
Abstract
Background
Diagnosis of hereditary myopathy is often challenging owing to overlapping clinical phenotypes and muscle histopathological findings. This retrospective study aimed to identify the phenotypic and genotypic spectra of hereditary myopathies at a tertiary hospital in Riyadh, Saudi Arabia.
Methods
We reviewed the medical records of patients with hereditary myopathy who were evaluated between January 2018 and December 2022.
Results
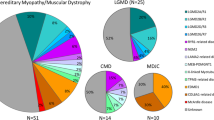
Eighty-seven patients (78 families) were included, two-thirds were men with a mean age of 35 (SD 14.2) years. Limb-girdle muscular dystrophy (LGMD) was the most prevalent clinical diagnosis (25 cases; 29%), of whom, a genetic diagnosis was achieved in 15 of 22 patients tested (68%). In genetically confirmed LGMD, the most prevalent disorders were dysferlinopathy (27%) followed by fukutin-related protein (FKRP) - related limb girdle muscular dystrophy (20%), sarcoglycanopathy (20%), lamin A/C related myopathy (13%), and calpain-3 myopathy (13%). In 26 patients with pathogenic/likely pathogenic variants, the genetic testing method was whole exome sequencing (WES) (42%), Next generation sequencing (NGS) (31%), and targeted single gene analysis (27%). The sensitivity of each genetic testing method was as follows: 100% for targeted single-gene analysis, 100% for targeted analysis of D4Z4 repeat array units, 88% for myotonic dystrophy protein kinase (DMPK) repeat expansion analysis, 42% for NGS-neuromuscular panel, and 46% for WES.
Conclusion
The prevalent types of hereditary myopathies were consistent with those reported locally and internationally. This study highlights the diagnostic yield of various molecular genetic tests for the diagnosis of hereditary myopathy in an adult cohort and the need for improved access to advanced molecular testing in cases suspected to have facioscapulohumeral muscular dystrophy (FSHD) or mitochondrial myopathies.
Similar content being viewed by others

Background
Hereditary myopathies are a heterogeneous group of disorders caused by mutations in the genes encoding proteins critical for muscle structure and function [1]. The diagnostic process can be complex and mostly includes a comprehensive clinical assessment, electromyography (EMG), laboratory testing, muscle histopathological evaluation, and molecular genetic testing. The clinical phenotype and muscle histopathological features can aid in narrowing the genetic differential diagnosis, however more commonly, these features overlap among hereditary myopathies [2], underscoring the need for early and wide-spectrum molecular analysis.
Despite recent advances and the declining costs of molecular genetic testing, the diagnostic yield of various molecular modalities across a wide spectrum of suspected hereditary myopathies has rarely been reported in Arab populations. A few studies from Saudi Arabia have reported the diagnostic yield of next-generation sequencing (NGS) in the molecular diagnosis of specific categories of hereditary myopathies in small patient cohorts [3, 4].
Establishing a definitive genetic diagnosis guides patient counselling and management, which can be lifesaving in some myopathies, such as prophylactic insertion of cardiac defibrillators to prevent fatal arrhythmias. Furthermore, a definite molecular diagnosis is a prerequisite for patient participation in clinical trials and is essential for accurate gene variant classification and diagnosis, as reported in national and global variant databases.
Herein, we report a retrospective, cross-sectional, single-center cohort of patients with hereditary myopathies who presented to our adult neuromuscular clinic. This study aimed to describe the phenotypic and genotypic spectrum of hereditary myopathies in a population with frequent consanguinity in addition to highlighting some of the limitations in achieving molecular diagnoses in a subgroup of our cohort.
Materials and methods
Study design and setting
This study is a retrospective chart review of patients with hereditary myopathy from a tertiary hospital (King Saud University Medical City, King Saud University) located in Riyadh, the largest city and capital of Saudi Arabia.
The study population included patients who were evaluated at our adult neuromuscular clinics between January 2018 and December 2022. The inclusion criteria were: (1) age ≥ 14 years, (2) persistent or transient muscle weakness attributed to myopathy by a neuromuscular specialist based on one or more of the following criteria: clinical features and electromyography (EMG) findings, muscle magnetic resonance imaging (MRI) features, or muscle histopathological findings, and (3) family history of myopathy or a phenotype consistent with hereditary myopathy such as myotonia, muscle rippling, early contractures, scapular winging, recurrent rhabdomyolysis, recurrent periodic paralysis not explained by acquired electrolyte disturbances, limb-girdle, facioscapulohumeral, humero-peroneal, or distal weakness, calf hypertrophy or atrophy, or scoliosis. We excluded patients with acquired myopathy or patients presenting with acute or subacute onset of weakness (with the exception of periodic paralysis or rhabdomyolysis), fatigability, sensory symptoms or signs, or upper motor neuron signs. Prior to genetic testing, most patients with suspected hereditary myopathy were given clinical diagnoses under general categories of myopathies (e.g., limb-girdle muscular dystrophy, mitochondrial myopathy, etc.) on the basis of age at onset, clinical phenotype, or family history.
