role in metabolic syndrome. Pharmacol Res. 2020;160:105174.
Rotterdam EA-SP. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod. 2004;19(1):41–7.
Ford ES, Giles WH. A comparison of the prevalence of the metabolic syndrome using two proposed definitions. Diabetes Care. 2003;26(3):575–81.
Joint Committee on the Chinese Guidelines for Lipid M. Chinese guidelines for lipid management (2023). Zhonghua Xin xue guan bing za zhi. 2023;51(3):221–55.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21(3):494–504.
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60.
Acknowledgements
We thank Editage (www.editage.com) for editing English language.
Funding
This work was funded by the National Natural Science Foundation of China (No. 82201777).
Author information
Authors and Affiliations
Contributions
YXC, XJH and BS designed the experiments, TJY contributed to the experiments and manuscript drafting. GJL contributed to the revision of the manuscript. YPX contributed to data collection. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The research has been performed in accordance with the Declaration of Helsinki, and this study was approved by the Ethics Committee of the First Affiliated Hospital of Anhui Medical University (PJ 2023-08-42). Each participant was informed about the sample collection and has provided a written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, T., Li, G., Xu, Y. et al. Characterization of the gut microbiota in polycystic ovary syndrome with dyslipidemia. BMC Microbiol 24, 169 (2024). https://doi.org/10.1186/s12866-024-03329-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-024-03329-x