
Abstract
Background
Sepsis remains a leading cause of mortality in intensive care units, and rapid and accurate pathogen detection is crucial for effective treatment. This study evaluated the clinical application of multi-site metagenomic next-generation sequencing (mNGS) for the diagnosis of sepsis, comparing its performance against conventional methods.
Methods
A retrospective analysis was conducted on 69 patients with sepsis consecutively admitted to the Department of Intensive Care Medicine, Meizhou People’s Hospital. Samples of peripheral blood and infection sites were collected for mNGS and conventional method tests to compare the positive rate of mNGS and traditional pathogen detection methods and the distribution of pathogens. The methods used in this study included a comprehensive analysis of pathogen consistency between peripheral blood and infection site samples. Additionally, the correlation between the pathogens detected and clinical outcomes was investigated.
Results
Of the patients with sepsis, 57.97% experienced dyspnea, and 65.2% had underlying diseases, with hypertension being the most common. mNGS demonstrated a significantly higher pathogen detection rate (88%) compared to the conventional method tests (26%). The pathogen consistency rate was 60% between plasma and bronchoalveolar lavage fluid samples, and that of plasma and local body fluid samples was 63%. The most frequently detected pathogens were gram-negative bacteria, and Klebsiella pneumonia. There were no significant differences in the clinical features between the pathogens.
Conclusion
mNGS is significantly superior to conventional methods in pathogen detection. There was a notable high pathogen consistency detection between blood and local body fluid samples, supporting the clinical relevance of mNGS. This study highlights the superiority of mNGS in detecting a broad spectrum of pathogens quickly and accurately.
Trial registration
Not applicable.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sepsis is a life-threatening condition characterized by organ dysfunction resulting from a dysregulated response to infection, including hypotension, elevated lactic acid, and oliguria [1]. It can rapidly progress to septic shock and multiple organ failure, leading to poor prognosis and high mortality rates. Of them, the mortality rate ranges from 25 to 30% for organ dysfunction and 40–50% for septic shock [2,3,4,5]. Bloodstream infection is common in severe pneumonia cases and is a significant cause of mortality in the intensive care unit (ICU) [6,7,8]. Traditional pathogen detection methods, such as blood cultures, are time-consuming and have low sensitivity, thus often failing to identify pathogens in patients meeting the criteria for sepsis [9]. These limitations are exacerbated by prior antibiotic treatment and slow-growing pathogens [10,11,12]. Early and accurate diagnosis is crucial for effective treatment.
Metagenomic next-generation sequencing (mNGS) testing is fast and sensitive, and it is less affected by previous treatments. mNGS can widely identify known and unknown pathogens [13]. Besides, mNGS can obtain quantitative data on pathogen concentration by analyzing sequencing readings, which can contribute to identifying mixed pathogen infections [14]. mNGS has been shown to have significant advantages over conventional methods for the detection of body fluids such as blood, sputum, and cerebrospinal fluid [15, 16]. Unlike conventional methods, mNGS does not rely on prior knowledge of the suspected pathogens and can detect low-abundance and fastidious organisms that may be missed by traditional cultures. Multi-site mNGS testing involves sampling from various infection sites such as blood, bronchoalveolar lavage fluid (BALF), and other body fluids, offering a more comprehensive view of the microbial landscape in patients with sepsis. This approach enhances the possibility of detecting pathogens that may be present at specific sites or at low levels in the bloodstream. Multi-site mNGS can guide targeted antimicrobial therapy, reduce the use of broad-spectrum antibiotics, and ultimately improve clinical outcomes by providing a broader and more accurate pathogen profile.
In this study, the purpose of this study was to evaluate the clinical application of multi-site mNGS testing in the diagnosis of sepsis. The performance of multi-site mNGS testing against conventional methods was compared to determine its effectiveness in improving pathogen detection rates and providing information for clinical decision-making.
Materials and methods
Sample and clinical data collection
A retrospective analysis was conducted on 69 patients with sepsis admitted to the ICU of Meizhou People’s Hospital from April 2022 to January 2023. Peripheral blood and infection site samples were collected for mNGS and routine pathogen detection. The samples were divided into two groups: the double-sample group (39 cases) and the single-sample group (30 cases). The distribution of sample sources and clinical suspected infection sites were shown in Fig. 1A.

