
Abstract
Background
Walnut is an oilseed tree species and an ecologically important woody tree species that is rich in oil and nutrients. In light of differences in the lipid content, fatty acid composition and key genes expression patterns in different walnut varieties, the key gene regulatory networks for lipid biosynthesis in different varieties of walnuts were intensively investigated.
Results
The kernels of two walnut varieties, ‘Xilin 3’ (X3) and ‘Xiangling’ (XL) were sampled at 60, 90, and 120 days post-anthesis (DPA) to construct 18 cDNA libraries, and the candidate genes related to oil synthesis were identified via sequencing and expression analysis. A total of 106 differentially expressed genes associated with fatty acid biosynthesis, fatty acid elongation, unsaturated fatty acid biosynthesis, triglyceride assembly, and oil body storage were selected from the transcriptomes. Weighted gene co-expression network analysis (WGCNA), correlation analysis and quantitative validation confirmed the key role of the FAD3 (109002248) gene in lipid synthesis in different varieties.
Conclusions
These results provide valuable resources for future investigations and new insights into genes related to oil accumulation and lipid metabolism in walnut seed kernels. The findings will also aid future molecular studies and ongoing efforts to genetically improve walnut.
Similar content being viewed by others


Introduction
Walnut (Juglans regia L.) is an ecologically important woody tree species that can be used for the production of oil. China is one of the centers of origin of walnut and has become the world’s largest walnut producer [1]. High-quality walnut germplasm resources have been cultivated for years [2, 3].
Walnuts are highly nutritious and offer several health benefits. They contain significant amounts of fat, protein, carbohydrates, trace elements, and minerals, which can help prevent cardiovascular diseases, they also provide a healthy food source for type II diabetics [4]. Oil is the most important component of walnut kernels, according to the American Nutritional Database, the fat content of walnut kernels is as high as 65.21 g/100 g, and it mainly consists of unsaturated fatty acids (more than 90%). Polyunsaturated fatty acids account for 47.17 g/100 g of the oil content of walnut kernels, and linoleic acid (38.90 g/100 g) and linolenic acid (9.08 g/100 g) are the main fatty acids [5, 6].
Lipids serve as the primary storage form of energy and carbon source in plants. They are predominantly stored in oil bodies in the form of triglycerides (TAGs) [7]. The TAG synthesis pathway has three stages [8]. The first stage is the synthesis of fatty acids in plastids. Pyruvate is converted to acetyl-CoA by the action of the dehydrogenase complex (PDHC). Acetyl-CoA is converted to malonyl-CoA by acetyl-CoA carboxylase (ACCase), and malonyl-CoA acyl protein transferase (MCAT) catalyzes the binding of malonyl-CoA to an acyl carrier protein (ACP) to generate the fatty acid carbon donor–malonyl-ACP. The fatty acid carbon chain undergoes a continuous polymerization cycle catalyzed by fatty acid synthase (FAS) complexes including KAR, HAD, EAR, and KAS, until the saturated fatty acids 16:0-ACP and 18:0-ACP are formed. C16:0-ACP continues to form C18:0-ACP via ketoacyl-ACP synthase (KAS). C18:0-ACP is then dehydrogenated by stearoyl-ACP desaturase (SAD) to form C18:1-ACP, which is further hydrolyzed by fatty acyl thioesterase A/B (FATA/B) to produce free fatty acids. Long-chain acyl-CoA synthase (LACS) then catalyzes the formation of acyl-CoA using fatty acid, and acyl-CoA is transported to the endoplasmic reticulum, where it participates in the synthesis of TAGs [9]. The second stage is the elongation and desaturation of carbon chains on the endoplasmic reticulum. Oleic acid is dehydrated by fatty acid desaturase 2 (FAD2) and 6 (FAD6) to form linoleic acid (C18:2). Linoleic acid is desaturated by fatty acid desaturase 3 (FAD3) and 7/8 (FAD7/8) to form linolenic acid. Fatty acid elongation enzymes (KCS, KCR, ECR, and HCD) produce ultra-long-chain fatty acids through a series of condensation, reduction, dehydration, and re-reduction processes [10].
TAG can be synthesized in plants through two different pathways. The first is the Kennedy pathway. That is, glycerol-3-phosphate acyltransferase (GPAT) catalyzes the transfer of a fatty acid chain from acyl-CoA to the sn-1 position of glycerol-3-phosphate (G3P) to generate lysophosphatidic acid (LPA). LPA is converted to phosphatidic acid (PA) by lysophosphatidic acid acyltransferase (LPAAT). Subsequently, PA is dephosphorylated by phosphatidylate phosphatase (PAP) to form diacylglycerol (DAG). Finally, DAG is further processed by diacylglycerol acyltransferase (DGAT) to form TAG, which is stored as an oil body [11]. Another pathway for TAG synthesis is independent of acyl coenzyme A. DAG in this pathway generates phosphatidylcholine (PC) in response to phosphatidylcholine: diacylglycerol phosphorylcholine transferase (PDCT). Modified PCs can shed CDP-choline to form DAG, which is catalyzed by PDCT, or they can release fatty acids at the sn-2 position to produce TAG with DAG, which is catalyzed by phospholipid: diacylglycerol acyltransferase (PDAT) [12].
