
Abstract
Background
Evidence suggests that coronavirus disease 2019 (COVID-19) is associated with the risk of cardiovascular diseases (CVDs). However, the results are inconsistent, and the causality remains to be established. We aimed to investigate the potential causal relationship between COVID-19 and CVDs by using two-sample Mendelian randomization (MR) analysis.
Methods
Summary‐level data for COVID-19 and CVDs including myocarditis, heart failure (HF), acute myocardial infarction (AMI), arrhythmia and venous thromboembolism (VTE) were obtained from the IEU OpenGWAS project, a public genome-wide association study (GWAS). Single nucleotide polymorphisms (SNPs) were used as instrumental variables. Five complementary MR methods were performed, including inverse variance weighted (IVW), MR-Egger, weighted median, weighted mode and simple mode methods. IVW method was considered as the primary approach. Besides, sensitivity analyses, including Cochran’s Q test, MR-Egger intercept test, and leave-one-out analysis, were performed to evaluate the robustness of the results.
Results
According to the IVW results, our MR study indicated that genetically predicted COVID-19 was not causally connected with the risk of CVDs [myocarditis: odds ratio (OR) = 1.407, 95% confidence interval (CI) = 0.761-2.602, p-value = 0.277; HF: OR = 1.180, 95% CI = 0.980-1.420, p-value = 0.080; AMI: OR = 1.002, 95% CI = 0.998-1.005, p-value = 0.241; arrhythmia: OR = 0.865, 95% CI = 0.717-1.044, p-value = 0.132; VTE: OR = 1.013, 95% CI = 0.997-1.028, p-value = 0.115]. The supplementary MR methods showed similar results. Sensitivity analyses suggested that the causal estimates were robust.
Conclusion
This two-sample MR analysis did not provide sufficient evidence for a causal relationship between COVID-19 and the risk of acute CVDs, which may provide new insights into the prevention of acute CVDs in COVID-19 patients.
Similar content being viewed by others

Introduction
Coronavirus disease 2019 (COVID-19), a global pandemic caused by severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2), can lead to acute respiratory distress syndrome (ARDS), multi-organ failure and even death, hence posing a major threat to the health of millions of people worldwide.
Increasing evidence from epidemiological studies showed that COVID-19 was associated with an increased risk of cardiovascular diseases (CVDs) [1]. Several studies demonstrated that COVID-19 increases the risk of CVDs such as myocarditis, acute coronary syndrome (ACS), heart failure (HF), and arrhythmia [2, 3]. COVID-19 patients accompanied by CVDs have increased mortality and may develop post-COVID syndrome, with symptoms persisting into the recovery period or even longer [4]. CVDs following vaccination have emerged as a new obstacle in our efforts to bring an end to the pandemic [5].
A comprehensive understanding of the relationship between COVID-19 and CVDs can guide prevention and treatment strategies. It is necessary to further explore the causal association between COVID-19 and CVDs. COVID-19 has long been recognized as an important risk factor for CVDs [6]. The associations between COVID-19 and CVDs have been observed, but the genetic association has not been proven.
The conventional observational studies were inevitably subject to various confounding factors and reverse causation. Indirect associations due to common confounders shared by COVID-19 and CVDs might trigger the association between COVID-19 and CVDs. COVID-19 patients are often accompanied by multiple metabolic disorders, including obesity, blood glucose disturbances, and dyslipidemia, which might synergistically increase the risk of CVDs [7]. In addition, several common pathophysiological mechanisms may contribute to the development of CVDs in COVID-19 patients, such as excessive inflammatory response, coagulation dysfunction, endothelial cell damage, and oxygen supply and demand imbalance [8,9,10]. Therefore, whether COVID-19 would causally contribute to CVDs is still inconclusive.
Mendelian randomization (MR) analysis is an epidemiological method that uses phenotypic-associated single nucleotide polymorphisms (SNPs) as instrumental variables to assess the causal relationship between exposure and outcomes [11]. As SNPs are randomly assigned at meiosis and fixed after fertilization, MR analyses are less likely to be influenced by confounding factors and reverse causality than traditional observational studies [12]. MR analysis has become an excellent and powerful tool for causal inference.
