
Abstract
Background
Arrhythmogenic cardiomyopathy (ACM) characterized by progressive myocardial loss and replacement with fibro-fatty tissue is a major cause of sudden cardiac death (SCD). In particular, ACM with predominantly left ventricular involvement, known as arrhythmogenic left ventricular cardiomyopathy (ALVC), has a poor prognosis.
Methods
The proband underwent whole-exome sequencing (WES) to determine the etiology of ALVC. Family members were then analyzed using PCR and Sanger sequencing. Clinical evaluations including 12-lead ECG, transthoracic echocardiography, and cardiac MRI were performed for all available first-degree relatives.
Results
WES identified two variants in the FLNC (c.G3694A) and JUP (c.G1372A) genes, the combination of which results in ALVC and SCD.
Conclusion
The present study comprehensively investigates the involvement of two discovered variants of FLNC and JUP in the pathogenesis of ALVC. More study is necessary to elucidate the genetic factors involved in the etiology of ALVC.
Similar content being viewed by others

Introduction
Arrhythmogenic cardiomyopathy (ACM) is an underdiagnosed genetic disorder marked by progressive replacement of myocardium with fibrofatty scar tissue that leads to rhythm disturbances and consequently, wall motion abnormalities. Initially termed arrhythmogenic right ventricular cardiomyopathy (ARVC) due to its predominant effect on the right ventricle (RV). the left ventricular (LV) involvement was largely thought of as the extension of the pathology originating from the RV [1]. However, two large forensic studies revealed that the majority (76%, and 87%) of sudden cardiac death (SCD) cases had fibrofatty lesions in the LV free wall and septum. Notably, in nearly a fifth of these cases, the RV was unaffected. [2]. Clinical presentations appear first between 10 and 50 years of age and include palpitations, syncope, chest pain, dyspnea, and SCD [3, 4]. About 30% of ARVC cases are estimated to be familial [3, 5] and penetrance is highly influenced by exercise level [6, 7]. The most prevalent pattern of inheritance is autosomal dominant [8]. Mutations responsible for ACM could be broadly categorized into those affecting the cell-to-cell adhesion structures most notably in genes encoding the desmosomes (JUP, DSP, PKP2, DSG2, DSC2) but in rare cases non-desmosomal genes (TMEM43, RYR2, TGF-beta-3, LMNA, PLN) are involved [8, 9]. Specific mutations in PKP2, DSG, DSC, and JUP were shown to have a tendency to cause the RV dominant phenotype of ACM while DSP, PLN, LMNA/C, TMMEM43, DES, and FLNC mutations are associated with LV-dominant ACM [10,11,12,13,14].
The FLNC gene encodes filamin-C, a muscle-specific actin cross-linking protein that plays a wide variety of roles in organization and integrity of cellular structures between sarcomere, cytoskeleton and cell membrane [15]. Filamin-C was also found to act as a hub in cell signaling, mechanotransduction, and cell repair [16]. Mutations in FLNC are linked to a variety of skeletal abnormalities and cardiac myopathies including dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM) [12, 17,18,19]. Filamin-C connects adhesion molecules to Desmin intermediate filaments through intracellular linker proteins like junction plakoglobin (JUP), plakophilin-2 (PKP2), and desmoplakin (DSP) [20]. The JUP gene encodes plakoglobin, a critical component of desmosomes and adherents’ junctions, which are essential for the structural integrity and function of cardiac tissues. Mutations in the JUP gene have been implicated in the development of ACM, a condition characterized by the replacement of myocardial tissue with fibrofatty tissue, leading to an increased risk of arrhythmias and SCD [21]. DSP contributes to the transcriptional regulation of the NKX2-5 gene in cardiac progenitor cells during a short period of cardiomyogenesis and in cardiac side population stem cells in the adult. Plays a role in maintaining an optimal conformation of nebulette (NEB) on heart muscle sarcomeres to bind and recruit cardiac alpha-actin [22]. Here, we report a complex co-occurrence of variants in FLNC and JUP genes in members of a large Iranian family with a history of several SCD, and confirmed diagnosis of Arrhythmogenic left ventricular cardiomyopathy (ALVC).
Materials and methods
Family recruitment and clinical evaluation
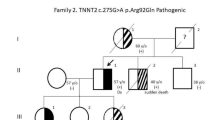
The proband, a 33-year-old Iranian man was referred to Rajaie Cardiovascular Medical and Research Center, Tehran, Iran, for cardiac workup due to multiple SCDs in his first-degree relatives (Fig. 1), including his mother (age 52) and two brothers (ages 38 and 16). Routine cardiovascular examinations, including a 12-lead electrocardiogram, transthoracic echocardiography, and cardiac magnetic resonance imaging (CMR), were performed for all available family members. Genetic studies were conducted to evaluate the etiology of the clinical features. The study adheres to the Declaration of Helsinki. Ethical approval was granted by Ethics Committees of Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran (IR.RHC.REC.1402.060). Written informed consent was obtained from the participants.