In recent years, various genes and transcription factors involved in walnut lipid metabolism have been identified following the publication of the walnut genome. Yang et al. [13]. sequenced the transcriptome of walnut kernels at three different stages of lipid transformation and found that the lipid transformation of walnut kernels was most active at the second stage of fatty acid synthesis, they also found that the expression of ACCA, ACCB, ACCC, FABF, FABG, FABI, and FAB2 in the fatty acid biosynthesis pathway related to lipid synthesis was up-regulated. Zhao et al. [14]. identified 4,817 differentially expressed genes (DEGs) based on transcriptome changes associated with lipid accumulation at different developmental stages of walnut kernels. The genes encoding ACCase, LACS, and FAD7 have been shown to play important roles in fatty acid synthesis. Zhao et al. [15]. comparatively analyzed the transcriptome and sRNA-seq data during three periods of kernel development, and they identified 204 conserved miRNAs and 554 novel miRNAs in walnut kernels, screened 104 key genes related to walnut oil accumulation and constructed 16 miRNA-mRNA regulatory modules related to walnut oil accumulation and fatty acid synthesis. Jre-miRn105, jre-miRn434, jre-miR477d, and jre-miR156a.2 are the key miRNAs regulating walnut FA synthesis. Jre-miRn411 and jre-miR399a.1 are closely related to oil and gas formation. Huang et al. [16]. identified 108 genes related to lipid biosynthesis, including 60 genes in the fatty acid biosynthesis pathway, 33 genes in the TAG biosynthesis pathway, 7 genes involved in oil body formation, and 8 transcription factors based on transcriptomic data. Yan et al. [17]. analyzed the lipid content and composition of ‘Qingxiang’ and ‘Xin-2’ walnut kernels using metabolomic data, and they constructed a preliminary regulatory network of the main lipid metabolism pathways in walnut kernels using transcriptomic data.
The demand for vegetable oils has increased steadily because of the continuous growth of the population and improvement in living standards. The rapid development of biofuels has also contributed to increases in the demand for vegetable oil products [18]. Walnut is an oilseed tree species that is highly valuable in the vegetable oil market because of its high oil content (> 60%). The quality and stability of edible oils are affected by their fatty acid composition, which has implications for assessing the nutritional value of vegetable fats and oils [19]. However, the key genes and transcription factors involved in the fatty acid metabolic pathway have not yet been identified, and the interactions between these key genes and transcriptional regulators have not been characterized. Given variation in the lipid content and fatty acid composition of different walnut varieties, as well as differences in key genes and expression patterns, additional research on the molecular mechanisms and gene regulatory networks relating to lipid biosynthesis in walnuts is needed.
In order to investigate the molecular mechanisms and gene regulatory network involved in walnut fruit lipid biosynthesis, and determine the oil content, fatty acid composition, and key gene expression patterns of two walnut varieties, ‘Xilin 3’ (X3) and ‘Xiangling’ (XL) were selected as research materials. XL is China’s nationally-validated walnut variety, and is the main cultivar in Shaanxi, and it is planted over a large area in the province, X3 is the Shaanxi Province-validated walnut variety, and it is protected by our intellectual property rights. X3 is characterized by large fruits and early ripening (ripening in mid- to late-August), this variety is a major component of the Shaanxi fresh walnut market. Measurements of the oil content and fatty acid compositions and analysis of the transcriptome of these two walnut varieties clarified the biological characteristics of the major walnut varieties in Shaanxi, the key genes involved in lipid biosynthesis in different varieties were identified, and the expression patterns of key genes in each variety were analyzed. The results of this study will provide a new insights for the genetic improvement of walnut fruit oil content and fatty acid fractions, these findings will also aid the study of walnut oil synthesis, fatty acid metabolism and the identification of related functional genes, and lay a solid foundation for further molecular breeding and creation of new germplasm in walnut.
Materials and methods
Plant materials
Fruits were collected from two walnut cultivars, X3 and XL, which were planted in the Walnut Experimental Station Northwest Agriculture and Forestry University in Shanyang (33.53°N, 109.89°E, 1,100 m above sea level), Shaanxi, China. Walnut samples were collected at 50 days post-anthesis (DPA), and then every 10 days until maturity at 130 DPA, which resulted in a total of nine sampling points (50, 60, 70, 80, 90, 100, 110, 120, and 130 DPA). At each stage, ten fruits were pooled into a single biological replicate for each sample, and there were three replicates for each stage. After quickly removing the peel and seed coat, the kernels were frozen in liquid nitrogen and stored in a refrigerator at -80 °C for lipid analysis and transcriptome sequencing [1, 16].
Determination of the content of lipids and fatty acids
Kernels from 60, 70, 80, 90, 100, 110, 120, and 130 DPA were dried to a constant weight at 45 °C and ground to powder. Oil was extracted from the powder at 80 °C for 5 h using petroleum ether as the solvent [20]. Fatty acid methyl esters were analyzed using GC-MS/MS (Trace 1610-TSQ9610, Thermo Fisher, USA) according to the GB 5009.168–2016 method. Three biological replicates for each developmental stage were performed.
RNA extraction, cDNA library construction, and RNA sequencing
Samples collected at T1 (60 DPA), T2 (90 DPA), and T3 (120 DPA) were used for transcriptome sequencing, these sampling points were selected based on changes in morphology and total oil content during the development of walnut kernels in the two walnut cultivars. Total RNA from the samples was extracted using an RNAprep Pure Plant Kit (QIAGEN, Germany). Total RNA integrity was determined using an Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). After samples were tested and qualified, a total of 18 cDNA sequencing libraries from three phases (two varieties per phase, three replicates per variety) were generated. A Qubit 2.0 fluorometer (Life Technologies, CA, USA) and Agilent 2100 bioanalyzer (Agilent Technologies, CA, USA) were then used to detect and accurately quantify the effective concentration of the libraries (the effective concentration of the libraries was greater than 2 nM) by qRT-PCR to ensure that the libraries were of high quality. Paired-end sequencing (150 bp) of the samples was conducted on the NovaSeq 6000 Sequencing System (Illumina) by Novogene Technology Co., Ltd (Beijing, China). Approximately 6 GB of raw data were sequenced for each sample.
RNA-Seq data analysis
Sequenced fragments were converted to raw sequence data by CASAVA. The raw data were filtered to eliminate reads containing adapters, reads containing N bases, and low-quality reads to enhance the accuracy of the data. Clean reads for further analysis were acquired after the raw data were filtered, the sequencing error rate was determined, and the GC content was calculated. Analysis of the clean data was performed using HISAT2 (v2.0.5) and StringTie (1.3.3b). HISAT2 v2.0.5 software was used to compare clean reads to the reference genome and determine the locations of reads in the genome [21]. StringTie was used to assemble the transcripts [22].