This study was approved by the Institutional Review Board of King Saud University, Saudi Arabia. Informed consent was waived. Data anonymization was maintained throughout data collection and analysis.
Study variables and analysis
Demographic data, onset, distribution, and pattern of weakness, family history, EMG features, and laboratory tests (including serum creatine phosphokinase) were reviewed. When available, results of pulmonary function testing (PFT), echocardiography, electrocardiogram (EKG), Holter monitoring, muscle biopsy, muscle MRI, and molecular genetic findings were collected. Continuous variables were expressed as mean and standard deviation (SD). Categorical data were presented as frequencies and percentages.
Molecular genetic testing
DNA was extracted from EDTA treated blood or from dried blood spots on filter cards using standard, spin column-based methods. DNA samples were shipped to the following Clinical Laboratory Improvement Amendments and College of American Pathologists (CLIA-CAP) certified laboratories: Centogene (Rostock, Germany), GenaTi (Jeddah, Saudi Arabia), or Center for Genomic Medicine (King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia). Molecular tests requested include Whole Exome Sequencing (WES), Whole Genome Sequencing (WGS), Next Generation Sequencing (NGS) neuromuscular panel (NGS-NM panel), and single gene analysis or targeted analysis. Choice of initial molecular testing method and genetic laboratory was at the discretion of the treating physician. In cases suspected to have Facioscapulohumeral muscular dystrophy (FSHD), testing for reduction of D4Z4 repeat array units on chromosome 4q35 was performed at Bioscientia (Ingelheim, Germany). Detailed methods for all molecular tests and genes included in the NGS-NM panel can be found in Appendix 1 of supplementary data.
Molecular genetic diagnosis
All potential patterns for mode of inheritance are considered. In addition, provided family history and clinical information are used to evaluate identified variants with respect to their pathogenicity and causality. Variant interpretation followed the American College of Medical Genetics (ACMG) guidelines, categorizing variants into five classes: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign, and benign [5].
Results
We identified 100 patients with suspected hereditary myopathy who attended our adult neuromuscular clinic between January 2018 and December 2022. Thirteen patients were excluded because of missing documentation or incomplete clinical evaluations. A total of 87 patients from 78 families, with 8 families having more than one person affected (Supplementary Table 5) were included in the analysis. The large majority of patients are Saudi (97%) males (66%) with a mean age of 35 (SD 14.2). Age at symptom onset was less than 10 years in 22 patients (25%), between 10 and 40 years in 41 patients (47%), and above 40 years in 12 patients (14%). Age at onset was not documented for 12 patients.
Family history of a similar disease was reported by 55% of patients and consanguinity was present in 38%. The most common pattern of weakness was limb-girdle distribution (55%). Less than two-thirds of patients were able to walk without assistance during their last clinic visit (Table 1). PFT was performed in 48 patients, 38 (79%) had abnormal results and 12 (25%) required bilevel positive airway pressure. Echocardiography was performed in 74 patients, 2 of whom (3%) had findings of dilated cardiomyopathy. Arrhythmia was detected by EKG or Holter monitoring in 7 (11%) of 65 tested patients. One patient with arrhythmia required pacemaker insertion. Further details are provided in Supplementary Table 1.
Prior to genetic testing, the most frequent clinical diagnoses were limb-girdle muscular dystrophy (LGMD) (29%) followed by facioscapulohumeral muscular dystrophy (FSHD) (10%), myotonic dystrophy type 1 (DM1) (10%), and dystrophinopathy (10%) (Table 2). A distinct clinical phenotype was lacking in 12 patients (14%).
A genetic test was requested in 63 of 87 (72%) patients suspected to have hereditary myopathy. The remaining 24 patients did not undergo genetic testing due to unavailability or undocumented reasons. In 7 patients, genetic testing method was not documented and access to original genetic reports was not possible, all of these patients were noted to have pathogenic/likely pathogenic variants by the treating physician in the following genes: 2 patients with LMNA and one patient each with MYO18B, DMD, SGCA, FKRP, and HSPG2. Original genetic reports were not available in 4 out of 7 patients with genetically confirmed myotonic dystrophy type 1, the presence of pathogenic repeat expansions in DMPK following targeted analysis of DMPK was documented in the electronic medical charts of all 4 patients.