There is evidence that COVID-19 may exacerbate pre-existing CVD symptoms in the acute phase, increase the risk of CVD onset, and even trigger CVD sequelae in the context of long COVID. The global burden of cardiovascular disease and its impact on healthcare systems will increase significantly. The causal relationship between COVID-19 and CVDs should be explored to develop evidence-based therapies or preventive strategies.
In this study, we performed a two-sample MR analysis to investigate the causal relationship between COVID-19 and the risk of CVDs, including myocarditis, heart failure (HF), acute myocardial infarction (AMI), arrhythmia and venous thromboembolism (VTE).
Methods
Study design and Instrumental variable selection
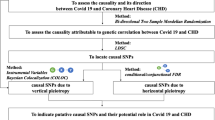
Publicly accessible GWAS summary data (https://gwas.mrcieu.ac.uk) were applied to assess the causal relationship between COVID-19 and CVDs by using a two-sample MR analysis. The flowchart of this study is presented in Fig. 1. In this MR analysis, COVID-19 and CVDs were served as exposure and outcome, respectively. Three assumptions must be satisfied when using the MR method: 1) the selected IVs must be strongly associated with the exposures; 2) the selected IVs should be independent of confounding factors that may affect the association between the exposure and the outcome; 3) the selected IVs could only influence the outcomes via the exposure of interest, not via other pathways [13].

Flowchart of this Mendelian randomization study. IVs, instrumental variables; COVID-19, coronavirus disease 2019; GWAS, genome-wide association study; CVDs, cardiovascular diseases; HF, heart failure; AMI, acute myocardial infarction; VTE, venous thromboembolism
SNPs strongly associated with COVID-19 were extracted as candidate IVs (p < 5.0 × 10−8) and SNPs were disregarded if they showed linkage disequilibrium (linkage disequilibrium r2 < 0.001, within 10-Mb distance), were palindromic with intermediate allele frequencies, or were not available in the outcome GWAS data. In addition, proxy SNPs were not included in the analysis. F statistics were calculated to assess the strength of IVs, and only SNPs with an F statistic > 10 were considered reliable IVs for COVID-19. The F-statistic is expressed as R2(n-1-k)/(1-R2)k. In the formula, R2, K, n refers to the interpreted variance of the IVs, the number of IVs for analysis, and the number of samples, respectively. Detailed information on those IVs is shown in Supplementary Table 1. The PhenoScanner database [14] was used to exclude instrumental variables related to various confounding factors.
A set of sensitivity analyses including Cochran’s Q statistic, MR‐Egger intercept tests, leave‐one‐out (LOO) analyses were performed to evaluate the robustness of the results [15]. All the original studies in this study obtained ethical approval and informed consent. This study was conducted based on the latest (STROBE-MR) guidelines [16].
COVID-19 and CVDs data sources
The GWAS summary-level data for COVID-19 and CVDs were extracted from the IEU open GWAS project (https://gwas.mrcieu.ac.uk). GWAS data for COVID-19 were derived from the COVID-19 Host Genetics Initiative, which conducted a meta-analysis of 1683,768 samples and 8660,177 SNPs [17]. For the CVDs datasets, GWAS data for HF was obtained from the European Bioinformatics Institute comprising 977,323 samples and 7,773,021 SNPs. Myocarditis data was retrieved from a public GWAS meta-analysis, including 218,792 samples and 16,380,466 SNPs. The summary-level data for AMI and VTE was obtained from the UK Biobank. The AMI dataset included 337,199 samples and 10,894,596 SNPs, while the VTE dataset included 361,194 samples and 11,901,177 SNPs. Summary statistics for arrhythmia were derived from East Asian descendants, comprising 212,453 samples and 8,885,805 SNPs. Detailed information of GWAS data was summarized in Table 1. Detailed information for the genetic variants was provided in Supplementary Table 1.
MR analysis
In this two-sample MR analysis, we used five methods [inverse-variance weighted (IVW), weighted-median estimation, MR-Egger, simple mode, and weighted mode] to investigate the causal relationship between COVID-19 and CVDs [18]. The IVW analysis is the primary method in our MR study because it provides the most convincing estimates when the directional pleiotropy of the IVs is absent [19]. The simple median predicts causal effects with less than 50% of the information comes from valid IVs, and the weighted median method requires more than 50% of valid IVs [20]. The MR-Egger method provides a consistent estimate of the causal effect under a weaker assumption-the InSIDE (INstrument Strength Independent of Direct Effect) assumption [21].
