
Abstract
Background
The genus Pulmonaria (Boraginaceae) represents a taxonomically complex group of species in which morphological similarity contrasts with striking karyological variation. The presence of different numbers of chromosomes in the diploid state suggests multiple hybridization/polyploidization events followed by chromosome rearrangements (dysploidy). Unfortunately, the phylogenetic relationships and evolution of the genome, have not yet been elucidated. Our study focused on the P. officinalis group, the most widespread species complex, which includes two morphologically similar species that differ in chromosome number, i.e. P. obscura (2n = 14) and P. officinalis (2n = 16). Ornamental cultivars, morphologically similar to P. officinalis (garden escapes), whose origin is unclear, were also studied. Here, we present a pilot study on genome size and repeatome dynamics of these closely related species in order to gain new information on their genome and chromosome structure.
Results
Flow cytometry confirmed a significant difference in genome size between P. obscura and P. officinalis, corresponding to the number of chromosomes. Genome-wide repeatome analysis performed on genome skimming data showed that retrotransposons were the most abundant repeat type, with a higher proportion of Ty3/Gypsy elements, mainly represented by the Tekay lineage. Comparative analysis revealed no species-specific retrotransposons or striking differences in their copy number between the species. A new set of chromosome-specific cytogenetic markers, represented by satellite DNAs, showed that the chromosome structure in P. officinalis was more variable compared to that of P. obscura. Comparative karyotyping supported the hybrid origin of putative hybrids with 2n = 15 collected from a mixed population of both species and outlined the origin of ornamental garden escapes, presumably derived from the P. officinalis complex.
Conclusions
Large-scale genome size analysis and repeatome characterization of the two morphologically similar species of the P. officinalis group improved our knowledge of the genome dynamics and differences in the karyotype structure. A new set of chromosome-specific cytogenetic landmarks was identified and used to reveal the origin of putative hybrids and ornamental cultivars morphologically similar to P. officinalis.
Similar content being viewed by others


Background
The genus Pulmonaria L. (Boraginaceae, sensu [1]) is a taxonomically complex group of species in which the rather similar morphology contrasts with striking karyological variation. In total, 16 different somatic chromosome counts ranging from 2n = 14 to 2n = 38 are currently reported in the genus [Kobrlová unpubl.], with x = 7 proposed as the basic chromosome number [e.g. 2–6]. According to the traditional morphology- and karyology-based taxonomy [cf. 3, 6], about 30 taxa are recognized (16 species and 14 subspecies), all found in Europe, with the exception of the P. mollis group, which extends as far as north-eastern and eastern Asia [5, 7,8,9]. Although almost nothing is known about the mechanisms of the chromosomal evolution of the genus, such a karyological diversity is indicative of extensive chromosomal rearrangements, i.e. the hypothesis of ancient episodes of polyploidization/hybridization and subsequent dysploidal chromosome reorganizations [cf. 6, 10].
To date, only a few attempts have been made to explore the evolutionary history of the genus Pulmonaria, focusing on a limited or specific group of species [10,11,12,13,14]. These studies have highlighted the significant role of hybridization (e.g. the recent hybrid origin of P. helvetica Bolliger [14] or the P. hirta complex [10]), and provided evidences for the involvement of introgression and dysploidy in the speciation process [10, 13]. In horticulture, Pulmonaria species are known to be able to cross (no geographical isolation, flowering synchrony, similar pollinator preferences), giving rise to a wide variety of cultivars [see 15]. Even in the contact zones of some taxa, intermediate chromosome numbers have rarely been documented [2, 3, 16, 17]. However, most of the evolutionary relationships are still unknown, and the question remains as to what lies behind the observed variability in their morphology and chromosome number.
Along with chromosome number, genome size is another relevant biodiversity trait that can indicate significant genomic events and evolutionary changes [18, 19]. Diversity in genome size is driven by multiple evolutionary processes such as polyploidy, the proliferation of repetitive DNA sequences and/or drastic reduction of non-genic DNA [18, 20,21,22]. Typically, post-polyploid diploidization is thought to be associated with extensive loss of DNA or ‘genome downsizing’ [23,24,25,26,27,28,29]. However, the mechanisms, rates and selection pressures driving these changes in DNA content remain unknown [cf. 28]. Although considerable karyological variation is evident in several Boraginaceae genera [e.g. 30–34], there have been almost no complex analyses of genome size variation and the evolutionary pathways behind the observed diversity [but see 35–37].
Our study is focused on the P. officinalis group, the most widespread European species complex [3, 38, 39]. This group contains two morphologically relatively distinct species differing in chromosome numbers, namely P. obscura Dumort. (2n = 14) and P. officinalis L. (2n = 16 [e.g. 3, 17, 40–42]). Within the latter, two subspecies are sometimes recognized [43,44,45]. As the P. officinalis complex has been cultivated for medicinal and ornamental purposes for a very long time [46, 47], it has probably become even more widespread. In some regions it may also be non-native as a result of frequent cultivation and possible garden escapes [42, 47]. “Pure” Pulmonaria species have often been crossed in cultivation to produce more attractive varieties [cf. 15], which are rarely found in nature. Many of these plants are listed under the name P. saccharata in the horticultural trade. Their origin is not clear, but it can be assumed that some of them are derived from P. officinalis (esp. plants with distinctly white-spotted leaves, cordate at the base). However, they are not “true” P. saccharata sensu Miller, whose taxonomic status has long been debated, and are most likely related to the P. hirta complex [cf. 10, 48, 49].
Despite the morphological and karyological differences observed, the origin of this species group remains unknown. To uncover the differences in the karyotype evolution, we used complex methodological approaches involving genome size estimation, genome skimming followed by bioinformatic analysis and characterization of repeatomes in the P. officinalis group. Specifically, we have analyzed repeatomes and karyotypes of P. obscura and P. officinalis s.str. individuals from pure separate populations, their putative hybrid accessions with 2n = 15, which have been collected in a mixed population where P. obscura and P. officinalis grew together, and three populations of ornamental cultivars, morphologically similar to P. officinalis, that have escaped into the wild where they were collected (here listed as P. saccharata-like). We aimed to identify a new set of chromosome-specific cytogenetic markers that can be used to identify individual chromosomes, and to compare karyotypes of closely related species and their inter-specific hybrids. We also studied the impact of repetitive DNA sequences on genome size in the P. officinalis group. This pilot study can serve as a springboard for future cytogenetic and genomic studies, to understand the role of chromosomal rearrangements in the evolution of this genus.
Results
Genome size and chromosome number variation in the P. officinalis group
A total number of 196 accessions from 65 populations of the Pulmonaria officinalis group were collected throughout Europe (Fig. 1A). The flow cytometry analyses resulted in good quality histograms with distinct peaks and low coefficients of variation (below 5%) for both PI and DAPI staining (Supplementary Table 1). The flow cytometry data were accompanied by chromosome counts, some of which were obtained de novo (see below), but previously published reports were also revised. A total of 758 published chromosome records were found and revised for the P. officinalis complex (Supplementary Table 2). Of these, 289 chromosome reports with 2n = 14 were related to P. obscura and 451 with 2n = 16 to P. officinalis s.str., respectively (Fig. 1A). Three records of 2n = 16 have been published for the subspecies P. officinalis subsp. marzolae G.Astuti, Peruzzi, Cristof. & P.Pupillo. In one case 2n = 17 have been reported for P. officinalis. In addition to this, there are even karyological records (2n = 15) that refer to hybrids of P. obscura and P. officinalis (Supplementary Table 2)

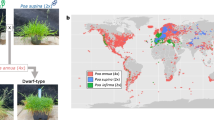
Distribution map of the Pulmonaria officinalis group. (A) Distribution map of the P. officinalis group sampled and analyzed in this study (large dots; see Supplementary Table 1), including published chromosome reports (758 records in total, small dots; see Supplementary Table 2): P. obscura in yellow (2n = 14) and P. officinalis s.str. (2n = 16) in red. Illustrative images of (B) P. obscura (B473.1), (C) P. officinalis s.str. (B470.1) and (D) P. saccharata-like accession (B15.1)
Pulmonaria obscura (2n = 14; mean 2C = 2.92 ± 0.10 pg) and P. officinalis s.str. (2n = 16; mean 2C = 3.21 ± 0.13 pg) differed in holoploid genome size (2C value: ANOVA, F = 93.69, P < 0.001, Fig. 2A) as well as in the genomic GC content (Fig. 2B). Their DNA amount ranges are listed in Table 1. The genome size value and GC content of the presumed hybrid plants (B481) with 2n = 15 (2C = 3.08 ± 0.02 pg and 35.42%, respectively) were between those of the supposed parents (i.e., within the theoretically expected intermediate genome size and GC content based on the genome sizes of P. obscura and P. officinalis; Table 1). Among P. saccharata-like populations, B465.1 with 2n = 16 had the genome size value (2C = 3.13 pg) in the range of P. officinalis. The remaining two populations (B15: 2C = 3.74 ± 0.03 pg; B472.1: 2C = 3.63 pg) had larger genome sizes, but differ in the number of chromosomes (B15: 2n = 15 vs. B472.1: 2n = 16) and GC content (Table 1).