The total number of tests with available documentation of genetic testing methods were 61 tests in 56 patients (Table 3). Frequencies of various genetic tests performed are summarized in Table 3. The most frequent genetic test requested was WES followed by NGS-NM panel. More than one test was requested in 5 patients; three patients underwent WES following a non-diagnostic NGS-NM panel, one patient underwent DMPK repeat expansion analysis following a non-diagnostic WES, and one patient underwent WGS following a non-diagnostic WES.
Pathogenic/likely pathogenic variants were detected in 26 patients, in whom genetic testing method was WES (42%), NGS (31%), and targeted single gene analysis (27%). The sensitivity of each genetic testing method was as follows: 100% for targeted single-gene analysis, 100% for targeted analysis of D4Z4 repeat array units, 88% for DMPK repeat expansion analysis, 42% for NGS-NM panel, and 46% for whole exome sequencing (WES).
Characteristics of pathogenic or likely pathogenic variants are summarized in Supplementary Table 2. A heterozygous variant in TTN and compound heterozygous variants in PGAM2 were detected in one patient each. All remaining pathogenic/likely pathogenic variants with autosomal inheritance detected in our cohort are present in a homozygous state with recessive inheritance (Supplementary Table 2).
Molecular diagnosis rates across specific hereditary myopathies are summarized in Table 2. Clinical phenotypes associated with various causative genes/molecular pathogenic changes are summarized in Table 4. Overall frequencies of various causative genes/molecular pathogenic changes are depicted in Fig. 1. Numbers of repeat expansions in 3 patients with genetically confirmed myotonic dystrophy type1 are summarized in Supplementary Table 6.

Genes/molecular pathogenic changes causative of hereditary myopathy in our cohort. Percentages indicate the proportion of patients with the corresponding genetic defect/molecular change among 42 patients with hereditary myopathy.
AGL, amylo-1,6-glucosidase, 4-alpha-glucanotransferase; ANO5, anoctamin-5; CAPN3, calpain-3; CLCN1, chloride voltage-gated channel 1; COL6A1, collagen Type VI Alpha 1 Chain; DMD, dystrophin; DMPK, dystrophia myotonica protein kinase; Reduction of D4Z4 repeat array units; DYSF, dysferlin; FKRP, fukutin-related protein; HSPG2, heparan sulfate proteoglycan 2; LAMP2, lysosomal associated membrane protein 2; LMNA, Lamin A/C; MYO18B, myosin XVIIIB; PGAM2, phosphoglycerate mutase 2; SGCA; sarcoglycan alpha; SGCD, sarcoglycan delta; TTN, titin; TYMP, thymidine phosphorylase
Twenty-one (33%) patients had indeterminate molecular genetic results (variants of uncertain significance or no variants detected). The clinical phenotypes, muscle histopathological diagnoses, and EMG findings of patients with indeterminate genetic testing are summarized in Supplementary Table 3. Characteristics of variants of uncertain significance and their respective clinical phenotypes are listed in Supplementary Table 4.
Discussion
The current cohort of adult patients with suspected hereditary myopathy provides insights into the clinical phenotypes, genetic testing outcomes, and diagnostic yield of various molecular testing methods in these conditions with overlapping clinical features. LGMD was the most common hereditary myopathy encountered in our clinics when considering both clinical and genetic diagnoses. A molecular diagnosis was achieved in 68% of patients clinically suspected to have LGMD (15 out of 22 tested), mostly by an NGS panel or WES (Table 2).
The choice of initial genetic testing method in our patients was at the discretion of the treating physician, mostly guided by a comprehensive clinical assessment. Targeted analysis is optimal when the clinical phenotype is characteristic (e.g., DM1, FSHD, or Duchenne muscular dystrophy), a familial mutation has been identified, or a founder mutation is common in an ethnic group [6]. Conversely, NGS panels are appropriate first-line genetic tests for cases with limb-girdle myopathies and overlapping clinical phenotypes, given their comprehensive coverage of associated genes, including testing for deletion/duplication [6]. WES is particularly useful when the clinical phenotype is complex or when targeted genes are not covered by an NGS panel [6].