Analysis of DEGs
DESeq2 software (1.20.0) was used to perform a differential expression analysis (|log2(FoldChange)|>=1 & padj = 0.05) [23]. ClusterProfiler (3.8.1) software was used to perform GO functional enrichment analysis and KEGG pathway enrichment analysis on the differential gene sets [24]. PlantTFDB 4.0 (http://planttfdb.gao-lab.org/) was used to predict transcription factors.
Coexpression network construction
Weighted gene co-expression network analysis (WGCNA) is a systems biology method used to characterize patterns of gene association between samples. The gene coexpression networks were constructed using the WGCNA package, and a network image was created using Cytoscape software [25, 26].
Quantitative real-time PCR (qRT-PCR) analysis
Total RNAs of walnut kernels from three developmental stages (T1, T2, and T3) in the two walnut cultivars were reverse-transcribed to synthesize cDNAs using the Evo M-MLV RT Mix Kit (Accurate Biotechnology, Hunan, China). qRT-PCR was performed in the LightCycler 96 Real-Time PCR system (Roche, Switzerland) using 2×Q3 SYBR qPCR Master Mix (Tolobio). Primer Premier 5.0 software was used to design qRT-PCR primers for fatty acid synthesis-related genes (Table S1). The expression level of each gene was normalized relative to the expression of the 18 S gene, and expression levels were calculated using the 2−△△CT method [27].
Results
Changes in the oil content in developing kernels
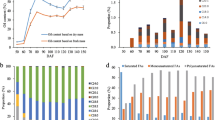
Consistent changes in the morphological characteristics of the two walnut cultivars, XL and X3, were observed (Fig. 1), the oil content increased gradually with fruit development, and the rate of change was initially rapid and then slow. At 50 DPA, the kernel comprised jelly-like substances and was relatively low in oil. At 60 DPA, walnut kernels began to solidify and were filled with white, firm cotyledon. The oil content of the walnut kernel increased dramatically between 60 and 90 DPA, by 90 DPA, the oil content of walnut kernels was nearly 60%. The increase in the oil content slowed between 90 and 120 DPA, at 120 to 130 DPA, walnuts were essentially mature, and oil accumulation increased slightly. At 130 DPA, walnuts were fully mature, the green pericarp detached from the nut, and the oil content of the two varieties was nearly identical (ca. 70%).
The oil content of X3 increased from less than 20–67.99%, and the dry kernel weight per nut increased from 0.65 g to 8.49 g. In contrast, the oil content of XL increased to 65.97%, and the dry kernel weight increased from 0.55 g to 6.76 g. The optimal periods for the transcriptome analysis were determined during the early stage of oil synthesis (60 DPA, T1), the peak of oil synthesis (90 DPA, T2), and the late stage of oil synthesis (120 DPA, T3) according to analysis of the oil content of fruits at different developmental stages of the two walnut species.

Changes in the morphological characteristics and oil content during the development of walnut fruit. X3 means ‘Xilin 3’ walnut kernels, XL means ‘Xiangling’ walnut kernels, data are displayed as mean ± SD (n = 3). The same below. (A) Morphological characteristics of longitudinal sections, (B) Morphological characteristics of walnut kernels, (C) Walnut kernel weight, (D) The oil content
Changes in fatty acid composition in developing kernels
The fatty acid composition of the walnut kernels was analyzed, the fatty acid composition of the kernels at 50–130 DPA is shown in Fig. 2. The fatty acid composition of the two walnut varieties differed, however, there were five dominant fatty acids in each walnut variety, including two saturated fatty acids (palmitic acid (16:0) and stearic acid (18:0)) and three unsaturated fatty acids (oleic acid (18:1), linoleic acid (18:2), and linolenic acid (18:3)). The relative content of these five fatty acids exceeded 98% in each period, with the exception of 50 DPA for X3.
At 60–130 DPA, linoleic acid was the fatty acid with the highest content, and the content of stearic acid was relatively low (< 2 g/100 g). The content of linoleic acid in the two varieties gradually increased from 50 DPA and was highest at 130 DPA and 110 DPA in X3 (34.20 g/100 g) and XL (37.30 g/100 g), respectively. The content of oleic acid in the kernel of X3 increased from 60 to 90 DPA, and it was highest at 90 DPA (16.57 g/100 g). XL increased from 50 to 100 DPA, and it was highest at 100 DPA (9.67 g/100 g). After peaking, the oleic acid content of the two varieties fluctuated slightly, the oleic acid content in X3 and XL was 11.02 g/100 g and 9.00 g/100 g at maturity, which corresponded to 19.72% and 17.45%, respectively. The content of palmitic acid, stearic acid and linolenic acid did not change significantly throughout development, however, the percentage of palmitic acid was higher at 50 DPA (18.29% and 20.49% in X3 and XL, respectively) than at all other sampling events. Throughout the development process, the content of total fatty acids, saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids increased, and the growth rate was faster in the early phase, the growth rate slowed and the content of fatty acids fluctuated after 90 DPA. However, fluctuations in the content of fatty acids were greater in X3 than in XL.