The pedigree of the ACM family. The phenotype of any individuals was mentioned under the symbol. + : mutant; -: wild type; NA: not available
Genetic investigation
Whole-exome sequencing and segregation analysis
After obtaining informed consent, whole blood samples were collected from proband and his family members. Following DNA isolation using salting-out method, the whole-exome sequencing (WES) was performed on the proband’s sample on an Illumina HiSeq 6000 platform (Macrogen, Amsterdam, Netherland). Raw data was analyzed at Cardiogenetic Research Center, Rajaie Cardiovascular Medical and Research Center Tehran, Iran. Identified variants were validated in all family members, affected and unaffected, by PCR and Sanger sequencing. Primer sequences and PCR protocols are available in Supplementary Table 1. The PCR products were sequenced using the ABI Sequencer 3500XL PE (Applied Biosystems) and analyzed with FinchTV 1.4.0 software.
Computational analysis
Bioinformatic tools including MutationTaster (www.mutationtaster.org/), PROVEAN (http://provean.jcvi.org/index.php), SIFT (https://sift.bii.a-star.edu.sg), CADD (https://cadd.gs.washington.edu/home), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/dbsearch.shtml), FATHMM (fathmm.biocompute.org.uk) were used to prediction of pathogenicity. Furthermore, the protein structure and function were investigated by UniProtKB/Swiss-Prot and PHYRE2.
Result
Clinical findings
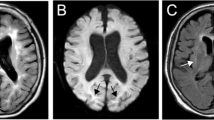
The proband patient, a 33-year-old male was referred to our arrhythmia clinic for cardiac workup due to frequent premature ventricular complexes (PVCs) on ECG and a strong family history of SCD including his mother at age 52, and two brothers at ages 38 and 16. The only reported cardiac symptoms were heart failure in patient I:1 (proband’s mother) and palpitation in patient II:4. Even in the cases of the now two deceased brothers, SCD was the first presentation. In addition to the utilization of echocardiography, CMR was employed to evaluate the extent of myocardial fibrosis in greater detail for the patients. The proband showed no sign of RV involvement. However, he was diagnosed as a definite case of arrhythmogenic left ventricular cardiomyopathy (ALVC) with intermediate risk based on LV systolic dysfunction (LVEF = 41%), abnormal LV lateral wall movement, late gadolinium enhancement (LGE) in five segment of LV lateral wall, and frequent PVCs (1.6% of total beats) (Fig. 2). Accordingly, ICD implantation was preformed and patient was put on betablockers therapy. While remaining asymptomatic, the proband showed significant clinical changes over the year following year. Even while on therapy with beta blockers, ambulatory electrocardiogram showed an increase in PVC heterogeneity and frequency compared to a year ago (1.6–5.6% of all heart beats). CMR also indicated doubling of scar tissue (from 24 to 47% of total LV mass) with an expansion from the lateral wall to the anterior segments of the left ventricle. 13 months after the diagnosis of ALVC, the patient experienced an aborted SCD as the first symptom of his disease. Notably, post discharge ambulatory electrocardiogram, showed an increase in variability of coupling intervals which also included short coupling intervals in the second PVC of some couplet PVCs. Additionally, alterations in ventricular repolarization were detected as evident in disparities in T-wave morphology before and after a PVC.

MRI Cardiac and ECG findings in the index patient. (A) Late gadolinium enhancement image at the mid-level of the left ventricle in the short-axis plane revealed transmural fibrotic replacement. (B) 12-lead Electrocardiogram indicating left anterior hemiblock and poor R-progression, pathologic Q wave in leads I and AVL
Overall, the diagnosis of ACM was ascertained in 5 out of the 7 siblings. The symptoms of arrhythmia were uncommon both in the proband and his affected relatives including the now 2 deceased brothers. CMR of other family members also had no symptoms except for palpitation in patients II:4. According to the interviews proband’s mother had suffered from heart failure for several years prior to SCD, while, the deceased brothers reported no symptoms prior to SCD. Clinical history was remarkable and no symptoms were experienced, however resting ECG, echocardiogram and CMR were indicative of ACM. The schematic clinical analysis of the pedigree individuals was shown in Fig. 3.