Genome size and GC content variation in Pulmonaria obscura and P. officinalis. (A) Variation in absolute genome size (2C-value); and (B) genomic GC content detected in the P. officinalis group: P. obscura (POBS), P. officinalis (POFF). Rectangles define the 25th and 75th percentiles, horizontal lines show median values, whiskers are 10–90 percentiles
Genome-wide repeatome analysis
To identify the major types of repetitive sequences and to compare their genome representation in P. officinalis group, the comparative mode of RepeatExplorer2 pipeline was used. The analysis was performed on genome skimming data of P. obscura (2n = 14; B473.1, Fig. 1B), P. officinalis (2n = 16; B470.3, Fig. 1C), their putative interspecific hybrid (2n = 15; B481.1) and three P. saccharata-like accessions (B15.1; 2n = 15, Fig. 1D and B465.1, B472.1; both 2n = 16). The identified repetitive sequences accounted for 58.48% of the P. obscura genome and 47.86% of the P. officinalis genome. A similar and overall highest proportion of repetitive sequences was found in repeatomes of the putative interspecific hybrid B481.1 (64.10%) and P. saccharata-like accession B15.1 (64.53%), while the repetitive DNA content of the other two P. saccharata-like accessions (B465.1 and B472.1) was around 50% (Table 2; Fig. 3, Supplementary Table 3).

Repeatome composition in analyzed Pulmonaria accessions. Genome proportion of individual repeat type was obtained as the ratio of reads specific to the individual repeat type to all reads used for the clustering analysis by the RepeatExplorer2 pipeline. P. obscura (POBS; B473.1); P. officinalis (POFF; B470.3); an interspecific natural hybrid P. obscura × P. officinalis (HYBR; B481.1); P. saccharata-like accessions (PSAC1–PSAC3; B465.1; B472.1; B15.1)
In all studied taxa, LTR retroelements were the most abundant repeats, accounting for 46.68% of P. obscura genome, 34.52% of P. officinalis genome and 33.80-49.96% of the other genomes. The genome proportion of Ty3/Gypsy elements, which were mainly represented by Tekay lineage, ranged from 23.90% in P. saccharata-like (B472.1) to 35.06% in putative interspecific hybrid (B481.1). Ty3/Gypsy elements were nearly twice abundant as Ty1/Copia superfamily, which was mostly represented by elements from SIRE and Angela clades (Table 2; Fig. 3). DNA transposons and long interspersed nuclear elements (LINE elements) were found in low copy numbers in all analyzed groups, with genome proportions ranging from 0.16 to 0.22% and from 0.14 to 0.23%, respectively (Table 2). rDNA sequences accounted for about 3.00–5.00%, and other tandem organized repeats represented 1.25% to more than 4.00% of the Pulmonaria genomes (Table 2).
Variation in the Satellite DNAs (satDNAs) and rDNA clusters
Five putative satellites (tandem organized repeats) were identified by TAREAN program [50] in individual P. obscura and P. officinalis datasets. Three of them (PulTR01_29, PulTR03_308, PulTR05_70) were shared by both species, whereas PulTR02_305 was found only in P. obscura, and PulTR04_420 was only detected in the P. officinalis genome. All identified tandem repeats produced chromosome-specific signals and were used together with rDNA sequences to create karyotypes of the Pulmonaria accessions studied.
The Pulmonaria 45S rDNA unit varied from 9.1 kb to 9.6 kb in length and was found to be highly conserved at the sequence level within all accessions studied (Supplementary Fig. 1). The intergenic spacer (IGS) region contained two different tandemly organized repeats (with repetitive units of 117 and 150-nt long) which were identical in all the accessions studied, with the exception of a P. saccharata-like plant B472.1, which contained tandem regions with shorter repetitive units (79 and 150-nt long). A higher variability was found in the IGS, which contained two relatively large INDEL regions that differed between P. obscura and P. officinalis (Supplementary Fig. 2A). To support this observation, we performed read mapping of Pulmonaria accessions to the assembled P. obscura 45S rDNA reference (Supplementary Fig. 2B). The differences in read coverage along the 45S rDNA unit are evident in the IGS, indicating the variability at the sequence level. It should be pointed out that the assembly of the 45S rDNA unit was obtained from short Illumina sequences, so the resulting consensus sequence may represent the most abundant sequence type.
The graph-based clustering algorithm of the RepeatExplorer2 pipeline allowed complete reconstruction of the 5S rDNA unit, even in the comparative analysis (Fig. 4). The graph region representing the 5S rRNA gene was shared by all Pulmonaria accessions studied (Fig. 4B). Three variable graph loops emanating from this conserved graph region correspond to three types of 5S rDNA unit, differing in the length of their IGS (Fig. 4A, B). Mapping of sample-specific sequencing reads onto the graph topology revealed the presence of different 5S rDNA units between P. obscura and P. officinalis (Fig. 4C, D). The cluster layout of the presumed hybrid between P. obscura and P. officinalis (B481.1), was represented by all three 5S rDNA graph loops specific to its putative progenitors (Fig. 4E). A similar situation was observed for the P. saccharata-like accession B15.1 (Fig. 4F). The other P. saccharata-like individuals (B465.1 and B472.1) shared the same graph layout as P. officinalis (i.e. B470.3; Fig. 4G, H).

Graph structure of 5S rDNA sequence reads from the comparative analysis of RepeatExplorer2. (A) Graph structure obtained from all sequence reads homologous to the 5S rDNA. (B) The position of the 5S genic region on the graph topology is highlighted in yellow. (C–H) Cluster graph with annotated read origin: P. obscura in green (C); P. officinalis in orange (D); reads specific for P. obscura × P. officinalis (B481.1) in blue (E); reads specific for P. saccharata-like accession B15.1 are highlighted in purple (F), B465.1 in pink (G), and B472.1 in red (C)
The analysis of 5S rDNA specific graph layouts observed by clustering analysis of individual accessions confirmed the observation of the comparative analysis. All Pulmonaria individuals studied were composed of at least two different types of 5S rRNA genes, which differed in the IGS (Supplementary Fig. 3).
Comparative karyotyping in the P. officinalis group
Molecular karyotyping was performed using newly identified satellites (PulTR01_29, PulTR02_305, PulTR03_308, PulTR04_420, and PulTR05_70) and 5S and 45S rDNA sequences. In general, in situ hybridization confirmed the results obtained by repeatome analysis. FISH analysis with the probes for rDNAs and four satDNAs resulted in well visible cluster signals on specific chromosomes in the genome of the analyzed plants of the P. officinalis complex.
P. obscura, P. officinalis and their putative natural interspecific hybrid
FISH analysis of P. obscura plants (all 2n = 14) collected from three different populations (B467, B469, B473; Supplementary Table 1) showed highly consistent results, with only minor differences observed in plants B473.1 - in the number of satDNA PulTR01_29 (Fig. 5A). The 45S rDNA was located into terminal NOR regions on four chromosome pairs, while 5S rDNA signals were detected on two chromosome pairs in pericentromeric regions (Figs. 5A, B and 6A, B, F and G). One chromosome pair contained signals of 5S, 45S rDNA and satellites PulTR02_305 and PulTR05_70 (Figs. 5A, B and 6B, D, F and G). In B473.1, the probe for PulTR01_29 provided very strong signals in subtelomeric regions only on one chromosome pair (Fig. 6E, G). The same chromosome pair also contained a signal of PulTR03_308 in the pericentromeric region. In B467.2 and B469.1, one additional subtelomeric signal of PulTR01_29 was found, located on a chromosome arm with 5S rDNA cluster (Figs. 5B and 6E and G). As expected from the results of the RepeatExplorer2 analysis, signals of PulTR04_420 were not detected in any of the P. obscura plants analyzed.