Overview of Design Study (A) Distribution of sample types. (B) Flowchart of the prospective study. BALF, bronchoalveolar lavage fluid; CSF, cerebrospinal fluid; mNGS, metagenomic next-generation sequencing; CMT, conventional method test; ICU, intensive care unit
This study was approved by the Ethics Committee of Meizhou People’s Hospital, and informed consent for all samples was obtained from all subjects. The inclusion criteria for this study was as follows: (1) the hospital length of stay of patients was over 24 h, (2) the Sepsis-3 criteria were met, which were based on the presence of suspected infection and clinical or microbiological evidence in the presence of at least two of the four systemic inflammatory response syndrome criteria. The following criteria were established: body temperature above 38 °C or below 36 °C, heart rate greater than 90 beats per minute, respiratory rate greater than 20 breaths per minute or carbon dioxide partial pressure below 4.3 kPa, and neutrophilia above 12,000/mm³ or neutropenia below 4000/mm³ with 10% or more immature neutrophils [17]. (3) pathogens were detected by blood culture and mNGS. The exclusion criteria included: (1) patients receiving immunosuppressive therapy; (2) patients with acquired immunodeficiency syndrome; (3) patients with incomplete clinical data. Clinical data collected for this study contained demographic information, immune deficiency status, underlying disease, computed tomography or magnetic resonance, laboratory tests, microbiological testing and treatment response judgments, length of stay, and in-hospital mortality. The study design was displayed in Fig. 1B. All data were de-identified and anonymized.
Traditional microbiological testing
All patients underwent conventional microbiological testing, including bacterial/fungal smears and cultures, fungal [1,2,3]-β-D-glucan assay, galactomannan, and grocott-gomori methenamine silver stain.
DNA extraction and mNGS detection
BALF and cerebrospinal fluid were collected by professional physicians in accordance with the standard procedures of Meizhou People’s Hospital. A 4 mL EDTA peripheral blood sample was taken from patients. DNA was extracted according to the instructions of the DNA extraction kit (TransGen Biotech, Beijing) and then used to construct a library. Quantification using Qubit 4.0. Sequencing was performed on the Illumina sequencing platform (BGISEQ200, SE75) with a read length of 150 base pairs and a depth of 30 million reads per sample. Human host sequences were removed from the data, and classification was performed using four microbial genome databases consisting of viruses, bacteria, fungi and parasites. Quality control measures included assessing the quality scores with FastQC and removing low-quality reads and adapters using Trimmomatic. Sequence reads were then processed using the Kraken2 software for quality filtering, trimming, and alignment, and high-quality reads were aligned to a comprehensive microbial database from the National Center for Biotechnology Information Reference Sequence.
To detect antimicrobial resistance genes (ARGs), sequence reads were also aligned to the Comprehensive Antibiotic Resistance Database (CARD). ARGs were detected based on the RGI bwt model [18]. Screening criteria: unique reads > 50, cover length > 200 bp, cover percentage > 70%. The abundance of ARGs was calculated as follows:
Note N mapped reads refers to the number of reads matched to the resistance genes, L reads refers to the sequencing length of the sequencer (SE75); L ARGs refers to the length of the resistance genes; S refers to the size of the data volume (M read).
The stringent mapped read number (SMRN) values were calculated by normalizing the read counts to the length of the reference genomes, allowing for standardized comparison across different pathogens. Additionally, reads per million (RPM) were calculated to provide a normalized measure of read counts, enabling comparison across samples by scaling the read counts to a standard of per million reads. SMRN and RPM, reported by bioinformatics software, quantify the abundance of microbial reads normalized by the length of the target genomes and the total read count, respectively. Therefore, such processes facilitated the comparison of relative abundance within and across samples.