The schematic clinical analysis of the pedigree individuals
Genetic findings
Heterozygote variants in exon 21 of FLNC gene (NM_001127487, c.G3694A: p.G1232R), exon 5 of DES gene (NM_001927, c.A977G: p.H326R) and exon 8 of JUP gene (NM_001352773, c.G1372A: p.A458T) were identified by WES (Fig. 4). The in-silico analysis of the variants is detailed in Table 1.

The chromatogram of Sanger sequencing for three identified heterozygous variants, FLNC (c.G3694A: p.G1232R), DES (c.A977G: p.H326R) and JUP (c.G1372A: p.A458T)
Discussion
In this study, we present a proband diagnosed of ALVC with history of three SCDs in his family. Three variants of FLNC (Fig. 5), DES, and JUP (Fig. 6) genes were seen in our proband. The loss-of-function variants in the FLNC gene has been acknowledged as a potential etiology for DCM, ACM, and heart failure [23,24,25]. FLNC gene encodes Gamma filamin, also known as filamin C, one of three filamin-related proteins [26]. Filamin C exhibits binding affinity towards multiple proteins located in the Z-disk region of the sarcomere and is known to play a crucial role in preserving the structural integrity of cardiac muscle cells [27, 28]. Recent studies have linked FLNC gene nucleotide malformations to ALVC, ARVC, and DCM [24, 29,30,31,32].

(A) The figure shows the schematic structures of FLNC mutation of the Glycine in position 1232 (left) and the mutant Arginine (right) amino acid. The backbone (red part), is same and the side chains (black part), is unique for each amino acid. There are differ in size (the mutant residue is bigger than the wild-type residue), charge (The wild-type residue charge was NEUTRAL, the mutant residue charge is POSITIVE) and hydrophobicity-value. The wild-type residue is more hydrophobic than the mutant residue. (B) The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named Filamin 10. The mutation into another residue might disturb this repeat and consequently any function this repeat might have. The wild-type residue is a glycine, the most flexible of all residues. This flexibility might be necessary for the protein’s function. Mutation of this glycine can abolish this function. The wild-type residue is very conserved, but a few other residue types have been observed at this position too. Based on conservation scores this mutation is probably damaging to the protein. The mutant residue is located near a highly conserved position.

(A) The figure shows the schematic structures of JUP mutation of the Alanine in position 458 (left) and the mutant Threonine (right) amino acid. The backbone (red part), is same and the side chains (black part), is unique for each amino acid. There are differ in size, charge (The wild-type residue is more hydrophobic than the mutant residue), The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named ARM 8. The mutation into another residue might disturb this repeat and consequently any function this repeat might have. (B) The mutation is located within a stretch of residues that is repeated in the protein, this repeat is named ARM 8. The mutation into another residue might disturb this repeat and consequently any function this repeat might have. Only this residue type was found at this position. Mutation of a 100% conserved residue is usually damaging for the protein. Based on this conservation information this mutation is probably damaging to the protein. The mutated residue is located in a domain that is important for binding, activity and connection with residues in another domain of other molecules. The mutation might affect this interaction and thereby disturb signal transfer from binding domain to the activity domain. In addition, it might disturb the interaction between domains and as such affect the function of the protein
Additionally, a growing body of evidence connects causal mutations in the JUP gene with ARVC [33]. Asimaki et al. first identified the JUP gene mutation in a German family diagnosed with ARVC [34]. To our knowledge, we report the JUP variant for the first time in patient with ALVC. Carruth et al. found that FLNC loss of function variants are linked with increased odds of ventricular arrhythmia and systolic dysfunction. Next-generation sequencing (NGS) analysis of 2,877 patients with inherited cardiovascular diseases identified twenty-three truncating mutations in the FLNC gene [32]. The phenotypes observed included LV dilation (68%), reduced ejection fraction (46%), myocardial fibrosis (68%), inferolateral inverted T waves and low voltage QRS complexes on ECG (33%), ventricular rhythm disturbances (82%) and frequent SCD (40 cases in 21 of 28 families) [32]. Brun et al. found two unique FLNC variants (c.6565G > T:p.Glu2189Ter and c.8107delG: p.Asp2703ThrfsTer69) in two families among 156 ARVC patients diagnosed according to 2010 ARVC task force criteria. These families exhibited ventricular arrhythmia and sudden cardiac death [29]. In our study, the proband was diagnosed with LV variant ACM based on LV systolic dysfunction (LVEF = 41%), abnormal LV lateral wall movement, and LGE in five LV lateral wall segments, but showed no signs of RV involvements. A pooled analysis of ACM probands with negative results for common ACM-related mutations, revealed that 4.4% had FLNC variants, with ALVC being the most common phenotype [31]. Additionally, a recent study by Gigli et al. on a large cohort of individuals with FLNC variants showed that 21% exhibited an ALVC phenotype, 42% had a DCM phenotype, and 3% displayed an ARVC phenotype [35]. Regarding the DES mutation identified in this study, although it has been interpreted as a pathogenic factor for sudden cardiac death in another study [36], in our research, there is no genotype-phenotype correlation with the DES mutation. The clinical presentation of our patients is due to the combined effect of the FLNC and JUP mutations.