Idiograms of analyzed Pulmonaria accessions

Chromosomal localization of newly identified satDNAs and rDNA sequences in P. obscura (2n = 14). (A, B, C, D) P. obscura (B473.1) with probes for: (A) 45S rDNA (yellow), PulTR01_29 (red) and 5S rDNA (green); (B) 45S rDNA (yellow), PulTR02_305 (red) and 5S rDNA (green); (C) PulTR01_29 (red), PulTR03_308 (orange), and 5S rDNA (green); (D) PulTR02_305 (yellow), 5S rDNA (red) and PulTR05_70 (green): yellow arrows indicate signals of PulTR02_305 and green arrows indicate signals of PulTR05_70. (E, F, G, H) P. obscura (B467.2) with probes for: (E) 45S rDNA (yellow), PulTR01_29 (red) and 5S rDNA (green): red arrow indicate subtelomeric signals of PulTR01_29; (F) 45S rDNA (yellow), PulTR02_305 (red) and 5S rDNA (green): red arrows indicate signals of PulTR02_305; (G) PulTR01_29 (red), PulTR03_308 (orange), and 5S rDNA (green); and (G) PulTR02_305 (yellow), 5S rDNA (red) and PulTR05_70 (green): yellow arrows indicate signals of PulTR02_305 and green arrows indicate signals of PulTR05_70. White arrows indicate 5S rDNA in all figures. Chromosomes were counterstained with DAPI (blue). Bars = 5 μm
The genome of P. officinalis contained one additional pair of chromosomes (i.e. 2n = 16) compared to P. obscura. The molecular karyotype was studied in individuals from two different populations (B100, B470; Supplementary Table 1) and, in contrast to P. obscura, a higher variation in the chromosomal distribution of the probes was found even between individuals within the same population (Fig. 5C, D; Supplementary Fig. 4). Terminal NORs (45S rDNA) were found only on three chromosome pairs in both analyzed individuals (B100.2, B470.1). These terminal 45S rDNA loci were often fragile and distended from the chromosomes (Fig. 7A, H, I). Interstitial 45S rDNA clusters were detected on one chromosome pair, on the same arm which also contained signals of satDNA PulTR04_420, PulTR01_29 and PulTR05_70 (Figs. 5C, D and 7B, E, H and I). Additional signals of PulTR05_70 were detected on two chromosome pairs that also contained 45S rDNA loci (Fig. 7E, I). The genome of B100.2 contained an additional weak signal of interstitial 45S rDNA (Figs. 5D and 7F, G and H). Variation in the number of signals specific to 5S rDNA was detected. While signals of 5S rDNA were identified in pericentromeric regions on seven chromosomes of B470.3 (Fig. 7A, E), only six chromosomes were bearing these signals in B100.2 (Fig. 7G, H). The signal of PulTR03_308 was detected on one NOR bearing chromosome pair in both individuals with various combinations of co-localization with PulTR04_420 and 5S rDNA (Figs. 5C, D and 7C, D and G). Signals of PulTR02_305 were not detected on chromosomes of P. officinalis, supporting the results of comparative repeatome analysis by RepeatExplorer2.

Chromosomal localization of newly identified satDNAs and rDNA sequences in P. officinalis (2n = 16). (A, B, C, D, E) P. officinalis (B470.3) with probes for: (A) 45S rDNA (yellow), PulTR01_29 (red) and 5S rDNA (green); (B) 45S rDNA (yellow), PulTR01_29 (red) and PulTR04_420 (green): green arrows indicate PulTR04_420 and; (C, D) the same plate with the signals for (C) 45S rDNA (red) and PulTR03_308 (green), and (D) co-localization of PulTR03_308 (green) and PulTR04_420 (red): green arrows indicate PulTR03_308, and red arrows indicate PulTR04_420; (E) 45S rDNA (yellow), PulTR05_70 (red), 5S rDNA (green): red arrows indicate signals of PulTR05_70. (F, G, H, I) P. officinalis (B100.2) with probes for: (F) 45S rDNA (yellow) and PulTR01_29 (red); (G) 45S rDNA (yellow), PulTR03_308 (red) and 5S rDNA (green): red arrows indicate PulTR03_308; and (H) 45S rDNA (yellow), PulTR04_420 (red), and 5S rDNA (green): red arrows indicate PulTR04_420; (I) 45S rDNA (yellow), PulTR05_70 (red) and PulTR03_308 (green): red arrows indicate signals of PulTR05_70 and green arrows point to PulTR03_308. White arrows indicate signals of 5S rDNA and yellow arrows point at interstitial 45S rDNA clusters. Chromosomes were counterstained with DAPI (blue). Bars = 5 μm
Karyotype analysis of the putative natural hybrid between P. obscura and P. officinalis (B481.1; Supplementary Table 1), confirmed the expected chromosome number 2n = 15., and the presence and the distribution pattern of P. obscura and P. officinalis species-specific satDNAs (Fig. 5E). In this case, eight 45S rDNA clusters were found, seven in terminal chromosomal regions and one in an interstitial position (Figs. 5E and 8A and C). 5S rDNA clusters were detected on five chromosomes in pericentromeric regions (Fig. 8A, B). One chromosome pair contained signals of PulTR01_29 and 5S rDNA (Fig. 8B, C), another individual chromosome contained 45S and 5S rDNA, PulTR02_305 (found only as one single locus in the genome) and PulTR05_70 (Fig. 8A, D). Additional two chromosomes contained one or two signals of 5S rDNA, respectively (Figs. 5E and 8A and B). Five signals of the most abundant satellite PulTR01_29 were detected on five chromosomes (Fig. 8B, C), one of which co-localized with the 45S rDNA locus, one signal was detected on the chromosome bearing weak interstitial signal of 45S and PulTR05_70 and one with signal of PulTR03_308 (Fig. 8B) and PulTR04_420 (Fig. 8C), respectively. One chromosome pair with the remaining two signals of PulTR01_29 also contained a signal of 5S rDNA (Fig. 8B). SatDNAs PulTR03_308 and PulTR04_420 co-localized on one chromosome pair, containing also PulTR01_29, or joint signal of 45S rDNA and PulTR01_29 (Fig. 8C). Finally, two additional signals of PulTR05_70 were found on chromosomes with terminal 45S rDNA clusters (Fig. 8D).

Chromosomal localization of new satDNAs and rDNA sequences in natural hybrid P. obscura × P. officinalis (B481.1; 2n = 15). (A) Probes for 45S rDNA (yellow), PulTR02_305 (red) and 5S rDNA (green): red arrow points at the locus of PulTR02_305; (B) PulTR01_29 (red), PulTR03_308 (orange) and 5S rDNA (green): orange arrows indicate PulTR03_308; (C) 45S rDNA (yellow), PulTR01_29 (red) and PulTR04_420 (green): green arrows indicate PulTR04_420; red arrow points to PulTR01_29 and yellow arrow points at 45S rDNA locus indicating co-localization of these two probes; (D) 45S rDNA (yellow), PulTR05_70 (red) and 5S rDNA (green): red arrows indicate signals of PulTR05_70. White arrows indicate 5S rDNA signals. Chromosomes were counterstained with DAPI (blue). Bars = 5 μm
Ornamental garden escapes, morphologically similar to P. Officinalis
Karyotype analysis of P. saccharata-like plants from three populations (B15, B465, B472; Supplementary Table 1) revealed variability in chromosome number, as well as variation in localization of satDNAs and rDNA sequences (Fig. 5F, G, H). While B465.1 and B472.1 were characterized by 2n = 16 and have very similar karyotypes resembling P. officinalis (Figs. 5F, G and 9A – I), 2n = 15 was detected in B15.1 with a pattern of cluster signals similar to that of the putative interspecific hybrid (Figs. 5H and 9J – M). The karyotypes of B465.1 and B472.1 were very similar, containing terminal NORs on three chromosome pairs and one interstitial 45S rDNA cluster on one additional chromosome pair (Fig. 9A, C, F – I). Two chromosome pairs were bearing PulTR01_29 in subtelomeric regions and other two chromosome pairs contained 5S rDNA clusters in pericentromeric regions (Fig. 9A, B, F). The signal of PulTR04_420 was found on two chromosome pairs (Fig. 9A, D, F, G). One of these chromosome pairs also contained signals of PulTR01_29, an interstitial signal of 45S rDNA and PulTR05_70 (Fig. 9E, I), while the other chromosome pair bearing terminal 45S rDNA and the PulTR04_420 co-localized with PulTR03_308 (Figs. 5F, G and 9A and D – G). Additional two chromosome pairs with terminal loci of 45S rDNA also contained signals of PulTR05_70 (Fig. 9E, I). However, these two accessions differed in the presence of an additional signal of 5S rDNA and PulTR01_29 (Figs. 5F, G and 9).