Stringency in this context refers to the criteria used to filter and validate the sequencing data, ensuring that only high-confidence reads were included in the final analysis. This minimized the risk of false positives by setting thresholds for read quality (Phred score > 30), alignment score, and removal of potential contaminants through negative control analysis.
mNGS data analysis
The analysis of mNGS data involved several computational steps and tools to ensure the accurate identification of microbial pathogens from the sequencing reads. Initially, bcl2fastq software was used to convert the raw sequencing data into FASTQ format for further processing. Next, fastp was employed to filter out low-quality reads, reads with low complexity, and short sequences to maintain high-quality data for analysis. Subsequent steps involved aligning the remaining high-quality reads to the human reference genome, GRCh38, using Bowtie2. This step was crucial for removing any human genome sequences from the data, ensuring that the subsequent analysis focused solely on microbial DNA. To identify microbial species, The Scalable Nucleotide Alignment Program was used to align the filtered reads to a comprehensive microbial genome database. This database, constructed by the National Center for Biotechnology Information, includes genomes of 9,855 bacteria, 6,926 viruses, 1,582 fungi, 312 parasites, 184 mycoplasmas, and 177 mycobacteria. This extensive database allowed for the broad-spectrum detection and accurate classification of microbial pathogens present in the samples.
The clinical relevance of mNGS results was meticulously evaluated by correlating these results with clinical manifestations, imaging findings, and conventional microbiological testing. For instance, in the case of Streptococcus pneumoniae detected by mNGS, the clinical relevance was confirmed if the patient exhibited fever and increased bilateral lower lobe inflammation on computed tomography scans and the Streptococcus pneumoniae was isolated from sputum cultures. Similarly, detection of the Mycobacterium tuberculosis complex by mNGS was deemed clinically relevant if accompanied by clinical signs of tuberculosis.
Statistical analysis
SPSS Statistics 22 (IBM, NY, USA) was used for statistical analysis. R project was employed for graphing. Categorical variables were expressed as numbers and percentages, and compared using the chi-square test or Fisher’s exact test. Continuous variables were presented as mean ± standard deviation. A t-test was adopted for comparison between the two groups if the data were normally distributed. If not, the median and interquartile range (IQR) were given and the Mann-Whitney U test, a non-parametric statistical test was performed. P < 0.05 was considered a statistically significant difference.
Results
Clinical features
A total of 69 patients with sepsis were included. The most common symptoms included dyspnea, fever, and cough. The baseline features of the patients, including demographic information, clinical symptoms, and laboratory findings, are detailed in Table 1.
Data were expressed as n (%) or median (IQR). APACHE II, acute physiology and chronic health evaluation II; WBC, white blood cells; PCT, plateletocrit; CRP, C-reactive protein; ICU, intensive care unit; IQR, interquartile range.
mNGS performed better than CMT in pathogen detection for sepsis
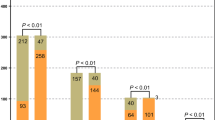
The sample types were mainly composed of blood samples and BALF samples. Of them, 78 samples were obtained from 39 patients for both sample types and 30 patients for one sample type, totaling 108 samples. All samples were subjected to mNGS and conventional method test (CMT), including 62 blood samples, 36 BALF samples, and 10 other samples. According to the results, a comparison was conducted on the positive rates of pathogens detected by mNGS and CMT in blood and BALF samples. The results revealed that pathogens were found in 28 samples in traditional culture. Of them, the detection rate was 15.0% in blood samples and 42.0% in BALF samples, with an overall positive rate of 26.0%. Pathogens were measured in 95 samples by mNGS, with a detection rate of 81.0% in blood samples and 97.0% in BALF samples. Besides, the overall positive rate was 88.0%. Therefore, the detection rate of mNGS was significantly higher than that of blood cultures (81.0% vs. 15.0%, P < 0.001) and BALF samples (97.0% vs. 42.0%, P < 0.001) (Fig. 2A). The proportions of 1 to 3 types of pathogens were 56% (35/62), 44.4% (16/36), 25% (1/4), and 33% (2/6) detected in blood, BALF, sputum, and other body fluids, respectively. The proportions of 4 to 6 types of pathogens detected were 19.0 (12/62), 42% (15/36), 75% (3/4) and 17% (1/6), respectively. The proportions of more than 6 types of pathogens detected were 3% (2/62), 11% (4/36), 0, and 50% (3/6), respectively (Fig. 2B).