Genetic testing methods with the highest diagnostic yields in our cohort were targeted single-gene analysis and targeted analysis of D4Z4 repeat array units (100% in both), although the number of tests is very small, 7 and 2 tests respectively. Sensitivity of repeat expansion analysis in DMPK followed at 88% and is not unexpected owing to the characteristic phenotype of myotonic dystrophy type 1. The high sensitivity of targeted single-gene analysis in 7 patients tested using this method is explained by presence of a highly characteristic phenotype in 5 patients (Duchenne muscular dystrophy) and family history of a specific gene defect in 2 patients (one patient with mitochondrial neurogastrointestinal encephalomyopathy [MNGIE] and one patient with Glycogen storage disease type 3).
Despite the relatively few patients, mutations in dysferlin (DYSF) comprised 27% (4 out of 15) of genetically confirmed LGMD, followed by fukutin-related protein (FKRP) (20%), sarcoglycanopathy (20%), lamin A/C related myopathy (13%), and calpain-3 myopathy (CAPN3) (13%). These findings are consistent with those of previous studies, Monies et al. [3]. reported a molecular diagnosis rate of 76% in patients suspected to have LGMD when testing with a NGS neurological disease panel (759 genes). Prevalent causative genes in their cohort were DYSF (20%), CAPN3 (16%), FKRP (14%), and sarcoglycanopathy (12%) [3]. Alharbi et al. [7, 8] reported a prevalence of 29.5% and 19.6% for DYSF and CAPN3 respectively in cohort of 112 Saudi Arabian families with LGMD. The contribution of ANO5 to the LGMD phenotype was minor in this study (7%) but slightly higher than that reported by Monies et al. (2%) [3] and Bohlega et al. (3.1%) [9].
Internationally, the frequency of genes affected are similar to those detected in cohorts from Saudi Arabia. In a report of 4,656 patients from the United States, the predominant contributors to LGMD phenotypes were CAPN3 (17%), DYSF (16%), and FKRP (9%), however the diagnostic yield of targeted NGS was lower (27%) than that of our cohort (42%) [10]. In an Australian cohort, the most common genes with recessive inheritance contributing to LGMD phenotypes were CAPN3 (5.9%), DYSF (4.2%), and FKRP (4.2%) genes [11]. WES achieved a diagnosis in 45% of patients in whom targeted gene sequencing was non-diagnostic, similar to the yield of WES in the current study (46%) [11]. In a Chinese study, DYSF (49.5%) and CAPN3 (24.8%) were the most common subtypes of LGMD when NGS was pursued as a first tier test [12]. Overall, the prevalent subtypes of LGMD are comparable internationally, but the yield of the NGS panels and WES is variable, possibly owing to differences in methodology, number of genes included in the NGS panels, or inclusion criteria of the reported cohorts [13,14,15,16,17]. The small number of cases limits comparison of the diagnostic yield of targeted analysis in our series to those reported in other studies (Table 3).
All but three pathogenic/likely pathogenic variants (COL6A1, TYMP, PGAM2) in our patients are previously published (Supplementary Table 2). Pathogenic variants in the current cohort that have been previously reported in the Saudi population include FKRP (c.941 C > T), DYSF (c.164dup & c.167dup), and SGCA (c.101G > A), reported by Monies et al. (2016), [3] in addition to the CAPN3 variant (c.1076 C > T) reported by Alharbi et al. (2021) [7] and ANO5 variant (c.172 C > T) reported by Bohlega et al. [9]. To our knowledge, none of the pathogenic/likely pathogenic variants detected in our patients arise as a founder effect.
Two of our patients presented with distal weakness and early respiratory failure, a clinical phenotype consistent with hereditary myopathy with early respiratory failure (HMERF). One patient carried a pathogenic variant in exon 344 of TTN, (NM_001267550.2) c.95,195 C > T p.(Pro31732Leu) (Tables 2 and Supplementary Table 2). The other patient carried a heterozygous VUS in exon 359 of titin (TTN), the first M-band exon (Mex1) of the TTN gene (c.106126G > A) (Supplementary Table 4). The patient carrying a VUS presented with distal leg weakness and respiratory insufficiency requiring non-invasive ventilation at age 38. A deltoid biopsy showed non-specific myopathic features such as endomysial fibrosis, type 1 fiber predominance, and internalized nuclei with no detected cytoplasmic bodies or rimmed vacuoles. MRI of the extremities was not performed; MRI can support the diagnosis in HMERF if a characteristic pattern of muscle involvement is seen (degeneration of semitendinosus and obturator muscles and anterolateral compartment of lower leg). To our knowledge, variants in exon 359 have not been previously reported in association with HMERF phenotype. Exon 344 (encoding the fibronectin-3 [FN3] domain in the A-band region of titin) has been identified as a mutational hotspot region in HMERF, with autosomal dominant inheritance of most variants affecting these exons [18, 19].