According to the ACM task force guideline, ICD implantation is warranted for ACM cases with at least one major risk factors including syncope, non-sustained VT or mild RV or LV dysfunction [37, 38]. In our study, the proband experienced ventricular fibrillation as the first symptom of ALVC, which occurred 13 months after the initial diagnosis. The potential sudden cardiac death was prevented by the implanted ICD. Corrado et al. studied the effects of ICD therapy on 134 ARVC patients, finding that approximately 50% experienced at least one ventricular tachyarrhythmia requiring ICD intervention over an average of 3.3 years post-implantation [39].
Conclusion
ALVC is an important phenotype of ACM patients. In this study we report and comprehensively discuss the role of two gene variants, FLNC, and JUP in the pathophysiology of ALVC. Further studies are warranted to clarify the role of genetics in pathogenesis of ALVC.
Data availability
The datasets generated and/or analyzed during the current study are available in the ClinVar repository [https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000661754.2]. The accession number of the variant in ClinVar is as follows: NM_001458.5(FLNC): c.3694G> A (p.Gly1232Arg): VCV000661754.2.
References
Sen-Chowdhry S, Lowe MD, Sporton SC, McKenna WJ. Arrhythmogenic right ventricular cardiomyopathy: clinical presentation, diagnosis, and management. Am J Med. 2004;117(9):685–95.
Groeneweg JA, Bhonsale A, James CA, Te Riele AS, Dooijes D, Tichnell C et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. 2015;8(3):437–46.
Corrado D, Link MS. Calkins HJNEjom. Arrhythmogenic Right Ventricular Cardiomyopathy. 2017;376(1):61–72.
Protonotarios N, Tsatsopoulou A, Patsourakos P, Alexopoulos D, Gezerlis P, Simitsis S et al. Cardiac abnormalities in familial palmoplantar keratosis. 1986;56(4):321–6.
Hermida J-S, Minassian A, Jarry G, Delonca J, Rey J-L, Quiret J-C et al. Familial incidence of late ventricular potentials and electrocardiographic abnormalities in arrhythmogenic right ventricular dysplasia. 1997;79(10):1375–80.
Dalal D, James C, Devanagondi R, Tichnell C, Tucker A, Prakasa K et al. Penetrance of mutations in plakophilin-2 among families with arrhythmogenic right ventricular dysplasia/cardiomyopathy. 2006;48(7):1416–24.
Saberniak J, Hasselberg NE, Borgquist R, Platonov PG, Sarvari SI, Smith HJ et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. 2014;16(12):1337–44.
Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. 2000;101(11):e101–6.
Fressart V, Duthoit G, Donal E, Probst V, Deharo J-C, Chevalier P et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. 2010;12(6):861–8.
Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. 2020;9(5):e014628.
Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. 2020;21(3):326–36.
Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. 2016;68(22):2440–51.
Hall CL, Akhtar MM, Sabater-Molina M, Futema M, Asimaki A, Protonotarios A et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. 2020;307:101–8.
Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. 2020;141(23):1872–84.
Mao Z, Nakamura FJI. Structure and function of filamin C in the muscle Z-disc. 2020;21(8):2696.
Leber Y, Ruparelia AA, Kirfel G, van der Ven PF, Hoffmann B, Merkel R et al. Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. 2016;25(13):2776–88.
Bönnemann C, Thompson T, Van der Ven P, Goebel HH, Warlo I, Vollmers B et al. Filamin C accumulation is a strong but nonspecific immunohistochemical marker of core formation in muscle. 2003;206(1):71–8.
Brodehl A, Ferrier RA, Hamilton SJ, Greenway SC, Brundler MA, Yu W et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. 2016;37(3):269–79.
Verdonschot JA, Vanhoutte EK, Claes GR, Helderman-van den Enden AT, Hoeijmakers JG, Hellebrekers DM et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. 2020;41(6):1091–111.
Saffitz JE. Desmosome mutations in arrhythmogenic right ventricular cardiomyopathy: important insight but only part of the picture. Circulation: Cardiovasc Genet. 2009;2(5):415–7.