mNGS is better than CMT for pathogen detection in sepsis (A) Comparison of the positive rates between mNGS and CMT for pathogen detection in the blood and BALF. Statistical histogram of positive proportion and total proportion results under different sample types and methods, and difference analysis by a chi-square test. (B) mNGS method, statistical histogram of infectious pathogens under different sample types. mNGS, Metagenomic next-generation sequencing; CMT, conventional method test; BALF, bronchoalveolar lavage fluid
Pathogen consistency of plasma and local body fluid sample by mNGS from patients with sepsis
We analyzed the proportion of the same pathogen detected in blood and BALF samples or other samples, and 39 patients underwent dual-sample mNGS and routine pathogen detection. Of these, 31 patients had both blood and BALF samples tested for mNGS and common pathogens, and 3% (1/31) had no pathogens detected. The consistency between blood and BALF samples was 60% (18/30), while the consistency between blood and local body fluid samples was 63% (5/8) (Fig. 3A). Due to the higher prevalence of secondary pulmonary infections in sepsis, we focused on analyzing the distribution of pathogens in peripheral blood and BALF samples. The distribution of the top 25 pathogens in these samples was analyzed, including 13 types of gram-negative bacteria, 6 types of gram-positive bacteria, 4 types of fungi, and 2 types of viruses (Fig. 3B). The top three pathogens consistently detected in both blood and BALF samples were Human alphaherpesvirus 1 (n = 10), Staphylococcus aureus (n = 9) and Klebsiella pneumonia (n = 9). These are also the most common cause of sepsis. In addition, the frequently detected pathogens included Haemophilus influenzae, Acinetobacter baumannii, Stenotrophomonas maltophilia and Streptococcus pneumoniae. The results of all pathogens detected in samples of different ages and genders were exhibited in the heat map (Fig. 3C). We observed that older patients had a higher number of pathogens detected in the BALF samples. Specifically, Fig. 3C illustrates that patients aged 70 and above had a median of 4 types of pathogens detected, compared to a median of 2 types in younger patients. This trend indicates that older patients might have a more diverse microbial profile, possibly due to age-related immunosenescence and increased exposure to healthcare settings. These findings emphasize the need to consider patient age when interpreting mNGS results and managing sepsis, as older patients may require broader-spectrum antimicrobial therapy.

Pathogen consistency of plasma and local body fluid sample mNGS from patients with septic (A) Analysis of the proportion of the same pathogen detected in blood and BALF; (B) The top 25 detected pathogens in Blood and BALF samples; (C) Comparison of co-detected pathogens in blood and BALF (each column representing a sample and each row indicating a potential pathogen; clinical phenotype accounting for the top, and the right color block representing the specific value of the clinical phenotype). mNGS, Metagenomic next-generation sequencing; CMT, conventional method test; BALF, bronchoalveolar lavage fluid
Clinical effect of blood and BALF mNGS testing
Clinical relevance refers to information, studies, or results that are closely related to clinical practice. Based on these, clinical relevance was used to assess whether the mNGS testing could provide an accurate basis for clinical judgment. Therefore, the clinical relevance was further evaluated. In 31 patients, peripheral blood and BALF samples were simultaneously tested for mNGS and conventional pathogens. At least one pathogen was detected in 97% of patients. A total of 60 pathogens were identified: more than 45% were bacteria (n = 27), 13.3% were fungi (n = 8) and 41.7% were DNA viruses (n = 25). Of these pathogens, 71.7% (n = 43) were ultimately deemed clinically relevant. Seven types of bacteria and two types of fungi were not clinically relevant, including Haemophilus influenzae, Staphylococcus aureus, Streptococcus pneumoniae and Klebsiella pneumoniae. However, 68% (17/25) of the viruses detected were clinically relevant, which was lower than that of bacteria (74.1%, 20/27) and fungi (75%, 6/8). Notably, no statistical difference was observed between the bacteria and fungi (Fig. 4A). Overall, the proportion of clinically relevant pathogens was high, with a higher number of clinically relevant pathogens detected in the pathogen group than in the discordant group.

Clinical effect of blood and BALF mNGS testing. (A) The bar chart of the clinical correlation and consistency of pathogens in the blood-BALF group in dual samples; (B) Violin plots of pathogen reads between clinically relevant and irrelevant groups. Sample types for statistical analysis, the normalized number of reads for each sample, and quartiles for plotting error bars. mNGS, Metagenomic next-generation sequencing; BALF, bronchoalveolar lavage fluid
Additionally, We also assessed the relation between SMRN values for pathogens and the identification of clinically relevant organisms. The median SMRN value for clinically relevant organisms was 50 (median RPM, 36; IQR = 2–210), while the median SMRN value for clinically irrelevant organisms was 36 (median RPM, 50; IQR = 4–2971). There was no statistical difference between the two groups (P = 0.11) (Fig. 4B).