Heterogeneity, pleiotropy and sensitivity analysis
In order to detect heterogeneity, IVW method and MR-Egger regression were adopted, and heterogeneity was quantified by Cochran’s Q statistic. If the P-value was greater than 0.05, indicating no heterogeneity, the fixed-effects IVW method was considered as the main method; otherwise, the random-effects model was used [22].
MR-Egger intercept tests was performed to evaluate the possibility of horizontal pleiotropy. The deviation of the MR-Egger intercept from zero determines whether there exists a horizontal pleiotropy, and p > 0.05 indicates no pleiotropy [23]. The MR Pleiotropy REsidual Sum and Outlier (MR-PRESSO) test was also conducted to assess the presence of horizontal pleiotropy [24].
Furthermore, we utilized the leave-one-out analysis to assess whether the causal effect was influenced by a single SNP, thereby assessing the robustness of the result.
Statistical analyses
All statistical analyses were carried out using the “MR-PRESSO” and “TwoSampleMR” packages in R software (Version 4.1.2). Bonferroni correction was applied to avoid false-positive results brought by multiple tests. P < 0.05 but above the Bonferroni corrected statistical significance was defined as suggestive evidence for potential causal associations. Two-tailed P-value < 0.05 was considered statistically significant if not otherwise stated. Power calculation for this MR study was obtained via an online web tool (https://sb452.shinyapps.io/power/).
Results
The results of instrumental variable selection
Following a series of screening criteria described above, a total of 7 myocarditis-associated SNPs, 5 HF-associated SNPs, 7 AMI-associated SNPs, 6 arrhythmia-associated SNPs and 7 VTE-associated SNPs were retained for the two-sample MR analyses. The F-statistic of all selected SNPs was greater than 10, indicating no weak instrument bias. Details of the selected IVs are shown in Supplementary Table 1.
Causal effect of COVID-19 on the five CVDs
The results based on the IVW method showed no evidence to support a causal relationship between COVID-19 and the risk of CVDs (myocarditis: OR 1.407, 95% CI 0.761–2.602, p-value = 0.277; HF: OR 1.180, 95% CI 0.980–1.420, p-value = 0.080; AMI: OR 1.002, 95% CI 0.999–1.005, p-value = 0.241; arrhythmia: OR 0.865, 95% CI 0.717–1.044, p-value = 0.132;VTE: OR 1.013, 95% CI 0.997–1.028, p-value = 0.115) (Table 2 and Fig. 2). Summary results of MR estimates are shown in Fig. 2. Detailed result of MR estimates was showed in Table 2 and Fig. 3. The results of the MR-Egger, weighted median, simple mode, and weighted mode analyses were consistent with the IVW method (Fig. 2). The scatter plot shows no causal relationship between COVID-19 and CVDs (Fig. 4).

The MR estimates of the causal effect of COVID-19 on CVDs

Forest plots of causal relationship of genetically predicted COVID-19 with the risk of CVDs

Scatter plots of the causal effect of COVID-19 on the risk of CVDs. A myocarditis; (B) heart failure(HF); (C) acute myocardial infarction (AMI); (D) arrhythmia; (E) venous thromboembolism (VTE). Each black dot represents a single nucleotide polymorphisms (SNP) significantly associated with COVID-19. The gray lines around the dot represent the 95% confidence intervals (CI) of each SNP. The slope of the line represents the causality of the different MR methods. The x‐axis represents the SNPs effects on COVID-19, and the y‐axis represents the SNPs effects on CVDs susceptibility
Heterogeneity, pleiotropy and sensitivity analysis
By combining Cochran’s Q p- value in IVW and MR‐Egger methodswith the funnel plot, we assessed the heterogeneity among the selected SNPs. The results showed that there was heterogeneity in the selected SNPS in the HF and VTE datasets, and the random-effects IVW model was used to investigate the causality (Table 3). No significant heterogeneity was observed among the selected SNPs in the myocarditis, AMI and arrhythmia datasets and fixed-effects IVW method was chosen to explore the causality (Table 3). The funnel plot showed the results of heterogeneity among the selected SNPs (Fig. 5).