Content and composition of fatty acids in walnut kernels. C14:0, myristic acid, C15:0, pentadecanoic acid, C16:0, palmitic acid, C17:0, heptadecanoic acid, C18:0, stearic acid, C20:0, arachidic acid, C22:0, behenic acid, C16:1, hexadecenoic acid, C17:1, heptadecenoic acid, C18:1, oleic acid, C18:2, linolenic, C18:3, linolenic acid, C20:2, arachidienoic acid. (A) Fatty acid components of X3 and XL, (B) Major fatty acid components (mean ± SD, n = 3), (C) Content of fatty acids, saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids
Transcriptome sequencing of developing kernels
The genes and their expression levels in walnut kernels were analyzed by RNA sequencing (RNA-seq) at different developmental stages (T1, T2, and T3) and in different varieties (X3 and XL). A total of 846,686,824 raw reads were obtained from 18 samples (2 varieties, 3 periods, and 3 replications). After removing reads with low quality, reads containing adapters, or reads with a high content of unknown bases, a total of 833,654,470 high-quality clean reads were obtained. The Q20 values of the high-quality sequences were above 97.27%, and the Q30 value was above 92.57%. The filtered clean data account for more than 97% of the total data, indicating that the data are reliable (Table S2).
All clean reads were compared against the walnut reference genome in the database. Approximately 94.90–96.50% of the reads of X3 in the samples from the three developmental periods were aligned against the walnut reference genome, and the average was 96.05%, approximately 94.48–96.66% of the reads were aligned to the walnut reference genome in the samples from the three periods in XL, and the average was 95.50%. The unique comparison rate between samples at different developmental stages and the reference genome ranged from 79.77 to 88.10%, with an average of 83.55%. The reads of different samples were evenly distributed in all regions of the transcripts, and most of the transcripts were completely covered. This indicates that the samples used for RNA-seq in this study were of high quality, and the data for subsequent analysis were reliable.
The DEGs in developing kernels
Principal component analysis (PCA) of all 18 samples based on Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) values from RNA-seq revealed that the two principal components explained 65.89% of the overall variance (Fig. 3A). PC1 explained the most variance (52.22%) in the datasets and could distinguish T3 samples from T1 and T2 samples. PC2 explained 13.67% of the variance in the datasets and could distinguish T1 samples from T2 samples. The biological replicates of the two walnut varieties were highly clustered within each period, and the data derived from different periods and varieties were separated.
The expression of genes is altered as the nucleolus develops. Results of the analysis of DEGs in different comparison groups during the entire development process from the initial formation of the nucleolus to maturity are shown in Fig. 3B. The number of DEGs in the T1 vs. T2 comparison group in the kernels of X3 and XL varieties was highest (14,835 and 14,615, respectively), followed by 13,227 and 12,637 in the T2 vs. T3 comparison group. The number of DEGs in the walnut kernels of the two varieties was lowest at T2 (1,173), the expression of 584 and 589 of these DEGs was up-regulated and down-regulated, respectively. The number of up-regulated and down-regulated genes varied among the comparison groups, suggesting that the regulatory effects of these genes on kernel development and fatty acid synthesis are achieved via different expression patterns, these finding highlight the complexity of lipid synthesis.

Differential expression of genes in various developmental stages in walnut kernels. T1, T2, and T3 means 60, 90, and 120 DPA, respectively, and 1, 2, and 3 denote three biological replicates for each period. The same below. (A) PCA based on RNA-seq FPKM, (B) Pairwise comparisons between samples
DEGs were identified using the following criteria: padj ≤ 0.05 and |log(Fold Change)| ≥ 1. We analysed the number of DEGs between different periods of the same species and between different species in the same period (Fig. S1). Venn diagrams of DEGs at different developmental stages showed that 1,477 and 1,490 DEGs were common to all stage comparisons in X3 and XL, respectively, suggesting that these genes might play a key role in the growth and development of walnut fruits. In addition, there are a number of genes that have been found to be independently expressed at different times or in different varieties, and it is hypothesized that they might have specific functions in kernel development and fatty acid synthesis.
We used hierarchical clustering to examine the clustering patterns of the FPKM values of the genes (Fig. 4A). Genes or samples with similar expression patterns were clustered in the heatmap, and the color in each square reflects the value obtained after normalizing the expression data. Redder colors indicate higher expression, and greener colors indicate lower expression.
Transcriptome sequencing revealed that some genes show similar expression patterns at different growth periods. Based on the expression information, genes with similar patterns were clustered into time-related gene clusters, and genes with consistent expression patterns were clustered into the same gene cluster.
All genes were categorized into four major expression patterns by cluster analysis in this study (Fig. 4B). The period of high expression and the number of genes differed among expression patterns. The highest number of genes were clustered in sub-cluster 2, with a total of 23,147 genes. However, the expression levels of the genes in this pattern did not change significantly during fruit ripening, suggesting that they were not involved in kernel development and fruit ripening. Sub-cluster 4 contained 247 genes, and the expression of these genes increased with fruit development, indicating that genes in this cluster were positively correlated with fruit development and oil synthesis. By contrast, the 172 genes in sub-cluster 3 were negatively correlated with fruit development, and their expression gradually decreased during the fruit ripening process. Sub-cluster 1 contained only four genes, but the XL kernel in this model had high expression at T3. High transcript levels were observed for each sub-cluster of genes, and strong clustering and differential expression patterns were observed at different stages of fruit development. These results further demonstrate the complexity of gene regulation during walnut kernel development.

Clustering analysis of DEGs during walnut kernel development. (A) Hierarchical cluster analysis of DEGs, (B) The four sub-clusters with different gene expression profiles
Functional and WGCNA of DEGs
To clarify the specific functions of DEGs, GO annotations in the molecular function (MF), cellular component (CC), and biological process (BP) categories were obtained for DEGs, and the threshold for significant enrichment was padj < 0.05. The number of GO terms for the different comparison groups ranged from 657 to 1,006 (Fig. S2). The enriched GO terms varied among the comparison groups of different varieties and developmental periods. Key genes involved in fatty acid metabolism are likely enriched in glyceraldehyde-3-phosphate metabolic process (GO:0019682), lipid metabolic process (GO:0006629), fatty acid metabolic process (GO:0006631), fatty acid biosynthetic process (GO:0006633), lipid catabolic process (GO:0016042), and other processes, additional research is needed to identify these key genes.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database is a comprehensive database that integrates genomic, chemical, and systemic functional information. Significantly enriched metabolic pathways during walnut kernel development were identified using a significance threshold of padj < 0.05 (Fig. S3). KEGG pathway analysis of DEGs revealed a total of 15 pathways related to lipid metabolism (Table S3), including Biosynthesis of unsaturated fatty acids (ko01040), Fatty acid metabolism (ko01212), Arachidonic acid metabolism (ko00590), Fatty acid biosynthesis (ko00061), alpha-linolenic acid metabolism (ko00592), and Fatty acid degradation (ko00071), etc. The greatest number of DEGs were enriched in Glycerophospholipid metabolism (ko00564), followed by Fatty acid metabolism. Among the different comparison groups, the highest number of DEGs was found in the T1 vs. T3 comparison group in X3 kernels, and the number of genes for Glycerophospholipid metabolism, Fatty acid metabolism, and Glycerolipid metabolism (ko00561) was 64, 56, and 51, respectively.