Clinical consistency analysis between the consistent and inconsistent groups of Blood-BALF pathogens
Based on the mNGS results, 18 patients (60%) were classified into the pathogen consistent group, and 12 patients (40%) were divided into pathogen inconsistent groups. The statistical clinical information showed that there were low levels in PCT (5.46 vs. 5.53, P = 0.673), WBC (10.7 vs. 14.65, P = 0.140) and CRP (98.04 vs. 128.35, P = 0.615) in the pathogen consistent group compared to the inconsistent group, but the difference was not significant. The pathogen detection rate was 78.95% (15/18) in the consistent group, which was significantly higher than that in the inconsistent group [50.0% (6/12)] (P = 0.053). There were no significant differences between the two groups in neutrophil ratio, length of stay, treatment improvement, and mortality (Table 2).
Data were presented as n (%) or median (IQR). PCT, procalcitonin; WBC, white blood cells; CRP, C-reactive protein; ICU, intensive care unit; IQR, interquartile range.
Discussion
Sepsis is a clinical syndrome of host dysregulation induced by infection [2], primarily caused by bacterial and fungal infections. A large number of cytokines and inflammatory mediators are released in patients with sepsis, causing a series of inflammatory reactions [19]. According to expert consensus, temperature fluctuation is the primary evidence for the diagnosis of infection, with more than 90% of infected patients potentially experiencing acute fever [20]. In this study, 53.62% of patients had fever, which could be attributed to decreased immunity and older age. PCT and CRP are closely related to the prognosis of patients with sepsis, and the serum PCT and CRP levels are less than 0.05ng/mL and 10 mg/L, respectively, in healthy individuals [21, 22]. The median levels of PCT and CRP in patients were 15.07 (0.29–233.64) ug/L and 129.71 (1.77–396.38) mg/L, respectively. PCT and CRP have diagnostic value in adults with sepsis [23]. More importantly, we found that mNGS significantly outperformed conventional methods in detecting pathogens from multiple sites in patients with sepsis. mNGS demonstrated a higher detection rate for pathogens in peripheral blood and BALF samples compared to traditional culture methods. This superior detection capability underscores the potential of mNGS to enhance the accuracy of sepsis diagnosis and improve clinical outcomes through more targeted and timely antimicrobial therapy.
To improve the positive rate and accuracy of pathogens, multiple suspected infection sites can be tested for pathogens [24]. The positive rate of mNGS in blood samples from critically ill patients is 5.2 times higher than that of conventional methods [25]. In this study, the positive rate of mNGS in blood samples was 5.4 times that of conventional methods (81.0% vs. 15.0%). The proportions of 1–3, 4–6 and over 6 pathogens detected were 56%, 19.0% and 3%, respectively. Besides, the positive rate of mNGS in BALF samples was 2.3 times that of conventional methods (97.0% vs. 42.0%), and the proportions of 1–3, 4–6 and over 6 pathogens detected were 44.4%, 42% and 11%, respectively. The proportion of mixed pathogens in BALF samples was higher than that in blood samples, which may be due to the contamination of BALF samples with normal oral flora and colonizing bacteria. Overall, the pathogen detection rate of mNGS was significantly higher than that of traditional culture.
At present, the main causes of sepsis are bacterial and fungal infections. While sepsis can be secondary to lung infection, it is not limited to this source. Blood samples are valuable for mNGS testing due to their ability to detect bloodstream infections, which are common in sepsis. However, direct testing of samples such as sputum or BALF is preferred for diagnosing pneumonia-related sepsis. This study explored the consistency between pathogens detected in blood and BALF samples. The results revealed a 60% consistency between plasma and BALF samples and a 63% consistency between plasma and local body fluid samples, suggesting that blood mNGS testing can serve as a valuable reference for identifying secondary infections.