Chromosomal localization of new satDNAs and rDNA sequences in P. saccharata-like accessions. (A, B, C, D, E) P. saccharata-like accession B465.1 (2n = 16) with probes for: (A) 45S rDNA (yellow), PulTR01_29 (red) and PulTR04_420 (green): green arrows indicate PulTR04_420; (B) PulTR01_29 (red) and 5S rDNA (green); (C) 45S rDNA (red) and PulTR03_308 (green): green arrows point at PulTR03_308; and (D) PulTR03_308 (green) and PulTR04_420 (red): green arrows point at PulTR03_308 and red arrows indicate PulTR04_420; (E) 45S rDNA (yellow), PulTR05_70 (red) and 5S rDNA (green): red arrows point to signals of PulTR05_70. (F, G, H, I) P. saccharata-like accession B472.1 (2n = 16) with probes for: (F) 45S rDNA (yellow), PulTR01_29 (green) and PulTR04_420 (red): red arrows point at PulTR04_420; (G) 45S rDNA (yellow), PulTR03_308 (green) and PulTR04_420 (red): green arrows point at PulTR03_308, and red arrows indicate PulTR04_420; (H) 45S rDNA (yellow), PulTR03_308 (red) and 5S rDNA (green); (I) 45S rDNA (yellow), PulTR05_70 (red) and PulTR03_308 (green): red arrows indicate signals of PulTR05_70 and green arrows point to PulTR03_308. (J, K, L, M) P. saccharata-like accession B15.1 (2n = 15) with probes for: (J) 45S rDNA (yellow), PulTR02_305 (red) and 5S rDNA (green): red arrow points at a signal of PulTR02_305; (K) 45S rDNA (yellow), PulTR01_29 (red) and PulTR04_420 (green): green arrows point at PulTR04_420; (L) PulTR03_308 (red) and 5S rDNA (green); (M) 45S rDNA (yellow), PulTR05_70 (red) and 5S rDNA (green): red arrows point to signals of PulTR05_70. White arrows indicate 5S rDNA signals and yellow arrows point at interstitial 45S rDNA clusters. Chromosomes were counterstained with DAPI (blue). Bars = 5 μm
The karyotype of B15.1 was similar to that of the putative interspecific hybrid (B481.1) with the same number of chromosomes (Fig. 5H). 45S rDNA clusters were located as strong signals in terminal regions of three chromosome pairs, one additional chromosome contained a weak terminal signal and another contained an interstitial signal (Fig. 9J, K). Odd number of signal localizations was detected for 5S rDNA (five chromosomes with interstitial signals) (Fig. 9J, K), PulTR01_29 (three chromosomes with the signals in terminal regions), PulTR04_420 (three chromosomes with interstitial signals), and for PulTR02_305 (found only on one chromosome) (Fig. 9H – J). PulTR03_308 provided signals on one chromosome pair co-localizing with PulTR04_420 (Figs. 5H and 9K and L). Finally, four signals of PulTR05_70 were found on three chromosomes with terminal 45S rDNA loci (on one of them co-localizing with 5S rDNA and PulTR02_305), and one chromosome with interstitial signal of 45S rDNA, PulTR04_420 and PulTR01_29 (Fig. 9M).
Discussion
The genus Pulmonaria is karyologically highly variable [e.g. 3, 6], but the origin and evolutionary consequences of genome size and karyotype variation remain unexplored [but see 10]. It appears that chromosomal rearrangements have played an important role in the evolution of this genus [cf. 10, 14, 51], but how and to what extent has never been clearly demonstrated. Therefore, we performed a pilot analysis of genome size and a comparative analysis of the repeatomes in the P. officinalis species group.
Impact of DNA repeats dynamics on genome size
Genome size can reflect some aspects of the evolutionary history of taxa by allowing us to understand the influence of DNA gain/loss between related species [e.g. 52, 53]. Our study represents the first large-scale investigation of interspecific genome size variation in Pulmonaria. As already shown in a pilot study by Kobrlová & Hroneš [31], genome size is effective in delimiting morphologically similar taxa of the Boraginaceae, which is also true for the P. officinalis group. The suitability of using flow cytometry to revise the distribution of the P. officinalis group (i.e. relative genome size) has already been documented in the Bohemian Forest and adjacent foothills [42].
So far, the genome size has only been estimated for eight Pulmonaria taxa, including P. officinalis and P. obscura, ranging from 2.27 to 4.27 pg (i.e. very small/small genomes according to the categories of Leitch et al. [54], Supplementary Table 5). Only minor differences were observed when comparing previously analyzed genome sizes of P. obscura and P. officinalis with our data, most likely due to different methodologies used (i.e. nuclei isolation buffer, reference standard, plant organ [cf. 31, 51]). The only exception is the study by Šmarda et al. [55], where almost the same 2C values are presented for P. obscura and P. officinalis, probably as a consequence of taxa misidentification.
There is enormous variation in the size of plant genomes, with much of this diversity driven by differences in the abundance of DNA transposons [e.g. 22, 56,57,58,59,60]. We found that most of the repetitive elements in the genomes of the Pulmonaria taxa studied were dispersed repeats represented by LTR retrotransposons [cf. 22, 58], with higher proportion of Ty3/Gypsy elements, which were twice more abundant than Ty1/Copia. Ty3/Gypsy elements represent one of the major classes of LTR retrotransposons and are dominant in many plant groups, such as the family Poaceae [e.g. 59, 61,62,63] or the tribe Fabeae [22, 64]. Unfortunately, a genome-wide analysis of DNA repeats and their impact on genome size has not been performed for any member of the Boraginaceae family. However, the higher proportion of Ty3/Gypsy retroelements have also been found in genera of the closely related Solanaceae family, such as Solanum, Nicotiana and Capsicum [65,66,67,68,69,70]. In contrast, recent studies in the genus Salvia, a member of the closely related Lamiaceae family, have shown that the nuclear genomes of different species contain different proportions of Ty3/Gypsy and Ty1/Copia retroelements [71,72,73], indicating a proliferation of different types of DNA repeats during the evolution of individual species. In comparison, the studied Pulmonaria species contained a similar proportion of the repeat lineages and individual clusters were represented by reads from all specimens analyzed. This indicates a high degree of genome homology within the P. officinalis complex, suggesting that the evolution of this species group was not accompanied by a dramatic diversification of DNA transposons, as previously shown in other plant species [e.g. 59]. To better understand the proliferation of DNA repeats during genome evolution and its impact on genome size variation and speciation, analysis of a larger data set of Pulmonaria species from different phylogenetic groups is required.
Satellite DNAs and their use in comparative karyotyping
The almost identical cytogenetic pattern of satDNAs and rDNA sequences in P. obscura (2n = 14), collected from three different populations, suggests karyotype stability in this diploid species. In comparison, the chromosome structure in P. officinalis appears to be more dynamic, as individuals from two different populations differ slightly in the cytogenetic pattern of the satDNAs and rDNA sequences. Odd number of signals of some satDNAs, rDNA sequences, and interstitial 45S rDNA clusters were found in both diploid accessions (2n = 16), indicating chromosomal structural changes involved in the origin and evolution of P. officinalis. It is generally accepted that n = 7 is the basic chromosome number in Pulmonaria [e.g. 2, 3, 4, 5, 6], which raises the question of how the species represented by different chromosome numbers arose. Unfortunately, the available data do not allow us to answer this question. Only a robust phylogeny of the whole genus and comparative analysis of genome structure would give a clearer idea of the chromosomal changes that could explain the origin of Pulmonaria species with different chromosome numbers.
Evidence of hybridization within the P. officinalis complex
Several molecular studies have been published highlighting the important role of hybridization and introgression in the evolution of the genus Pulmonaria [10, 13, 14]. Some species groups exhibit weak ecological and geographic isolation [see 56, 75], near-synchronous phenology and pollinator sharing, all of which may facilitate the hybridization [cf. 13]. So far, however, natural hybrids have only occasionally been identified by chromosome counting [3, 16] or distinguished on the basis of intermediate morphology [e.g. 74]. This is particularly true for the P. officinalis complex, which is widespread in Europe and therefore often in secondary contact with other Pulmonaria species [cf. 38, 75]. As the ranges of P. obscura and P. officinalis partly overlap (Fig. 1A), the co-occurrence of both species in the same habitat can be expected. Some authors have occasionally reported mixed populations, with morphological intermediates rarely observed [16, 17]. However, the extent of hybridization between these two species is still controversial and has not been clearly confirmed. Nevertheless, several karyological data referring to as P. obscura × P. officinalis with an intermediate number of chromosomes (i.e. 2n = 15), may provide convincing evidence of an ongoing hybridization between these two species [13, 16, 17].
In this study, we analyzed presumed hybrids from a mixed population (B481) of P. obscura and P. officinalis. Chromosome counting in all three analyzed individuals confirmed 2n = 15, and their hybrid origin was also supported by their genome sizes, halfway between those of the parents (Table 1). The cytogenetic mapping of the set of satDNAs and rDNA sequences also supports their hybrid origin, by the presence of P. obscura and P. officinalis species-specific satDNAs in haploid state, as well as their pattern on chromosomes, which was further supported by a detailed analysis of 5S rDNA sequences. As recently shown, graph-based clustering of the RepeatExplorer pipeline enables reconstruction of complete 5S rDNA sequences from genome skimming data and provides clues to the evolutionary history of interspecific hybrids and allopolyploids [76, 77].