Gerull B, Brodehl A. Insights into genetics and pathophysiology of arrhythmogenic cardiomyopathy. Curr Heart Fail Rep. 2021;18:378–90.
Hnia K, Ramspacher C, Vermot J, Laporte JJC. research t. Desmin in muscle and associated diseases: beyond the structural function. 2015;360:591–608.
Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, et al. The Geisinger MyCode community health initiative: an electronic health record–linked biobank for precision medicine research. Genet Sci. 2016;18(9):906–13.
Sveinbjornsson G, Olafsdottir EF, Thorolfsdottir RB, Davidsson OB, Helgadottir A, Jonasdottir A, et al. Variants in NKX2-5 and FLNC cause dilated cardiomyopathy and sudden cardiac death. Circulation: Genomic Precision Med. 2018;11(8):e002151.
Povysil G, Chazara O, Carss KJ, Deevi SV, Wang Q, Armisen J, et al. Assessing the role of rare genetic variation in patients with heart failure. JAMA Cardiol. 2021;6(4):379–86.
Maestrini E, Patrosso C, Mancini M, Rivella S, Rocchi M, Repetto M et al. Mapping of two genes encoding isoforms of the actin binding protein ABP-280, a dystrophin like protein, to Xq28 and to chromosome 7. 1993;2(6):761–6.
Wu T, Xu Y, Zhang L, Liang Z, Zhou X, Evans SM et al. Filamin C is essential for mammalian myocardial integrity. 2023;19(1):e1010630.
Ohashi K, Oshima K, Tachikawa M, Morikawa N, Hashimoto Y, Ito M et al. Chicken gizzard filamin, retina filamin and cgABP260 are respectively, smooth muscle-, non‐muscle‐and pan‐muscle‐type isoforms: distribution and localization in muscles. 2005;61(4):214–25.
Brun F, Gigli M, Graw SL, Judge DP, Merlo M, Murray B, et al. <em > FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy</em >. J Med Genet. 2020;57(4):254–7.
Carruth ED, Qureshi M, Alsaid A, Kelly MA, Calkins H, Murray B et al. Loss-of-Function < i > FLNC Variants Are Associated With Arrhythmogenic Cardiomyopathy Phenotypes When Identified Through Exome Sequencing of a General Clinical Population. Circulation: Genomic and Precision Medicine. 2022;15(4):e003645.
Celeghin R, Cipriani A, Bariani R, Bueno Marinas M, Cason M, Bevilacqua M, et al. Filamin-C variant-associated cardiomyopathy: a pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022;19(2):235–43.
Ortiz-Genga MF, Cuenca S, Ferro MD, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating < i > FLNC mutations are Associated with High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016;68(22):2440–51.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355(9221):2119–24.
Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81(5):964–73.
Gigli M, Stolfo D, Graw SL, Merlo M, Gregorio C, Nee Chen S, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation. 2021;144(20):1600–11.
Brodehl A, Dieding M, Klauke B, Dec E, Madaan S, Huang T et al. The novel desmin mutant p. A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. 2013;6(6):615–23.
Corrado D, Leoni L, Link MS, Bella PD, Gaita F, Curnis A et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. 2003;108(25):3084–91.
Fulgenzi CA, D’Alessio A, Airoldi C, Scotti L, Demirtas CO, Gennari A et al. Comparative efficacy of novel combination strategies for unresectable hepatocellular carcinoma: a network metanalysis of phase III trials. 2022;174:57–67.
Corrado D, Leoni L, Link MS, Bella PD, Gaita F, Curnis A, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084–91.
Acknowledgements
The authors wish to acknowledge the kind contribution of the family described herein. The Cardiogenetic Research Center, Rajaie Cardiovascular Medical and Research Center, Tehran, Iran, funded this research.
Funding
The authors received no specific funding for this research.
Author information
Authors and Affiliations
Contributions
SK designed the project and performed WES. KM, AA, and AE evaluated the patients clinically. KM, MS, TM, and AA prepared the first version of manuscript and performed wet lab. MM, AF, and SA confirmed the clinical finding of the patient and made complementary revision of the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study complies with the Declaration of Helsinki. Ethical approval was obtained from the Ethics Committees of Rajaie Cardiovascular Medical and Research Center, Iran University of Medical Sciences, Tehran, Iran (IR.RHC.REC.1402.060). Written informed consent was obtained from the participants.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mehdizadeh, K., Soveizi, M., Askarinejad, A. et al. Combination of FLNC and JUP variants causing arrhythmogenic cardiomyopathy in an Iranian family with different clinical features. BMC Cardiovasc Disord 24, 442 (2024). https://doi.org/10.1186/s12872-024-04126-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12872-024-04126-0