Funnel plots of the causal effect of COVID-19 on the risk of CVDs. A myocarditis; (B) heart failure(HF); (C) acute myocardial infarction (AMI); (D) arrhythmia; (E) venous thromboembolism (VTE)
The results of MR-Egger regression and MR-PRESSO test showed that there was no horizontal pleiotropy across SNPs in the causal estimates (myocarditis: intercept = 0.100, p-value = 0.339; HF: intercept = 0.008, p-value = 0.822; AMI: intercept = − 8.16 × 10–5, p-value = 0.876; arrhythmia: intercept = 0.0287, p-value = 0.375; VTE: intercept = -0.001, p-value = 0.672) (Table 4).
Leave-one-out analysis indicated that causal estimates between COVID-19 and CVDs were not driven by any single SNP, confirming the reliability of the MR analysis (Fig. 6).

Leave-one-out plots of causal estimates of COVID-19 on the risk of CVDs. A myocarditis; (B) heart failure(HF); (C) acute myocardial infarction (AMI); (D) arrhythmia; (E) venous thromboembolism (VTE)
The statistical power for the CVDs, including myocarditis, heart failure, AMI, arrhythmias, and venous thromboembolism ranged between 80 and 100%, thus affirming the robustness of the MR results.
Discussion
In this study, we performed a two-sample MR analysis to explore the potential causal effects of COVID-19 on the risk of CVDs. Our study provided no evidence of causal relationships between genetically predicted COVID-19 and CVDs such as myocarditis, heart failure, AMI, arrhythmias, and venous thromboembolism. The sensitivity analyses showed no heterogeneity and horizontal pleiotropy, demonstrating the robustness of the causal estimation.
How the COVID-19 pandemic affects the prevention and management of CVD is not fully understood. It is urgent to identify individuals at high risk of CVDs in order to avoid a large number of excess future CVD events [25]. Several studies have been conducted to assess the risk of CVDs in COVID-19 survivors after SARS-CoV-2 infection [26, 27]. The most common CVDs among COVID-19 patients are HF, AMI, arrhythmias, VTE and myocarditis [6, 28,29,30]. The results of a systematic review and meta-analysis suggested an increased risk of AMI after COVID-19 recovery [31]. The incidence of DVT in COVID-19 patients varies from 14.8% to 27% [32, 33]. According to a retrospective cohort study from 23 hospitals in the United States and Europe, the prevalence of acute myocarditis was 2.4 cases per 1000 hospitalized COVID-19 patients [29]. Arrhythmias have been recognized as common features of COVID-19 and are associated with poorer outcomes [34]. Heart failure was observed in 23% of COVID-19 patients, and the proportion of HF was higher in non-survivors than in survivors [35, 36].
Several possible mechanisms associated with CVDs in COVID-19 patients include oxidative stress, dysfunction of the renin–angiotensin–aldosterone system (RAAS), endothelial dysfunction, and systemic immunoinflammatory response [37]. Further research is needed to identify potential causative relationships and guide surveillance strategies to address COVID-19 related cardiovascular sequelae.
Our study did not support causal relationship between genetically determined COVID-19 and CVDs. There might be other mechanisms rather than genetics that play an important role in the increased susceptibility to CVDs in COVID-19 patients. The increased susceptibility to CVDs in COVID-19 patients may be related to hyperinflammatory response, endothelial dysfunction and immunothrombosis [38]. SARS-CoV-2 infection causes a characteristic hyperinflammatory response referred to as the “cytokine storm” [39]. Systemic inflammation triggered by inflammatory mediators, neutrophils, neutrophil extracellular traps (NETs), and proteolytic enzymes can lead to multiple organ failure syndrome and even death in COVID-19 patients [40].
Endothelial dysfunction and immunothrombosis were suggested to be the key pathogenic mechanisms of COVID-19 [38]. It has been reported that endothelial dysfunction may be the potential pathogenesis of cardiovascular dysfunction induced by COVID-19 [41]. SARS-CoV-2 infection and the consequent endotheliitis cause multiple instances of endothelial dysfunction, including altered vascular tone, oxidative stress, inflammation/leukocyte adhesion, endothelial mesenchymal transition (EndoMT) and mitochondria dysfunction [42, 43]. Exacerbation of endothelial dysfunction in COVID-19 may act as a trigger for immunothrombosis, leading to organ hypoperfusion and cardiovascular thrombotic events [44].