Origin of ornamental cultivars morphologically similar to P. officinalis
As a valuable medicinal and ornamental plant, P. officinalis is represented in horticulture by several cultivars and has also been used to generate new artificial hybrids [cf. 15]. This seems to be the case for plants with distinctly white-spotted leaves, cordate at the base, which are sometimes offered commercially as P. saccharata. However, they are not “true” P. saccharata sensu Miller [see 10, 48, 49]. These plants often escape into the wild and are sometimes confused with P. officinalis. The origin of these cultivars is unknown, they only resemble P. officinalis complex in their morphology.
Our cytogenetic analysis and detailed examination of the reconstructed 5S rDNA sequence indicate that two analyzed P. saccharata-like accessions with 2n = 16 (B465.1 and B472.1) are probably derived from the P. officinalis.
On the other hand, an interesting cytogenetic pattern was observed in the third P. saccharata-like plant analyzed (B15.1). The karyotype and reconstructed 5S rDNA units of this plant were similar to that of the interspecific hybrid B481.1, with the same chromosome number. In contrast, the genome size of B15.1 (the whole population, respectively) was the largest in the whole data set presented (Table 1). However, unlike the population B481, population B15 was collected in the area where only P. obscura occurs naturally and where no population of P. officinalis has been confirmed (Kobrlová, pers. obs.). The morphology of the plants was also typical for cultivated P. saccharata-like plants. Their origin therefore requires further investigation, although the cytogenetic data presented suggest a hybrid origin between P. obscura and P. officinalis (e.g. phylogenetic revision and analysis of a larger data set of Pulmonaria species from different phylogenetic groups). As this population is a garden escape, its geographical origin is unclear and it cannot be ruled out that it was originally collected from a mixed population of both species.
Conclusions
Our study provides comprehensive information on genome size variability and repeatome dynamics of the two morphologically similar species of the P. officinalis group. Large-scale genome size analysis using flow cytometry confirmed a significant difference in DNA content between P. obscura and P. officinalis, corresponding to the number of chromosomes. Genome skimming of six accessions, including putative natural hybrid of P. obscura and P. officinalis, and ornamental garden escapes resembling P. officinalis, showed that a large proportion of their genomes is represented by various types of DNA transposons, with Ty3/Gypsy elements being the most abundant. Comparative analysis of the repeatomes revealed no species-specific retrotransposons or striking differences in their copy number between the species, suggesting a common evolutionary history. Comparative karyotyping supported the hybrid origin of putative hybrids with 2n = 15, collected from a mixed population of P. obscura and P. officinalis, and also outlined the origin of ornamental garden escapes morphologically similar to the P. officinalis complex. Finally, databases of repeats were created, and can be used for repeat identification (or masking) in future sequencing projects.
Materials and methods
Plant material
A total of 196 plants from 65 populations of the Pulmonaria officinalis group (Fig. 1A), representing typical populations of P. obscura and P. officinalis s. str. (Figure 1B and C), including their potential hybrids and several garden escapes of cultivars morphologically similar to P. officinalis (in horticulture often referred to as P. saccharata, here listed as P. saccharata-like, Fig. 1D), were included in this study (see Supplementary Table 1). An Italian taxon, P. officinalis subsp. marzolae, was not part of this study and is therefore not discussed further, only mentioned in the karyological review (see Supplementary Table 2). These samples were collected between 2014 and 2023 from natural populations across Europe, some of which were cultivated in the experimental garden of Palacký University in Olomouc, Czech Republic, or deposited in the Herbarium of Palacký University in Olomouc (OL).
Flow cytometry: genome size and GC content
Estimation of the nuclear DNA content, i.e. absolute genome size (AGS [78]), and DNA base composition (GC content [79]) were estimated using Partec PAS and Partec ML instruments, with PI (propidium iodide) and DAPI (4,6-diamidino-2-phenylindole) staining. The same methodology as in Kobrlová and Hroneš [31] was followed, using fresh, rarely silica dried, leaves for sample preparation. Pisum sativum L. ‘Ctirad’ (2C = 9.09 pg [80]; GC content = 38.5% [81]) was selected as a primary internal standard, since it has non-overlapping genome size with neither G1 and nor G2 phase of all studied samples. The conversion from picograms (pg) to base pairs (bp) followed Doležel et al. [82], using 1 pg DNA = 978 Mbp. DNA base content was estimated using the protocol and GC content calculation tool of Šmarda et al. [79]. One-way ANOVA was used to test for differences between population means of genome size/GC content of P. obscura and P. officinalis. The data analyses were performed using the NCSS 9 statistical software [83].
DNA extraction and sequencing
Genomic DNA was isolated using alkyltrimethylammonium bromide (MATAB) lysis: after sorbitol washes, the ground plant material was incubated in 2% (w/v) MATAB for 20 min at 65 °C, immediately after the incubation, the same volume of chloroform: isoamyl alcohol (24:1) was added, gently but thoroughly mixed and centrifuged at 10,000 g for 3 min at 4 °C. After centrifugation, aqueous upper phase was collected to a new tube and this step was repeat until the upper phase was clear. Genomic DNA was precipitated by adding 0.7 volume of isopropanol, centrifuged at 10,000 g for 3 min at 4 °C. Finally, the pellet was washed by cold 70% and 96% ethanol, air dried and diluted in TE buffer, pH 8.
Genomic DNA was sheared by Bioruptor Plus (Diagenode, Liege, Belgium) to achieve an insert size of about 500 bp. Libraries for sequencing were prepared from 2 µg of fragmented DNA using TruSeq® DNA PCR-free kit (Illumina) and sequenced on a NovaSeq 6000 (Illumina), producing 2 × 100-bp or 2 × 150-bp paired-end reads to achieve at least 3Gb of nucleotide sequence per each genotype. Raw data were trimmed for low-quality bases and adapter sequences and to the same length using fastp v.0.20.1 [84].
Analysis and characterization of DNA repeats
Random datasets corresponding to 0.1× coverage (Supplementary Table 6) of the individual accessions were used for reconstruction and characterization of DNA repeats using RepeatExplorer2 (long queue was used for comparative analysis as well as for single species clustering: -l select = 1:ncpus = 16:mem = 112gb: scratch_local = 50gb -l walltime = 336:00:00 -q elixirre@pbs.elixir-czech.cz -v TAREAN_MAX_MEM = 64000000,TAREAN_CPU = 150) [85], that includes TAREAN analysis tool for identification of tandemly organized repeats [50]. RepeatExplorer2 and TAREAN analyses were also used to perform comparative analysis of Pulmonaria repeatomes on a merged dataset containing all studied individuals (1 mil. reads per accession; Supplementary Table 6), marked by specific prefixes. In both cases, the resulting clusters of repeats were characterized by various tools, including BLASTN and BLASTX, and phylogenetic analysis of the repetitive elements’ coding domains [86, 87]. The presence of tandemly organized repeats within the clusters identified by TAREAN was confirmed with Dotter [88].
The results of the clustering were then used to create repetitive databases. Databases of Illumina reads were deposited in the Sequence Read Archive (project number: PRJNA1076467). Assembled contigs from different types of repetitive DNA elements are publicly available online (https://olomouc.ueb.cas.cz/en/content/dna-repeats). The sequences of newly identified tandem organized repeats and 5S rDNA which were used as cytogenetic markers were deposited in GenBank (accessions: PP457292–PP457296). Cluster graphs of 5S rDNA sequences were visualized using SeqGrapheR visualization tool [85]. The reconstruction of the whole 45S rDNA unit was performed according to Kapustová et al. [89].
Preparation of chromosome spreads
Mitotic metaphase chromosome spreads were prepared from root meristems by a dropping method according to Šimoníková et al. [90]. Briefly, actively growing root tips of Pulmonaria were collected and pre-treated in 0.05% (w/v) colchicine for three hours at room temperature, fixed in 3:1 ethanol: acetic acid fixative overnight at 4 °C and stored in 70% ethanol at − 20 °C. Chromosome preparations were prepared using the drop technique according to Kato et al. [91, 92], with minor modifications: After washing in 75 mM KCl and 7.5 mM EDTA (pH 4), root tip segments were digested in a mixture of 2% (w/v) cellulase and 2% (w/v) pectinase in 75 mM KCl and 7.5 mM EDTA (pH 4) for 45 min at 37 °C. The cell suspension was dropped onto glass slides in a box lined with wet paper towels and let dried.