In terms of the types of pathogens detected, gram-negative bacteria are significant contributors to sepsis. Among these, Klebsiella pneumoniae, along with other common pathogens such as Staphylococcus aureus and Escherichia coli, frequently causes sepsis. It is important to note that the prevalence of specific pathogens can vary based on patient populations and clinical settings. Klebsiella pneumoniae is a common pathogen associated with sepsis, especially in patients with compromised immune systems. When the body’s immune resistance is weakened due to factors such as advanced age, chronic illness, or immunosuppressive therapy, the risk of opportunistic infections by Klebsiella pneumoniae increases significantly [26]. This pathogen is known to cause severe sepsis and multiple site infections, which further complicates the clinical management of these patients. In addition, Haemophilus influenzae and Acinetobacter baumannii were frequently detected. Haemophilus influenzae is prone to suppurative infection, and is responsible for 10–20% of community-acquired pneumonia cases [27]. Bacteremia and secondary meningitis can be caused by Acinetobacter baumannii, posing a significant threat to critically ill patients and patients in the cardiac care unit and ICU. Additionally, this study displayed that more types of detected pathogens increased with patient age. Furthermore, Human alphaherpesvirus was the largest number of viruses detected in the mNGS results, particularly prevalent in immunodeficient or immunosuppressed populations.
In this study, 74.1% of the bacteria detected were clinically relevant, indicating that mNGS could provide an accurate basis for clinical judgment. mNGS detection significantly increased the rate of pathogen detection in the samples. However, 28.3% of the pathogens were clinically irrelevant, suggesting the importance of interpreting data and incorporating clinical features. In addition, it is particularly important to determine whether the pathogen is an infectious bacterium or a colonizing bacterium in immunocompromised patients. The read values from mNGS can be used to identify different pathogen infections [28, 29]. A higher threshold filters out a smaller number of pathogens. False positives can also be generated due to the limitations of mNGS technology itself [30, 31]. In this study, the median SMRN value was higher in the clinically relevant group as opposed to the clinically irrelevant group (50 vs. 36), with no statistically significant difference between the two groups (P = 0.11). Therefore, it is still necessary to comprehensively determine whether the detected pathogen is the cause of sepsis. Culture or polymerase chain reaction methods can be used to verify the microorganisms that do not reach the reporting threshold but are consistent with clinical manifestations.
Moreover, multi-group comparisons of clinical features and pathogens were carried out in each group. The samples were divided into pathogen consistent group and pathogen inconsistent group according to whether the pathogens detected in the blood were consistent with those detected in BALF. The degree of clinical compliance, defined as the alignment between pathogen detection results and clinical presentation, was higher in the pathogen consistent group compared to the pathogen inconsistent group (78.95% vs. 50%, P = 0.053). This suggests that the pathogens detected by mNGS in the consistent group were more likely to be clinically relevant, thus supporting the utility of mNGS in guiding appropriate clinical interventions. PCT is associated with the abundance of pathogens detected in the blood of patients with sepsis [32]. PCT monitoring improves the possibility of distinguishing patients with sepsis from those with non-infectious systemic inflammatory response syndrome [33]. In this study, there was no significant difference in median PCT, WBC, and CRP levels between the pathogen consistent group and the pathogen inconsistent group. This observation suggests that the inflammatory response, as measured by these biomarkers, was similar regardless of whether the same pathogens were detected in both blood and BALF samples by mNGS. The lack of difference in these inflammatory markers indicates that the presence of consistent pathogens in both sample types does not significantly alter the overall inflammatory state of the patient.
In addition, the absence of significant differences in neutrophil ratio, length of hospital stay, treatment improvement, and mortality between the two groups further supports the clinical relevance of mNGS findings. The consistency in pathogen detection between blood and BALF samples highlights the reliability of mNGS in identifying relevant pathogens across different sample types, thereby providing a comprehensive picture of the patient’s infection status. This reinforces the utility of mNGS as a valuable diagnostic tool in the management of sepsis, ensuring accurate and timely identification of pathogens to guide appropriate clinical interventions.
The rapid and accurate diagnosis of sepsis is crucial for effective treatment and improved patient outcomes. Although traditional diagnostic methods are the basis for identifying pathogens, they often fall short in speed and sensitivity, particularly in patients with polymicrobial infections or those treated with empirical antibiotic therapy. Recent advancements in mNGS have shown promise in addressing these limitations. Multi-site mNGS testing analyzed samples from different infection sites and provided a comprehensive view of the microbial landscape, potentially leading to more targeted and timely interventions [34, 35]. This approach not only enhanced pathogen detection rates but also provides crucial data on antimicrobial resistance, thereby guiding more precise antimicrobial therapy. In this study, the application of multi-site mNGS testing in patients with sepsis was explored, aiming to demonstrate its utility in improving diagnostic accuracy and patient management compared to conventional methods.