An important pathophysiological feature of COVID-19 is the development of a prothrombotic state. SARS-CoV-2 infection induces immunothrombosis, in which activated neutrophils and monocytes interact with platelets and the coagulation cascade, leading to microvascular and macrovascular thrombosis [45, 46]. NETs can promote thrombosis by triggering platelet activation and adhesion, activating complement, binding to fibrinogen and von willebrand factor (vWF) [47, 48]. In addition, NETs can initiate thrombosis by activating the extrinsic coagulation pathway through tissue factor (TF) production and the contact-dependent coagulation pathway via the activation of coagulation factor XII (FXII). Furthermore, NETs contribute to thrombosis through inhibition of TF pathway inhibitor (TFPI) and fibrinolysis. Hypoxia-induced release of P-selectin and vWF by endothelial cells contributes to the recruitment and activation of neutrophils, whereby released NETs form a scaffold for binding of platelets, erythrocytes, fibrin, and coagulation factors, further promote thrombosis [49, 50]. Platelet-neutrophil aggregates (PNAs) express high TF levels and are linked to intravascular coagulation and thrombosis in COVID-19 [51]. Excessive immunothrombosis can lead to a vicious cycle of microthrombosis and inflammation, which eventually progresses to thromboinflammation in COVID-19 [52].
The long-term cardiovascular effects of COVID-19 remain incompletely understood. A comprehensive assessment of the causality between COVID-19 and CVDs can improve risk stratification, develop targeted therapies and optimize the application of healthcare services. A coordinated and interdisciplinary approach may be needed to manage long COVID-19 patients, such as close follow-up by healthcare professionals.
The present study has several strengths. The main advantage is that we performed a two-sample MR analysis that provided new insights into the causal relationship between COVID-19 and acute CVDs. The application of the MR method can reduce bias from confounding factors and reverse causality, thereby providing genetic estimates of causality. Additionally, we used five complementary MR methods to thoroughly explore the causality and performed sensitivity analyses to verify the robustness of the results. Of note, the GWAS summary statistics for COVID-19 and CVDs improved the statistic power for this study.
However, there are also some limitations to our study. First, as severity-stratified information were not provided by the COVID-19 Host Genetics Initiative, we were unable to identify a causal relationship between hospitalized COVID-19/critical respiratory COVID-19 and CVDs. The lack of subgroup data for COVID-19 may limit the generalizability of our findings. Secondly, GWAS data from the COVID-19 Host Genetics Initiative in this study were mainly derived from European ancestry. As ethnic and racial minorities are disproportionately affected by the COVID-19 pandemic, the population stratification may increase bias in estimates of causality. Non-European descent are needed to clarify the causal relationship between COVID-19 and CVDs in the future studies. Thirdly, the relatively small sample size, such as cases of myocarditis, may have influence on the inference of causality between COVID-19 and CVDs. Finally, the causality between COVID-19 and CVDs inferred by MR analysis needs to be validated in experimental and clinical studies in the future. A larger, multicenter, and longer follow-up study may provide new insights into the causal relationship between COVID-19 and CVDs.
Conclusion
In conclusion, using a two-sample MR analysis, our study provided no evidence of a causal relationship between genetically predicted COVID-19 and the risk of CVDs such as myocarditis, HF, AMI, arrhythmia and VTE. Other factors, other than genetics, may contribute to the risk of CVDs in COVID-19 patients.
Availability of data and materials
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
References
Chung MK, Zidar DA, Bristow MR, Cameron SJ, Chan T, Harding CV 3rd, Kwon DH, Singh T, Tilton JC, Tsai EJ, et al. COVID-19 and cardiovascular disease: From Bench to Bedside. Circ Res. 2021;128(8):1214–36.
Luo J, Zhu X, Jian J, Chen X, Yin K. Cardiovascular disease in patients with COVID-19: evidence from cardiovascular pathology to treatment. Acta Biochim Biophys Sin. 2021;53(3):273–82.
Cersosimo A, Cimino G, Amore L, Calvi E, Pascariello G, Inciardi RM, Lombardi CM, Vizzardi E, Metra M. Cardiac biomarkers and mortality in COVID-19 infection: A review. Monaldi Arch Chest Dis. 2022;93(1).
Tajbakhsh A, Gheibi Hayat SM, Taghizadeh H, Akbari A, Inabadi M, Savardashtaki A, Johnston TP, Sahebkar A. COVID-19 and cardiac injury: clinical manifestations, biomarkers, mechanisms, diagnosis, treatment, and follow up. Expert Rev Anti-Infective Ther. 2021;19(3):345–57.