Probe design and fluorescence in situ hybridization
Consensus sequences of TAREAN analysis which contained tandemly organized repeats were used for specific primer design using the Primer3 program [93]. PCR products were sequenced using the BigBye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, USA) according to the manufacturer’s instructions and run on ABI 3730xl DNA analyzer (Applied Biosystems) to confirm the accuracy of the sequences used for FISH analyses. Sequences are publicly available online (https://olomouc.ueb.cas.cz/en/content/dna-repeats). Probes for newly identified tandem repeats were labeled by PCR either directly with Cy5 fluorochrome (Thermo Fisher Scientific), DEAC (Jena Biosciences, Jena, Germany), or indirectly with biotin-dUTP or digoxigenin-dUTP (Sigma Aldrich/Roche Applied Science, Mannheim, Germany) using primers listed in Supplementary Tables 4 and P. obscura DNA as template. The 25 µl of PCR mix contained 30 ng of genomic DNA, 200 µM dNTPs including directly- or indirectly-labeled dUTP, 1 µM primers and 0.5 U of Q5 High-Fidelity DNA polymerase and appropriate reaction buffer (New England Biolabs, Massachusetts, USA). Plasmid pTa71 (45 S rDNA) containing 9-kb fragment from Triticum aestivum with 18S-5.8S-26S rDNA and intergenic spacers [94] was labeled by nick translation (Sigma Aldrich) using Cy5 fluorochrome (Thermo Fisher Scientific).
Hybridization mixture containing 50% (v/v) formamide, 10% (w/v) dextran sulfate in 2 × SSC and 10 ng/µl of labeled probes was added onto slide and denatured for 30s at 80 °C, followed by overnight hybridization performed in a humid chamber at 37 °C. If the chromosome structure was damaged after the denaturation step, the slides with chromosome spreads were post-fixed in 4% (v/v) formaldehyde in 2 × saline-sodium citrate (SSC) for 10 min at room temperature, washed in 2 × SSC for 2 × 5 min, and dehydrated using ethanol series. The sites of digoxigenin- and biotin-labeled probes were detected using anti-digoxigenin-FITC (Sigma Aldrich/Roche Applied Science) and streptavidin-Cy3 (Thermo Fisher Scientific/Invitrogen, Carlsbad, CA, USA), respectively. Chromosomes were counterstained with DAPI and mounted in Vectashield Antifade Mounting Medium (Vector Laboratories, Burlingame, CA, USA).
Microscopic and image analysis
Slides were examined using Axio Imager Z.2 Zeiss microscope (Zeiss, Oberkochen, Germany) equipped with a Cool Cube 1 camera (Metasystems, Altlussheim, Germany) and appropriate optical filters, and a PC running ISIS software 5.4.7 (Metasystems). The final image adjustment was performed in Adobe Photoshop CS5, and idiograms and final pictures were created in Adobe Photoshop CS5 and GIMP (GNU Image Manipulation Program) v2.10.34. A minimum of ten preparations with mitotic metaphase chromosome spreads and different probe combinations were used for the final karyotype reconstruction of each genotype.
Data availability
Sequence data that support the findings of this study have been deposited in the European Nucleotide Archive with the primary accession code PRJNA1076467. The sequences of newly identified tandem organized repeats were deposited in GenBank (accessions: PP457292-PP457296). Assembled contigs from different types of repetitive DNA elements are publicly available online (https://olomouc.ueb.cas.cz/en/content/dna-repeats).
References
Chacón J, Luebert F, Hilger HH, Ovchinnikova S, Selvi F, Cecchi L, et al. The borage family (Boraginaceae s.s.): a revised infrafamilial classification based on new phylogenetic evidence, with emphasis on the placement of some enigmatic genera. Taxon. 2016;65(3):523–46. https://doi.org/10.12705/653.6.
Merxmüller H, Grau J. Dysploidie Bei Pulmonaria. Rev Roum Biol-Botanique. 1969;14(1):57–63.
Sauer W. Karyo-Systematische Untersuchungen an Der Gattung Pulmonaria (Boraginaceae). Chromosomen-Zahlen, Karyotyp-Analysen und allgemeine Hinweise Auf die Entwicklungsgeschichte. Bibliot Bot. 1975;131:1–85.
Sauer W. A phylogenetic model for North-Eurasiatic Tubifloras on the base of comparative morphology, zytogenetics and ecology. Acta Bot Yunnanica. 1986;8(4):383–95.
Májovský J, Murín A. Cytotaxonomy of the genus Pulmonaria L. in Slovakia. Bol Soc Brot Ser 2. 1980;53:25–739.
Bolliger M. Die Gattung Pulmonaria in Westeuropa. Liechtenstein, Vaduz: J. Cramer; 1982.
Sauer W. The Pulmonaria dacica group: its affinities with central and south-east European allies and with the genus paraskeria (Boraginaceae). PIant Syst Evol. 1987;55:257–76. https://doi.org/10.1007/BF00936303.
Ge-ling Z, Riedl H, Kamelin R. Boraginaceae. In: Wu ZY, Raven PH, eds. Flora of China, Vol. 16: Gentianaceae through Boraginaceae. China, Beijing: Science Press and USA, St. Louis: Missouri Botanical Garden Press; 1995. pp. 329–427.
Baasanmunkh S, Oyuntsetseg B, Tsegmed Z, Oyundelger K, Urgamal M, Gantuya B, et al. Distribution of vascular plants in Mongolia – I Part. Mong J Biol Sci. 2022;20(1):3–28. https://doi.org/10.22353/mjbs.2022.20.01.
Liu L, Astuti G, Coppi A, Peruzzi L. Different chromosome numbers but slight morphological differentiation and genetic admixture among populations of the Pulmonaria hirta complex (Boraginaceae). Taxon. 2022;71(5):1025–43. https://doi.org/10.1002/tax.12721.
Kirchner D. Molekulare Phylogenie und Biogeographie Der Gattung Pulmonaria L. (Boraginaceae). Germany, Aachen: Verlag Mainz; 2004.
Şuteu D, Băcilă I, Coldea G. Detection of barcode markers able to differentiate the endemic taxon Pulmonaria Filarzskyana Jáv. From Pulmonaria Rubra Schott. Contrib Bot. 2016;51:7–16.
Meeus S, Janssens S, Helsen K, Jacquemyn H. Evolutionary trends in the distylous genus Pulmonaria (Boraginaceae): evidence of ancient hybridization and current interspecific gene flow. Mol Phylogenet Evol. 2016;98:63–73. https://doi.org/10.1016/j.ympev.2015.11.022.
Grünig S, Fischer M. Recent hybrid speciation at the origin of the narrow endemic Pulmonaria helvetica. Ann Bot. 2021;127(1):21–31. https://doi.org/10.1093/aob/mcaa145.
Bennett M. Pulmonarias and the borage family. UK, London: BT Batsford; 2003.
Bolliger M. Die Pulmonaria Obscura-Officinalis-Gruppe in Der Schweiz. Ber Schweiz Bot Ges. 1978;88(1/2):30–62.
Dersch G. Über Pulmonaria officinalis L., Pulmonaria obscura Dum. Und ihren Bastard in Nordhessen Und Südniedersachsen. Hessische Floristische Briefe. 1994;43:49–63.
Pellicer J, Hidalgo O, Dodsworth S, Leitch IJ. Genome size diversity and its impact on the evolution of land plants. Genes. 2018;9:88. soltis10.3390/genes9020088.
Soltis PS, Soltis DE. Plant genomes: markers of evolutionary history and drivers of evolutionary change. Plants People Planet. 2021;3(1):74–82. https://doi.org/10.1002/ppp3.10159.
Wendel JF. Genome evolution in polyploids. Plant Mol Biol. 2000;42(1):225–49. Doi:.
Ibarra-Laclette E, Lyons E, Hernández-Guzmán G, Pérez-Torres CA, Carretero-Paulet L, Chang T-H, et al. Architecture and evolution of a minute plant genome. Nature. 2013;498:94–8. https://doi.org/10.1038/nature12132.
Macas J, Novák P, Pellicer J, Čížková J, Koblížková A, Neumann P, et al. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE. 2015;10(11):e0143424. https://doi.org/10.1371/journal.pone.0143424.
Leitch IJ, Bennet MD. Genome downsizing in polyploid plants. Biol J Linn Soc. 2004;82(4):651–63. https://doi.org/10.1111/j.1095-8312.2004.00349.x.
Bennetzen JL, Ma J, Devos KM. Mechanisms of recent genome size variation in flowering plants. Ann Bot. 2005;95:127–32. https://doi.org/10.1093/aob/mci008.
Renny-Byfield S, Kovarik A, Kelly LJ, Macas J, Novak P, Chase MW, et al. Diploidization and genome size change in allopolyploids is associated with differential dynamics of low- and high-copy sequences. Plant J. 2013;74(5):829–39. https://doi.org/10.1111/tpj.12168.