WGCNA was used to classify genes into different groups according to their expression patterns. We identified gene sets showing highly synergistic changes and analyzed correlations between modules and changes in nucleolus lipids. We also constructed a gene regulatory network for the different gene sets and identified key regulatory genes involved in lipid synthesis. WGCNA categorized the genes into 16 modules (Fig. 5A, Fig. S4, and Table S4). The largest module was Turquoise, which contained 5,507 DEGs and the largest number of gene clusters. The number of DEGs clustered in various modules differed, but they also displayed distinct expression patterns at different developmental periods.
To better understand the relationship between the gene expression patterns of the modules and physiological traits, we performed a correlation analysis of modules and traits using WGCNA (Fig. 5B).The content of the five dominant fatty acids was highly positively correlated with the Magenta module and significantly negatively correlated with the Yellow module. In the KEGG enrichment analysis, genes in the Magenta module were related to Cutin, suberine and wax biosynthesis (ko00073), Ether lipid metabolism (ko00565), Sphingolipid metabolism (ko00600), and Glycerophospholipid metabolism, and genes in the yellow module were related to Steroid biosynthesis (ko00100), Fatty acid metabolism, Fatty acid biosynthesis, and Ether lipid metabolism (ko00565).

WGCNA of genes in developing walnut kernels. TFA, total fatty acid content, SFA, saturated fatty acids, MUFA, monounsaturated fatty acids, PUFA, polyunsaturated fatty acids, PA, palmitic acid, SA, stearic acid, OA, oleic acid, LA, linoleic acid, LNA, linolenic acid. (A) Module clustering and correlation heat map, (B) Heat map of correlation coefficients between modules and traits
Identification of genes related to lipid biosynthesis during kernel development
To clarify the molecular mechanism of lipid accumulation in walnuts and identify key genes involved in lipid biosynthesis, we constructed a transcription model of lipid biosynthesis to visualize changes in gene expression levels during kernel development in the two walnut varieties (Fig. 6). We identified 106 lipid biosynthesis-associated unigenes, of which 40, 18, 16, 15, and 17 were associated with fatty acid biosynthesis in plastids, fatty acid elongation, unsaturated fatty acid biosynthesis, triglyceride (TAG) assembly in the endoplasmic reticulum, and lipid storage, respectively. Of the 106 unigenes, 30 belong to the Turquoise module, 15 belong to the Yellow module, 13 belong to the Brown module, 12 belong to the Green module, 11 belong to the Blue module, and 17 belong to the other modules.
The expression abundances of genes varied among developmental stages (Table S5). In our transcriptome data, some genes, such as those encoding FAD, oleosin (OLE), and caleosin (CLO), had FPKM values > 1,000. These highly expressed genes might play an important role in the oil biosynthesis of kernels. In addition, genes with up-regulated expression from T1 to T3 in both varieties were correlated with the pattern of lipid accumulation, and genes with significantly higher expression at T2 than at T1 merit increased attention because lipids accumulated rapidly from T1 to T2.

Transcriptional model of lipid biosynthesis in developing walnut kernels. The six squares in each horizontal row correspond to the three stages of the two varieties, the data were homogenized for each horizontal row. G-3-P, glycerol-3-P, LPA, 1-acylglycerol-3P, PA,1,2-diacylglycerol-3P, DAG, 1,2-diacylglycerol, TAG, triacylglycerol, PDH, pyruvate dehydrogenase, ACC1, acetyl-CoA carboxylase 1, BCCP, biotin carboxyl carrier protein, α-CT, α-carboxyltransferase, MCAT, malonyl-CoA ACP transacylase, KAS, 3-oxoacyl-ACP synthase, KAR, 3-oxoacyl-ACP reductase, HAD, 3-hydroxyacyl-ACP dehydratase, EAR, enoyl-ACP reductase, FAT, fatty acyl-ACP thioesterase, SAD, stearoyl-ACP desaturase, LACS, long-chain acyl-CoA, KCS, 3-ketoacyl-CoA synthase, KCR, very-long-chain 3-oxoacyl-CoA reductase, HCD, very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase, ECR, very-long-chain enoyl-CoA reductase, FAD, fatty acid desaturase, PDAT, phospholipid: diacylglycerol acyltransferase, PDCT, phosphatidylcholine: diacylglycerol choline phosphotransferase, DGAT, diacylglycerol acyltransferase, PAP, phosphatidate phosphatase, LPAAT, lysophosphatidic acid acyltransferase, GPAT, glycerol-3-phosphate acyltransferase
To further reveal the key genes involved in lipid biosynthesis, Pearson correlation analyses were performed between the expression levels of 106 candidate genes and the lipid content and fatty acid components (Fig. S5). A total of 20 unigenes were found to be significantly correlated with the oil content or fatty acid composition (Fig. 7A) using Pvalue < 0.05 and correlation > 0.8 as thresholds. Thirteen of them were correlated with the lipid content, 10 with the total fatty acid content, and 12, 5, 13, and 13 genes were related to stearic, oleic, linoleic and linolenic acids, respectively. Based on differences in the oleic and linoleic acid content between X3 and XL varieties, the FATB (108992677), SAD (108984606), FAD2 (109002707/109011954), FAD3 (109002248) and FAD6 (108993197) genes were selected for quantitative validation. Correlation analyses of phenotypic traits and relative gene expression levels (Fig. 7B) showed that FAD3 (109002248) was significantly positively correlated with the oil content, as well as with the content of palmitic acid, stearic acid, oleic acid, and linolenic acid. The positive correlation with the total fatty acid content and linoleic acid was particularly strong. This gene was further confirmed to play a critical role in lipid synthesis and fatty acid component formation.