Patone M, Mei XW. Risks of myocarditis, pericarditis, and cardiac arrhythmias associated with COVID-19 vaccination or SARS-CoV-2 infection. Nat Med. 2022;28(2):410–22.
Liu F, Liu F. COVID-19 and cardiovascular diseases. J Mol Cell Biol. 2021;13(3):161–7.
Chang WT, Toh HS, Liao CT, Yu WL. Cardiac involvement of COVID-19: A comprehensive review. Am J Med Sci. 2021;361(1):14–22.
Dou Q, Wei X, Zhou K, Yang S, Jia P. Cardiovascular manifestations and mechanisms in patients with COVID-19. Trends Endocrinol Metab. 2020;31(12):893–904.
Cimino G, Vizzardi E, Calvi E, Pancaldi E, Pascariello G, Bernardi N, Cersosimo A, Amore L, Inciardi RM, Raddino R, et al. Endothelial dysfunction in COVID-19 patients assessed with Endo-PAT2000. Monaldi Arch Chest Dis. 2022;92(4).
Pancaldi E, Pascariello G, Cimino G, Cersosimo A, Amore L, Alghisi F, Bernardi N, Calvi E, Lombardi CM, Vizzardi E, et al. Thrombotic risk in patients with COVID-19. Rev Cardiovasc Med. 2021;22(2):277–86.
Birney E. Mendelian randomization. Cold Spring Harb Perspect Med. 2022;12(4):a041302.
Sheehan NA, Didelez V, Burton PR, Tobin MD. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 2008;5(8):e177.
Boef AG, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. 2015;44(2):496–511.
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35(22):4851–3.
Chen X, Hong X, Gao W, Luo S, Cai J, Liu G, Huang Y. Causal relationship between physical activity, leisure sedentary behaviors and COVID-19 risk: a Mendelian randomization study. J Transl Med. 2022;20(1):216.
Skrivankova VW, Richmond RC, Woolf BAR, Yarmolinsky J, Davies NM, Swanson SA, VanderWeele TJ, Higgins JPT, Timpson NJ, Dimou N, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: The STROBE-MR Statement. JAMA. 2021;326(16):1614–21.
The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet. 2020;28(6):715–718.
Bowden J, Holmes MV. Meta-analysis and Mendelian randomization: A review. Res Synth Methods. 2019;10(4):486–96.
Burgess S, Davey Smith G. Guidelines for performing Mendelian randomization investigations: update for summer 2023. Wellcome Open Res. 2019;4:186.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14.
Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–89.
Bowden J, Spiller W, Del Greco MF, Sheehan N, Thompson J, Minelli C, Davey Smith G. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int J Epidemiol. 2018;47(4):1264–78.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Verbanck M, Chen CY. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8.
Dale CE, Takhar R, Carragher R. The impact of the COVID-19 pandemic on cardiovascular disease prevention and management. Nat Med. 2023;29(1):219–25.
Raisi-Estabragh Z, Cooper J, Salih A, Raman B, Lee AM, Neubauer S, Harvey NC, Petersen SE. Cardiovascular disease and mortality sequelae of COVID-19 in the UK Biobank. Heart. 2022;109(2):119–26.
Aleksova A, Fluca AL, Gagno G, Pierri A, Padoan L, Derin A, Moretti R, Noveska EA, Azzalini E, D'Errico S, et al. Long-term effect of SARS-CoV-2 infection on cardiovascular outcomes and all-cause mortality. Life Sci. 2022;310:121018.
Tobler DL, Pruzansky AJ, Naderi S, Ambrosy AP, Slade JJ. Long-Term Cardiovascular Effects of COVID-19: emerging data relevant to the cardiovascular clinician. Curr Atheroscler Rep. 2022;24(7):563–70.
Ammirati E, Lupi L, Palazzini M, Hendren NS. Prevalence, characteristics, and outcomes of COVID-19-associated acute myocarditis. 2022;145(15):1123–1139.
Liu PP, Blet A, Smyth D, Li H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation. 2020;142(1):68–78.
Zuin M, Rigatelli G, Battisti V, Costola G, Roncon L, Bilato C. Increased risk of acute myocardial infarction after COVID-19 recovery: A systematic review and meta-analysis. Int J Cardiol. 2023;372:138–43.