Zenil-Ferguson R, Ponciano JM, Burleigh JG. Evaluating the role of genome downsizing and size thresholds from genome size distributions in angiosperms. Am J Bot. 2016;103(7):1175–86. https://doi.org/10.3732/ajb.1500408.
Simonin KA, Roddy AB. Genome downsizing, physiological novelty, and the global dominance of flowering plants. PLoS Biol. 2018;16(1):e2003706. https://doi.org/10.1371/journal.pbio.2003706.
Wang X, Morto JA, Pellicer J, Leitch IJ, Leitch AR. Genome downsizing after polyploidy: mechanisms, rates and selection pressures. Plant J. 2021;107:1003–15. https://doi.org/10.1111/tpj.15363.
Chase MW, Samuel R, Leitch AR, Guignard MS, Conran JG, Nollet F, et al. Down, then up: non-parallel genome size changes and a descending chromosome series in a recent radiation of the Australian allotetraploid plant species, Nicotiana section suaveolentes (Solanaceae). Ann Bot. 2023;131(1):123–42. https://doi.org/10.1093/aob/mcac006.
Weigend M, Selvi F, Thomas DC, Hilger HH. Boraginaceae. In: Kadereit JW, Bittrich V, editors. The families and genera of vascular plants, Vol. 14. Flowering plants. Eudicots. Aquifoliales, Boraginales, Bruniales, Dipsacales, Escalloniales, Garryales, Paracryphiales, Solanales (except Convolvulaceae), Icacinaceae, Metteniusaceae, Vahliaceae. China, Cham: Springer; 2016. pp. 41–102.
Kobrlová L, Hroneš M. First insights into the evolution of genome size in the borage family–complete data set of Boraginaceae in the Czech Republic. Bot J Linn Soc. 2019;189(2):115–31. https://doi.org/10.1093/botlinnean/boy079.
Selvi F, Coppi A, Bigazzi M. Karyotype variation, evolution and phylogeny in Borago (Boraginaceae), with emphasis on subgenus buglossites in the Corso-Sardinian system. Ann Bot. 2006;98:857–68. https://doi.org/10.1093/aob/mcl167.
Bigazzi M, Selvi F. Karyotype morphology and cytogeography in Brunnera and Cynoglottis (Boraginaceae). Bot J Linn Soc. 2001;136(4):365–78. https://doi.org/10.1111/j.1095-8339.2001.tb00580.x.
Bigazzi M, Selvi F. Chromosome variation in Anatolian species of Nonea Medik. (Boraginaceae), with special reference to endemics and N. persica. Caryologia. 2003;56(4):509–19. https://doi.org/10.1080/00087114.2003.10589364.
Kobrlová L, Hroneš M, Koutecký P, Štech M, Trávníček B. Symphytum tuberosum complex in central Europe: cytogeography, morphology, ecology and taxonomy. Preslia. 2016;88:77–112.
Kobrlová L, Duchoslav M, Hroneš M. Morphological, ecological and geographic differences between diploids and tetraploids of Symphytum officinale (Boraginaceae) justify both cytotypes as separate species. AoB Plants. 2022;144:plac028. https://doi.org/10.1093/aobpla/plac028.
Kolarčik V, Kocová V, Cakovic´ D, Kačmárová T, Piovár J, Mártonfi P. Nuclear genome size variation in the allopolyploid Onosma Arenaria – O. pseudoarenaria species group: methodological issues and revised data. Botany. 2018;96:397–410. https://doi.org/10.1139/cjb-2017-0164.
Merxmüller H, Sauer W. Pulmonaria L. In: Tutin TG, Heywood VH, Burges NA, Moore DM, Valentine DH, Walters SM, Webb DA, editors. Flora Europaea. Volume 3. Cambridge: Cambridge University Press;: UK; 1972. pp. 100–2.
Hultén E, Fries M. Atlas of North European vascular plants north of the Tropic of Cancer. Germany, Königstein: Koeltz Scientific Books; 1986.
Sauer W, Gruber G. Beitrag Zur Kenntnis Der Gattung Pulmonaria in Bayern: Kritische Sippen, Verbreitung und allgemeine Hinweise Auf die Arealbildung. Bayerische Bot Gesellschaft Zur Erforschung Der Heimischen Flora. 1979;50:127–60.
Hroneš M, Kobrlová L. 12. Pulmonaria L. In: Kaplan Z, Danihelka J, Chrtek J Jr, Kirschner J, Kubát K, Štech M eds. Key to the Flora of the Czech Republic. Ed.2. CZ, Praha: Academia; 2019. pp. 815–816.
Vejvodová K, Kúr P, Koutecký P, Diewald W, Grulich V, Máchalová K, et al. Distribution and identification of Pulmonaria officinalis and P. obscura in the Bohemian Forest and its adjacent foothills. Silva Gabreta. 2021;27:97–119.
Astuti G, Cristofolini G, Peruzzi L, Pupillo P. A new subspecies of Pulmonaria officinalis (Boraginaceae) from the southern Alps. Phytotaxa. 2014;186(3):148–57. https://doi.org/10.11646/phytotaxa.186.3.3.
Cecchi L, Selvi F. Synopsis of Boraginaceae subfam. Boraginoideae tribe Boragineae in Italy. Plant Biosyst. 2015;149(4):630–77. https://doi.org/10.1080/11263504.2015.1057261.
POWO. Plants of the World Online Kew (UK): The Royal Botanic Gardens. 2024. http://www.plantsoftheworldonline.org/. Accessed 3 March 2024.
Scott P. Physiology and behaviour of plants. UK. Chichester: John Wiley & Sons Ltd.; 2008.
Meeus S, Brys R, Honnay O, Jacquemyn H. Biological Flora of the British Isles: Pulmonaria officinalis. J Ecol. 2013;101(5):1353–68. https://doi.org/10.1111/1365-2745.12150.
Puppi G, Cristofolini G. Sul Significato Del Binomio Pulmonaria saccharata Miller. Webbia. 1991;45(2):221–34.
Puppi G, Cristofolini G. Systematics of the complex Pulmonaria saccharata, Pulmonaria vallarsae and related species (Boraginaceae). Webbia. 1996;51(1):1–20.
Novák P, Ávila Robledillo L, Koblížková A, Vrbová I, Neumann P, Macas J. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017;45(12):e111. https://doi.org/10.1093/nar/gkx257.
Koprivý L, Fráková V, Kolarčik V, Mártonfiová L, Dudáš M, Mártonfi P. Genome size and endoreplication in two pairs of cytogenetically contrasting species of Pulmonaria (Boraginaceae) in Central Europe. AoB PLANTS. 2022;14(5):plac036. https://doi.org/10.1093/aobpla/plac036.
Leitch IJ, Soltis DE, Soltis PS, Bennett MD. Evolution of DNA amounts across land plants (Embryophyta). Ann Bot. 2005;95(1):207–17. https://doi.org/10.1093/aob/mci014.
Hawkins JS, Proulx SR, Rapp RA, Wendel JF. Rapid DNA loss as a counterbalance to genome expansion through retrotransposon proliferation in plants. Proc Natl Acad Sci USA. 2009;106(42):17811–6. https://doi.org/10.1073/pnas.0904339106.
Leitch IJ, Chase MW, Bennett MD. Phylogenetic analysis of DNA C-values provides evidence for a small ancestral genome size in flowering plants. Ann Bot. 1998;82:85–94.
Šmarda P, Knápek O, Březinová A, Horová L, Grulich V, Danihelka J, et al. Genome sizes and genomic guanine + cytosine (GC) contents of the Czech vascular flora with new estimates for 1700 species. Preslia. 2019;91(2):117–42. https://doi.org/10.23855/preslia.2019.117.
Piednoel M, Aberer AJ, Schneeweiss GM, Macas J, Novak P, Gundlach H, et al. Next-generation sequencing reveals the impact of repetitive DNA across phylogenetically closely related genomes of Orobanchaceae. Mol Biol Evol. 2012;29(11):3601–11. https://doi.org/10.1093/molbev/mss168.
Novák P, Hřibová E, Neumann P, Koblížková A, Doležel J, Macas J. Genome-wide analysis of repeat diversity across the family Musaceae. PLoS ONE. 2014;9(6):e98918. https://doi.org/10.1371/journal.pone.0098918.
McCann J, Macas J, Novák P, Stuessy TF, Villasenor JL, Weiss-Schneweiss H. Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 2020;11:362. https://doi.org/10.3389/fpls.2020.00362.