Correlation analysis of the oil content and fatty acid composition with genes related to lipid synthesis. TFA, total fatty acid content, SFA, saturated fatty acids, MUFA, monounsaturated fatty acids, PUFA, polyunsaturated fatty acids, PA, palmitic acid, SA, stearic acid, OA, oleic acid, LA, linoleic acid, LNA, linolenic acid, *P < 0.05, **P < 0.01, ***P < 0.001
Validation of the target genes related to lipid synthesis
qRT-PCR was used to analyze the expression patterns of key genes related to oil biosynthesis in two walnut varieties: PAP, CLO, DGAT, KAS II, KAS III, PDH, SAD, HAD, and ACCase for X3 kernels and PAP, CLO, DGAT, KAS II, EAR, KAR, FATA, LACS, and PDAT for XL kernels. The expression patterns of these genes were analyzed in walnut kernels at different periods to assess the accuracy of the FPKM value derived from the transcriptomic data (Fig. 8). The results showed that the expression patterns of these genes inferred by qRT-PCR and transcriptome sequencing were consistent, indicating that the expression data obtained by RNA-Seq were reliable.

qPCR validation of differential expression of nine important genes associated with lipid biosynthesis. Data are means ± SD (n = 3). X3 stands for ‘Xilin 3’ walnut kernel and XL stands for ‘Xiangling’ walnut kernel
Discussion
The quantity of differentially expressed genes is impacted by fruit growth
To investigate the transcriptional expression of key genes for fruit oil synthesis in different walnut varieties, X3 and XL kernels were subjected to RNA-seq. Analysis of the number of DEGs in different varieties and developmental stages of walnut kernels revealed that the number of DEGs was higher in the early stages of kernel development. When the kernels initially formed, the expression of genes involved in biological processes related to early development might be activated to meet the developmental needs of the kernels. This was followed by a gradual increase in the number of DEGs during maturation, which presumably maintained normal development within the kernel, and this results in the activation and gradual increase in the expression of genes related to oil synthesis. However, the number of DEGs in the two varieties was relatively low in the early to middle development stages. It is possible that differences in the number of DEGs between X3 and XL are small at the early stage of kernel development and the period of rapid oil transformation, and the increase in the difference in the number of DEGs in T3 might be explained by differences in the rate at which X3 and XL mature, as X3 matures a little earlier than XL.
The expression of ACC gene promotes rapid accumulation of oil
Analysis of the relevant pathways and regulatory network of lipid synthesis from the transcriptome data and identification of key genes affecting fatty acid synthesis and metabolism can provide valuable insights with implications for the molecular breeding of walnut. ACCase plays an important role in regulating the flow of carbon into lipid synthesis, and it is the first key and rate-limiting enzyme in the entire synthesis pathway. Roesler et al. [28]. specifically overexpressed the homogeneous ACCase gene of Arabidopsis thaliana in an oilseed rape plasmid, and this increased the oil content of transgenic oilseed rape seeds by 5%, which might be attributed to the suppressed expression of other genes involved in fatty acid synthesis by ACCase. This suggests that ACCase plays a key role in the feedback regulation of fatty acid synthesis. Andre et al. [29]. showed that ACCase can catalyze the conversion of short-chain fatty acids into long-chain fatty acids, and the gradual increase in fatty acids might have a negative regulatory effect on ACCase. The expression of all the ACCase genes in the X3 kernels in this study was up-regulated from T1 to T2, and the expression of three genes was maintained at a high level at T2 (KPFM > 70). The up-regulated expression of the ACCase genes and their high expression levels at T2 promoted the rapid accumulation of oil during this period. The expression of some genes was down-regulated in the T2 vs. T3 comparison group, which might be due to the negative feedback regulatory effect of increases in fatty acids.
The content of unsaturated fatty acids in kernels is attributed to the high expression of FAD family
Fatty acid desaturases play a key role in the de novo synthesis of fatty acids. SAD catalyzes the conversion of 18:0-ACP into 18:1-ACP, it thus plays a role in regulating the conversion of saturated fatty acids to unsaturated fatty acids. The suppression of SAD gene expression in oilseed rape resulted in a significant increase in the stearic acid content from 2 to 40% in seeds [30]. Cloning of the soybean SAD gene and its transient expression in tobacco leaves resulted in an increase in the oleic acid content and a decrease in the stearic acid content [31]. The expression of three SAD genes was high in our study, and the expression of all three of these SAD genes was highest at T2. This finding is consistent with the rate of increase in the oil content and changes in the oleic acid content, the high expression of SAD genes during this period might have promoted the regulation of unsaturated fatty acid synthesis. FAD2 catalyzes the conversion of oleic acid into linoleic acid, whereas FAD3 regulates the conversion of linoleic acid into linolenic acid. Overexpression of FAD2 and FAD3 in Arabidopsis thaliana increases the linoleic and linolenic acid content and regulates the final ratio of fatty acid fractions [32]. The content of oleic acid in soybean oil was as high as 85%, and the polyunsaturated fatty acid content was extremely low when the expression of FAD2 was inhibited [33]. High expression of the FAD3 gene in Perilla is induced when the unsaturated fatty acid content in the body is high [34]. During the development of X3 and XL walnut kernels, similar expression patterns were observed for some FAD2, FAD3, and SAD genes, and the expression levels of these genes were significantly higher than those of other structural genes, the highest expression level detected was 9853.48. This result suggests that FAD genes play an important role in lipid synthesis and explains the high content of unsaturated fatty acids in walnut kernels. Zhang et al. [28]. explored the molecular mechanism underlying variation in the content of α-linolenic acid in peony seeds and showed that the content of linolenic acid was higher than the content of linoleic acid in the seeds when the expression of the FAD3 gene was higher than the expression of the FAD2 gene. The expression of the FAD3 gene was higher than that of FAD2 in the walnut kernels of XL and X3. However, additional research will be required to determine why the linolenic acid content in the kernels was much lower than the linoleic content. The high linoleic acid content might require the consumption of more FAD3 genes in the desaturation process, or there might be differences in the RNA content [35] and protein catalytic efficiency [36], alternatively, the high expression of PDAT and PDCT in walnuts might facilitate the accumulation of linoleic acid and the depletion of linolenic acid [37].