There are some limitations in this study. The actual impact of blood mNGS testing results may be overestimated or underestimated due to the clinical complexity of patients. In addition, the relatively small number of clinical samples included may introduce potential selection bias, which could affect the research findings. In the future, the number of enrolled patients will be expanded and further comparative analysis will be conducted to verify the results. Although mNGS provides valuable insights into the microbial landscape, the overlap in SMRN values between clinically relevant and irrelevant organisms highlights the need for a multifaceted approach to interpreting these results. Clinicians should combine SMRN data with comprehensive clinical assessments to make informed decisions regarding pathogen relevance and appropriate treatment strategies.
Conclusion
Overall, this study explored the accuracy and clinical applications of multi-site mNGS detection in improving the diagnosis of patients with sepsis. Special attention should be paid to interpreting the clinical applications of pathogens with high numbers of specific reads. Comparisons of the pathogen consistency and clinical consistency were conducted on peripheral blood and other samples to provide evidence for the value of mNGS in pathogen detection in patients with sepsis.
Data availability
Datasets used in this article are available from the corresponding author on reasonable request.
References
Cohen J, Vincent J-L, Adhikari NKJ, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis. 2015;15(5):581–614.
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus definitions for Sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–10.
Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369(9):840–51.
Kaukonen K-M, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA. 2014;311(13):1308–16.
Vincent J-L, Marshall JC, Namendys-Silva SA, François B, Martin-Loeches I, Lipman J, et al. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med. 2014;2(5):380–6.
Huang M, Cai S, Su J. The pathogenesis of Sepsis and potential therapeutic targets. Int J Mol Sci. 2019;20(21).
Lee JH, Kim YH. Predictive factors of true bacteremia and the clinical utility of blood cultures as a prognostic tool in patients with community-onset pneumonia. Med (Baltim). 2016;95(41):e5058.
Søgaard M, Nørgaard M, Dethlefsen C, Schønheyder HC. Temporal changes in the incidence and 30-day mortality associated with bacteremia in hospitalized patients from 1992 through 2006: a population-based cohort study. Clin Infect Dis. 2011;52(1):61–9.
Duncan CF, Youngstein T, Kirrane MD, Lonsdale DO. Diagnostic challenges in Sepsis. Curr Infect Dis Rep. 2021;23(12):22.
Fenollar F, Raoult D. Molecular diagnosis of bloodstream infections caused by non-cultivable bacteria. Int J Antimicrob Agents. 2007;30(Suppl 1):S7–15.
Lamy B, Roy P, Carret G, Flandrois J-P, Delignette-Muller ML. What is the relevance of obtaining multiple blood samples for culture? A comprehensive model to optimize the strategy for diagnosing bacteremia. Clin Infect Dis. 2002;35(7):842–50.
Jain S, Self WH, Wunderink RG, Fakhran S, Balk R, Bramley AM, et al. Community-Acquired Pneumonia requiring hospitalization among U.S. adults. N Engl J Med. 2015;373(5):415–27.
Chiu CY. Viral pathogen discovery. Curr Opin Microbiol. 2013;16(4):468–78.
Salipante SJ, Hoogestraat DR, Abbott AN, SenGupta DJ, Cummings LA, Butler-Wu SM, et al. Coinfection of Fusobacterium nucleatum and Actinomyces israelii in mastoiditis diagnosed by next-generation DNA sequencing. J Clin Microbiol. 2014;52(5):1789–92.
Miller S, Naccache SN, Samayoa E, Messacar K, Arevalo S, Federman S, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019;29(5):831–42.
Zinter MS, Dvorak CC, Mayday MY, Iwanaga K, Ly NP, McGarry ME, et al. Pulmonary metagenomic sequencing suggests missed infections in Immunocompromised Children. Clin Infect Dis. 2019;68(11):1847–55.
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644-55.
Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M, Edalatmand A, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48(D1):D517–25.
Tacke F, Roderburg C, Benz F, Cardenas DV, Luedde M, Hippe H-J, et al. Levels of circulating miR-133a are elevated in sepsis and predict mortality in critically ill patients. Crit Care Med. 2014;42(5):1096–104.