Zwyrtková J, Němečková A, Čížková J, Holušová K, Kapustová V, Svačina R, et al. Comparative analyses of DNA repeats and identification of a novel Fesreba centromeric element in fescues and ryegrasses. BMC Plant Biol. 2020;20(1):280. https://doi.org/10.1186/s12870-020-02495-0.
Ferraz ME, Ribeiro T, Sader M, Nascimento T, Pedrosa-Harand A. Comparative analysis of repetitive DNA in dysploid and non-dysploid Phaseolus beans. Chromosome Res. 2023;31(4):30. https://doi.org/10.1007/s10577-023-09739-3.
International Rice Genome Sequencing Project, Sasaki T. The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. https://doi.org/10.1038/nature03895.
Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–5. https://doi.org/10.1126/science.1178534.
Papon N, Lasserre-Zuber P, Rimbert H, De Oliveira R, Paux E, Choulet F. All families of transposable elements were active in the recent wheat genome evolution and polyploidy had no impact on their activity. Plant Genome. 2023;16(3):e20347. https://doi.org/10.1002/tpg2.20347.
Kreplak J, Madoui MA, Cápal P, Novák P, Labadie K, Aubert G, et al. A reference genome for pea provides insight into legume genome evolution. Nat Genet. 2019;51:1411–22. https://doi.org/10.1038/s41588-019-0480-1.
Kim S, Park M, Yeom SI, Kim YM, Lee JM, Lee AH, et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat Genet. 2014;46:270–8. https://doi.org/10.1038/ng.2877.
Sierro N, Battey J, Ouadi S, Bakaher N, Bovet L, Willig A, et al. The tobacco genome sequence and its comparison with those of tomato and potato. Nat Commun. 2014;5:3833. https://doi.org/10.1038/ncomms4833.
Dodsworth S, Jang TS, Struebig M, Chase MW, Weiss-Schneweiss H, Leitch AR. Genome-wide repeat dynamics reflect phylogenetic distance in closely related allotetraploid Nicotiana (Solanaceae). Plant Syst Evol. 2017;303:1013–20. https://doi.org/10.1007/s00606-016-1356-9.
Gaiero P, Vaio M, Peters SA, Schranz ME, de Jong H, Speranza PR. Comparative analysis of repetitive sequences among species from the potato and the tomato clades. Ann Bot. 2019;123(3):521–32. https://doi.org/10.1093/aob/mcy186.
Song B, Song Y, Fu Y, Kizito EB, Kamenya SN, Kabod PN, et al. Draft genome sequence of Solanum aethiopicum provides insights into disease resistance, drought tolerance, and the evolution of the genome. Gigascience. 2019;8(10):giz115. https://doi.org/10.1093/gigascience/giz115.
Li N, He Q, Wang J, Wang B, Zhao J, Huang S, et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat Genet. 2023;55:852–60. https://doi.org/10.1038/s41588-023-01340-y.
Song Z, Lin C, Xing P, Fen Y, Jin H, Zhou C, et al. A high-quality reference genome sequence of Salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome. 2020;13(3):e20041. https://doi.org/10.1002/tpg2.20041.
Jia KH, Liu H, Zhang RG, Xu J, Zhou S-S, Jiao S-Q, et al. Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Hortic Res. 2021;8:177. https://doi.org/10.1038/s41438-021-00614-y.
Muravenko OV, Yurkevich OY, Kalnyuk JV, Samatadze TE, Zoshchuk SA, Amosova AV. Integration of repeatomic and Cytogenetic Data on Satellite DNA for the Genome Analysis in the Genus Salvia (Lamiaceae). Plants. 2022;11(17):2244. https://doi.org/10.3390/plants11172244.
Gams H, Pulmonaria L. In: Hegi G, editor. Illustrierte Flora Von Mitteleuropa. Volume V/3. Germany, München: J.F. Lehmanns; 1927. pp. 2209–21.
Meusel H, Jäger EJ, Rauschert S, Weinert E. Vergleichende Chorologie Der zentraleuropäischen Flora. Band II. Germany, Jena: Gustav Fischer; 1978.
Garcia S, Wendel JF, Borowska-Zuchowska N, Aïnouche M, Kuderova A, Kovarik A. The utility of graph clustering of 5S ribosomal DNA homoeologs in plant allopolyploids, homoploid hybrids, and cryptic introgressants. Front Plant Sci. 2020;11:41. https://doi.org/10.3389/fpls.2020.00041.
Vozárová R, Herklotz V, Kovařík A, Tynkevich YO, Volkov RA, Ritz CM, et al. Ancient origin of two 5S rDNA families dominating in the genus Rosa and their behavior in the Canina-type meiosis. Front Plant Sci. 2021;12:643548. https://doi.org/10.3389/fpls.2021.643548.
Greilhuber J, Doležel J, Lysák M, Bennett MD. The origin, evolution and proposed stabilization of the terms ‘Genome size’ and ‘C-Value’ to describe nuclear DNA contents. Ann Bot. 2005;95(1):255–66. https://doi.org/10.1093/aob/mci019.
Šmarda P, Bureš P, Horová L, Foggi B, Rossi G. Genome size and GC content evolution of Festuca: ancestral expansion and subsequent reduction. Ann Bot. 2008;101(3):421–33. https://doi.org/10.1093/aob/mcm307.
Doležel J, Greilhuber J, Lucretti S, Meister A, Lysák MA, Nardi L, et al. Plant genome size estimation by flow cytometry: inter-laboratory comparison. Ann Bot. 1998;82(Supplement A):17–26. https://doi.org/10.1093/oxfordjournals.aob.a010312.
Barow M, Meister A. Lack of correlation between AT frequency and genome size in higher plants and the effect of nonrandomness of base sequences on dye binding. Cytometry A. 2002;47(1):1–7. https://doi.org/10.1002/cyto.10030.
Doležel J, Bartoš J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry A. 2003;51(2):127–8. https://doi.org/10.1002/cyto.a.10013. author reply 129.
Hintze J. NCSS 9.0.15. 2013. https://www.ncss.com/
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Novák P, Neumann P, Macas J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat Protoc. 2020;15(11):3745–76. https://doi.org/10.1038/s41596-020-0400-y.
Novák P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29(6):792–3. https://doi.org/10.1093/bioinformatics/btt054.
Neumann P, Novák P, Hoštáková N, Macas J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob DNA. 2019;10:1. https://doi.org/10.1186/s13100-018-0144-1.
Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167(1–2):GC1–10. https://doi.org/10.1016/0378-1119(95)00714-8.
Kapustová V, Tulpová Z, Toegelová H, Novák P, Macas J, Karafiátová M, et al. The dark matter of large cereal genomes: long tandem repeats. Int J Mol Sci. 2019;20(10):2483. https://doi.org/10.3390/ijms20102483.
Šimoníková D, Němečková A, Čížková J, Brown A, Swennen R, Doležel J, et al. Chromosome painting in cultivated bananas and their wild relatives (Musa spp.) reveals differences in chromosome structure. Int J Mol Sci. 2020;21:7915. https://doi.org/10.3390/ijms21217915.
Kato A, Lamb JC, Birchler JA. Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc Natl Acad Sci USA. 2004;101(37):13554–9. https://doi.org/10.1073/pnas.0403659101.
Kato A, Albert PS, Vega JM, Birchler JA. Sensitive fluorescence in situ hybridization signal detection in maize using directly labeled probes produced by high concentration DNA polymerase nick translation. Biotech Histochem. 2006;81(2–3):71–8. https://doi.org/10.1080/10520290600643677.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115. https://doi.org/10.1093/nar/gks596.
Gerlach WL, Bedbrook JR. Cloning and characterization of ribosomal-RNA genes from wheat and barley. Nucleic Acids Res. 1979;7(7):1869–85. https://doi.org/10.1093/nar/7.7.1869.
Acknowledgements
We thank Eva Jahnová, Radomíra Tušková, and Petr Navrátil for excellent technical assistance. The computing was supported by the e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic, and the ELIXIR-CZ project (ID:90255), part of the international ELIXIR infrastructure.
Funding
This study was financially supported by an internal grant from Palacký University, Olomouc (IGA PrF-2024-001).
Author information
Authors and Affiliations
Contributions
E.H. and L.K. conceived the project. K.V. provided plant material from the B481 population. L.K. performed flow cytometry and analyzed the results. J.Č., L.K. and E.H. provided and analyzed sequencing data. J.Č. and V.Z. performed the cytogenetic part of the work. L.K., J.Č. and E.H., wrote the original draft, and V.Z. and K.V. revised and edited the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kobrlová, L., Čížková, J., Zoulová, V. et al. First insight into the genomes of the Pulmonaria officinalis group (Boraginaceae) provided by repeatome analysis and comparative karyotyping. BMC Plant Biol 24, 859 (2024). https://doi.org/10.1186/s12870-024-05497-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05497-4