Suh YJ, Hong H. Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta- analysis. Radiology. 2021;298(2):E70–80.
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7.
Bhatla A, Mayer MM, Adusumalli S, Hyman MC, Oh E, Tierney A, Moss J, Chahal AA, Anesi G, Denduluri S, et al. COVID-19 and cardiac arrhythmias. Heart Rhythm. 2020;17(9):1439–44.
Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, Ma K, Xu D, Yu H, Wang H, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: retrospective study. BMJ. 2020;368:m1091.
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–62.
Bohmwald K, Diethelm-Varela B, Rodríguez-Guilarte L, Rivera T, Riedel CA, González PA, Kalergis AM. Pathophysiological, immunological, and inflammatory features of long COVID. Front Immunol. 2024;15:1341600.
Bonaventura A, Vecchié A, Dagna L, Martinod K. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021;21(5):319–29.
Tomar B, Anders HJ, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells. 2020;9(6):1383
Wu S, Liao G, Mao J, Yan H, Chen J, Peng J. Factors associated with mortality among severe omicron patients for COVID-19. Infect Drug Resist. 2024;17:1309–19.
Yang K, Holt M, Fan M, Lam V, Yang Y, Ha T, Williams DL, Li C, Wang X. Cardiovascular dysfunction in COVID-19: Association between endothelial cell injury and lactate. Front Immunol. 2022;13:868679.
Nägele MP, Haubner B, Tanner FC, Ruschitzka F, Flammer AJ. Endothelial dysfunction in COVID-19: Current findings and therapeutic implications. Atherosclerosis. 2020;314:58–62.
Xu SW, Ilyas I, Weng JP. Endothelial dysfunction in COVID-19: an overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol Sin. 2023;44(4):695–709.
Otifi HM, Adiga BK. Endothelial Dysfunction in Covid-19 Infection. Am J Med Sci. 2022;363(4):281–7.
Thakur M, Junho CVC, Bernhard SM, Schindewolf M, Noels H. NETs-induced thrombosis impacts on Cardiovascular and chronic kidney disease. Circ Res. 2023;132(8):933–49.
Janiuk K, Jabłońska E, Garley M. Significance of NETs formation in COVID-19. Cells. 2021;10(1):151.
Gould TJ, Vu TT, Swystun LL, Dwivedi DJ, Mai SH, Weitz JI, Liaw PC. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34(9):1977–84.
Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, Ntinopoulou M, Sertaridou E, Tsironidou V, Tsigalou C, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Investig. 2020;130(11):6151–7.
Norooznezhad AH, Mansouri K. Endothelial cell dysfunction, coagulation, and angiogenesis in coronavirus disease 2019 (COVID-19). Microvasc Res. 2021;137:104188.
Jahani M, Dokaneheifard S, Mansouri K. Hypoxia: A key feature of COVID-19 launching activation of HIF-1 and cytokine storm. J Inflamm (Lond). 2020;17:33.
Nicolai L, Leunig A, Brambs S, Kaiser R, Weinberger T, Weigand M, Muenchhoff M, Hellmuth JC, Ledderose S, Schulz H, et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation. 2020;142(12):1176–89.
Behzadifard M, Soleimani M. NETosis and SARS-COV-2 infection related thrombosis: a narrative review. Thromb J. 2022;20(1):13.
Acknowledgements
The authors want to thank the IEU OpenGWAS Project for providing summary-level GWAS data.
Funding
This work was supported by the Natural science foundation of Liaoning province (LZ2020068).
Author information
Authors and Affiliations
Contributions
Funan Liu and Liwang Cui designed the study. Yuling Li wrote the original manuscript. Jian Kang and Yaming Cao contributed to literature search. Dongliang Yang contributed to methodology and visualization. All authors have reviewed the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Summary-level data for the studies used for analysis in this study were obtained from published studies. All studies were conducted in accordance with the Declaration of Helsinki and were conducted with the approval of institutional ethics committees, and therefore did not require additional ethical approval.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Yang, D., Kang, J. et al. COVID-19 and the risk of acute cardiovascular diseases: a two-sample Mendelian randomization study. BMC Cardiovasc Disord 24, 389 (2024). https://doi.org/10.1186/s12872-024-04066-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-024-04066-9