The high expression of OLEs might be closely related to the high oil content
TAGs in seeds are ultimately stored as oil bodies, and OLE, which controls the structure of oil bodies and lipid accumulation, is an important protein in seeds [38]. Our study showed that almost all OLE genes were highly expressed in the two walnut varieties, and their expression levels were significantly higher than those of CLO and STE. The high expression of the two OLE genes was consistent with patterns of lipid accumulation, the expression of the rest of the OLE genes was consistent with the rate of fatty acid accumulation. This suggests that these genes might play an important role in oil accumulation, the high expression of OLEs might be closely related to the high oil content of walnut kernels.
Conclusion
In this study, the excellent walnut varieties XL and X3, which are mainly planted in Shaanxi Province, were utilized as research materials, and both varieties had an oil content of greater than 65% at maturity and an unsaturated fatty acid content of approximately 90%. Transcriptome analyses indicate that the oil content and fatty acid composition were associated with the specific expression of genes related to lipid biosynthesis during kernel development. The key role of the FAD3 (109002248) gene in lipid synthesis in different varieties was confirmed based on WGCNA, correlation analysis and quantitative validation. Our findings enhance our understanding of the molecular mechanism of walnut lipid synthesis; they also provide candidate genes for improving the quality of walnut lipids, new approaches that could be employed to increase the walnut lipid content and optimize the fatty acid composition, and insights that could aid the selection of high-quality walnut varieties and the comprehensive use of walnut germplasm resources.
Data availability
Sequence data that support the findings of this study have been deposited in the SRA with the primary accession code PRJNA1114912.
References
Wang WQ, Wen H, Jin Q, Yu WJ, Li G, Wu MY, Bai HJ, Shen LR, Wu CY. Comparative transcriptome analysis on candidate genes involved in lipid biosynthesis of developing kernels for three walnut cultivars in Xinjiang. Food Sci Hum Wellness. 2022;11(5):1201–14.
Bernard A, Barreneche T, Lheureux F, Dirlewanger E. Analysis of genetic diversity and structure in a worldwide walnut (Juglans regia L.) germplasm using SSR markers. PLoS ONE. 2018;13(11):e0208021.
Shahi Shavvon R, Qi HL, Mafakheri M, Fan PZ, Wu HY, Bazdid Vahdati F, Al-Shmgani HS, Wang YH, Liu J. Unravelling the genetic diversity and population structure of common walnut in the Iranian Plateau. BMC Plant Biol. 2023;23(1):201.
Zibaeenezhad MJ, Farhadi P, Attar A, Mosleh A, Amirmoezi F, Azimi A. Effects of Walnut oil on lipid profiles in hyperlipidemic type 2 diabetic patients: a randomized, double-blind, placebo-controlled trial. Nutr Diabetes. 2017;7(4):e259.
Ghafoor K, Juhaimi FA, Geçgel Ü, Babiker EE, Özcan MM. Influence of roasting on Oil Content, Bioactive Components of different Walnut Kernel. J Oleo Sci. 2020;69(5):423–8.
Song H, Cong Z, Wang C, He M, Liu C, Gao P. Research progress on Walnut oil: bioactive compounds, health benefits, extraction methods, and medicinal uses. J Food Biochem. 2022;46(12):e14504.
Yang Y, Benning C. Functions of triacylglycerols during plant development and stress. Curr Opin Biotechnol. 2018;49(3):191–8.
Baud S, Lepiniec L. Regulation of de novo fatty acid synthesis in maturing oilseeds of Arabidopsis. Plant Physiol Biochem. 2009;47(6):448–55.
Saha S, Enugutti B, Rajakomari S, Rajasekharan R. Cytosolic triacylglycerol biosynthetic pathway in oilseeds: molecular cloning and expression of peanut cytosolic diacylglylglycerol acyhransferase. Plant Physiol. 2006;141:1533–43.
Yang Y, Kong Q, Lim ARQ, Lu S, Zhao H, Guo L, Yuan L, Ma W. Transcriptional regulation of oil biosynthesis in seed plants: current understanding, applications, and perspectives. Plant Commun. 2022;3(5):100328.
Chapman KD, Ohlrogge JB. Compartmentation of triacylglycerol accumulation in plants. J Biol Chem. 2012;287(4):2288–94.
Yang J, Liu J, Pan Y, Maréchal E, Amato A, Liu M, Gong Y, Li Y, Hu H. PDAT regulates PE as transient carbon sink alternative to triacylglycerol in Nannochloropsis. Plant Physiol. 2022;189(3):1345–62.
Yang L, Chen H, Pan CD, Shang LQ, Fang M, Zhao MM. Transcriptome analysis for walnut seed during oil synthesis period. J Fruit Sci. 2017;34(09):1084–94.
Zhao S, Zhang X, Su Y, Chen Y, Liu Y, Sun M, Qi G. Transcriptome analysis reveals dynamic Fat Accumulation in the Walnut Kernel. Int J Genomics. 2018: 8931651.