Pei F, Yao R-Q, Ren C, Bahrami S, Billiar TR, Chaudry IH, et al. Expert consensus on the monitoring and treatment of sepsis-induced immunosuppression. Mil Med Res. 2022;9(1):74.
Xu X, Nie S, Liu Z, Chen C, Xu G, Zha Y, et al. Epidemiology and clinical correlates of AKI in Chinese hospitalized adults. Clin J Am Soc Nephrol. 2015;10(9):1510–8.
Schultz DR, Arnold PI. Properties of four acute phase proteins: C-reactive protein, serum amyloid A protein, alpha 1-acid glycoprotein, and fibrinogen. Semin Arthritis Rheum. 1990;20(3):129–47.
Tan M, Lu Y, Jiang H, Zhang L. The diagnostic accuracy of procalcitonin and C-reactive protein for sepsis: a systematic review and meta-analysis. J Cell Biochem. 2019;120(4):5852–9.
Chien J-Y, Yu C-J, Hsueh P-R. Utility of Metagenomic Next-Generation sequencing for Etiological diagnosis of patients with Sepsis in Intensive Care Units. Microbiol Spectr. 2022;10(4):e0074622.
Geng S, Mei Q, Zhu C, Fang X, Yang T, Zhang L, et al. Metagenomic next-generation sequencing technology for detection of pathogens in blood of critically ill patients. Int J Infect Dis. 2021;103:81–7.
Li S, Yu S, Peng M, Qin J, Xu C, Qian J, et al. Clinical features and development of Sepsis in Klebsiella pneumoniae infected liver abscess patients: a retrospective analysis of 135 cases. BMC Infect Dis. 2021;21(1):597.
Shoar S, Musher DM. Etiology of community-acquired pneumonia in adults: a systematic review. Pneumonia (Nathan). 2020;12:11.
Zhang X-X, Guo L-Y, Liu L-L, Shen A, Feng W-Y, Huang W-H, et al. The diagnostic value of metagenomic next-generation sequencing for identifying Streptococcus pneumoniae in paediatric bacterial meningitis. BMC Infect Dis. 2019;19(1):495.
Ren D, Ren C, Yao R, Zhang L, Liang X, Li G, et al. The microbiological diagnostic performance of metagenomic next-generation sequencing in patients with sepsis. BMC Infect Dis. 2021;21(1):1257.
McIntyre ABR, Ounit R, Afshinnekoo E, Prill RJ, Hénaff E, Alexander N, et al. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017;18(1):182.
Chattaway MA, Schaefer U, Tewolde R, Dallman TJ, Jenkins C. Identification of Escherichia coli and Shigella species from whole-genome sequences. J Clin Microbiol. 2017;55(2):616–23.
Yan G, Liu J, Chen W, Chen Y, Cheng Y, Tao J, et al. Metagenomic next-generation sequencing of Bloodstream Microbial Cell-Free Nucleic Acid in Children with suspected Sepsis in Pediatric Intensive Care Unit. Front Cell Infect Microbiol. 2021;11:665226.
Mustafić S, Brkić S, Prnjavorac B, Sinanović A, Porobić Jahić H, Salkić S. Diagnostic and prognostic value of procalcitonin in patients with sepsis. Med Glas (Zenica). 2018;15(2).
Grumaz S, Stevens P, Grumaz C, et al. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med. 2016;8(1):73.
Langelier C, Zinter MS, Kalantar K, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic Cellular Transplant patients. Am J Respir Crit Care Med. 2018;197(4):524–8.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
T.J.P designed the study and wrote the frst draft of the manuscript. W.W.L.,S.S.Z.,and J.Y.X collated the data, carried out data analyses. Z.X.,Y.Y.Z.,X.F.Z, H.J.G.,and M.L.Y were responsible for laboratory testing and analysis. All authors have read and approved the final submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Meizhou People’s Hospital, and informed consent for all samples was obtained from the subjects. This study was reported in accordance with ARRIVE guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, Tj., Luo, Ww., Zhang, Ss. et al. The clinical application value of multi-site mNGS detection of patients with sepsis in intensive care units. BMC Infect Dis 24, 920 (2024). https://doi.org/10.1186/s12879-024-09822-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09822-y