Zhao X, Yang G, Liu X, Yu Z, Peng S. Integrated Analysis of seed microRNA and mRNA transcriptome reveals important functional genes and microRNA-Targets in the process of Walnut (Juglans regia) seed oil Accumulation. Int J Mol Sci. 2020;21(23):9093.
Huang R, Zhou Y, Zhang J, Ji F, Jin F, Fan W, Pei D. Transcriptome Analysis of Walnut (Juglans regia L.) embryos reveals Key Developmental stages and genes involved in lipid biosynthesis and polyunsaturated fatty acid metabolism. J Agric Food Chem. 2021;69(1):377–96.
Yan S, Wang X, Yang C, Wang J, Wang Y, Wu B, Qiao L, Zhao J, Mohammad P, Zheng X, Xu J, Zhi H, Zheng J. Insights into Walnut lipid metabolism from Metabolome and Transcriptome Analysis. Front Genet. 2021;12:715731.
Poljšak N, Kreft S, Kočevar Glavač N. Vegetable butters and oils in skin wound healing: scientific evidence for new opportunities in dermatology. Phytother Res. 2020;34(2):254–69.
Zhou F, Liu Y, Liang C, Wang W, Li C, Guo Y, Ma J, Yu Y, Fan L, Yao Y, Zhao D, Liu X, Huang X. Construction of a high-density genetic linkage map and QTL mapping of oleic acid content and three agronomic traits in sunflower (Helianthus annuus L.) using specific-locus amplified fragment sequencing (SLAF-seq). Breed Sci. 2018;68(5):596–605.
Yuan Y, Liu C, Zhao G, Gong X, Dang K, Yang Q, Feng B. Transcriptome analysis reveals the mechanism associated with dynamic changes in fatty acid and phytosterol content in foxtail millet (Setaria italica) during seed development. Food Res Int. 2021;145:110429.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Love MI, Huber W, Anders S. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Yu G, Wang L, Han Y, He Q, ClusterProfiler. An R package for comparing biological themes among gene clusters. OMICS: J Integr Biol. 2012;16:284–7.
Linn J, Ren M, Berkowitz O, Ding W, Der Merwe MJV, Whelan J, Jost R. Root cell-specific regulators of phosphatedependent growth. Plant Physiol. 2017;174:1969–89.
Smoot M, Ono K, Ideker T, Maere S. PiNGO: a Cytoscape plugin to find candidate genes in biological networks. Bioinformatics. 2011;27:1030–1.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–△△CT method. Methods. 2001;25:402–8.
Roesler K, Shintani D, Savage L, Boddupalli S, Ohlrogge J. Targeting of the Arabidopsis homomeric acetyl-coenzyme A carboxylase to plastids of rapeseeds. Plant Physiol. 1997;113(1):75–81.
Andre C, Haslam RP, Shanklin J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc Natl Acad Sci USA. 2012;109(25):10107–12.
Jia YL, Wu L, Lu CM. Stearoyl- acyl carrier protein desaturase (SAD) gene cloning and expression in Brassica napus. Chin J Oil Crop Sci. 2014;36(2):135–41.
Deng MM, Liu BL, Wang ZL, Xue JA, Zhang HM, Li RZ. Identification and functional analysis of soybean stearoyl-ACP ∆9 desaturase (GmSAD) gene family. Chin J Biotechnol. 2020;36(04):716–31.
Zhang Q, Yu R, Sun D, Rahman MM, Xie L, Hu J, He L, Kilaru A, Niu L, Zhang Y. Comparative transcriptome analysis reveals an efficient mechanism of α-Linolenic acid in Tree Peony seeds. Int J Mol Sci. 2018;20(1):65.
Fan R, Li L, Cai G, Ye J, Liu M, Wang S, Li Z. Molecular cloning and function analysis of FAD2 gene in Idesia polycarpa. Phytochemistry. 2019;168:112114.
Wang JP, Liang Q, Zhou YL, Li RZ, Xue LF, Hao XJ. Effects of exogenous MeJA on the regulation of fatty acid synthesis and FAD3 gene expression in Perilla frutescens under low temperature. J Shanxi Agricultural Univ (Natural Sci Edition). 2019;39(03):1–6.
Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–23.
Pan X, Siloto RM, Wickramarathna AD, Mietkiewska E, Weselake RJ. Identification of a pair of phospholipid:diacylglycerol acyltransferases from developing flax (Linum usitatissimum L.) seed catalyzing the selective production of trilinolenin. J Biol Chem. 2013;288(33):24173–88.
Marmon S, Sturtevant D, Herrfurth C, Chapman K, Stymne S, Feussner I. Two acyltransferases Contribute differently to linolenic acid levels in seed oil. Plant Physiol. 2017;173(4):2081–95.
Siloto RM, Findlay K, Lopez-Villalobos A, Yeung EC, Nykiforuk CL, Moloney MM. The accumulation of oleosins determines the size of seed oilbodies in Arabidopsis. Plant Cell. 2006;18(8):1961–74.
Funding
This work was supported by the Research Fund of the Shaanxi Provincial Key Research and Development Program [2023-ZDLNY-18] and the National Key Research and Development Program of China [2023YFD2200300].
Author information
Authors and Affiliations
Contributions
HYR and ZMZ conceived and designed the study; HYR, GXR, XR and ZXN prepared experimental samples; HYR and GXR performed the experiments; GXR and ZXN helped to analyze the data; HYR wrote the manuscript. ZMZ modified the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, Y., Ge, X., Xu, R. et al. Transcriptome analysis of lipid biosynthesis during kernel development in two walnut (Juglans regia L.) varieties of ‘Xilin 3’ and ‘Xiangling’. BMC Plant Biol 24, 828 (2024). https://doi.org/10.1186/s12870-024-05546-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-024-05546-y