No cases of myotonic dystrophy type 2 (DM2) were diagnosed in our cohort, DM2 is frequently under-recognized clinically as this condition may lack evidence of percussion or grip myotonia. Moreover, molecular diagnosis of DM2 requires targeted analysis of CNBP which is not routinely included in NGS panels, contributing further to under recognition of this disorder. A total of 9 patients in this cohort are suspected to have FSHD based on characteristic clinical phenotype and autosomal dominant inheritance pattern, albeit targeted analysis of D4Z4 repeat array units was performed in only 2 of these patients due to restricted access to laboratories performing this test at our institution [20].
In cases of indeterminate molecular diagnosis following testing with NGS, WES, or targeted analysis, the approach for further evaluation should be tailored to individual patients. The 2021 American Academy of Electrodiagnostic and Neuromuscular Medicine guidelines recommend testing for 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) or signal recognition particle (SRP) antibodies in genetically undetermined cases of limb-girdle weakness with moderate or severe creatinine kinase elevation [6], in light of recent studies reporting immune mediated necrotizing myositis mimicking clinical phenotypes of LGMD and facial-sparing FSHD. [21,22,23,24] None of the indeterminate cases with the limb-girdle phenotype in our cohort underwent testing for SRP or HMGCR antibodies.
Histopathological evaluation of the muscles was performed in the majority of genetically indeterminate cases, none of which revealed unique pathological features that guide additional genetic testing (e.g., mitochondrial features) or suggest pathogenicity of variants of uncertain significance (e.g., structural defects in implicated proteins). Muscle pathology techniques at our institution are not advanced (limited availability of immunohistochemical stains), therefore this may have resulted in underestimation of the value of muscle pathology in guiding further genetic testing.
Limitations of our analysis include the retrospective nature of the study with incomplete clinical documentation. Limited access to mitochondrial genome sequencing at our institution may have resulted in underrepresentation of mitochondrial myopathies in our patients (2 patients with ptosis and ophthalmoplegia with indeterminate genetic diagnosis). Access to original genetic reports was not possible for 7 patients noted to have pathogenic/likely pathogenic variants (2 patients with LMNA, and one patient each with MYO18B, DMD, SGCA, FKRP, and HSPG2) in addition to 4 patients noted to have pathogenic repeat expansions in DMPK. Distribution patterns of myopathic changes on EMG, as well as detailed muscle pathologic features, are not elucidated in our cohort, the authors deemed these to be beyond the scope and objectives of the study. Furthermore, referral bias limits the generalizability of the results as the findings were reported from a single tertiary referral hospital.
Conclusions
Observations from this cohort shed light on the clinical and genetic outcomes of adult patients with hereditary myopathy in central Saudi Arabia and underscore the significance of tailoring molecular genetic analysis to each clinical phenotype to guide cost-effective, early diagnosis of these conditions. In limb-girdle pattern of weakness, molecular diagnosis is frequently achieved through NGS panels or WES, whereas myopathies associated with a unique phenotype were best approached using targeted analysis (FSHD, DM1). The high rate of indeterminate molecular genetic findings in our cohort warrants further research to identify the potential diagnostic value of advanced muscle histopathological evaluations or whole-genome sequencing. Moreover, improved access to advanced molecular testing for FSHD and mitochondrial genome sequencing is of critical importance.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Dowling JJ, Weihl CC, Spencer MJ. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat Rev Mol Cell Biol. 2021;22(11):713–32. https://doi.org/10.1038/s41580-021-00389-z.
Mercuri E, Muntoni F. Muscular dystrophies. Lancet. 2013;381(9869):845–60. https://doi.org/10.1016/S0140-6736(12)61897-2.
Monies D, Alhindi HN, Almuhaizea MA, et al. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum Genomics. 2016;10:32. https://doi.org/10.1186/s40246-016-0089-8.
Bohlega SA, Alfawaz S, Abou-Al-Shaar H, et al. LGMD1D myopathy with cytoplasmic and nuclear inclusions in a Saudi family due to DNAJB6 mutation. Acta Myol. 2018;37(3):221–6.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Nicolau S, Milone M, Liewluck T. Guidelines for genetic testing of muscle and neuromuscular junction disorders. Muscle Nerve. 2021;64(3):255–69. https://doi.org/10.1002/mus.27337.
Alharbi N, Matar R, Cupler E, et al. Clinical, neurophysiological, radiological, pathological, and genetic features of Dysferlinopathy in Saudi Arabia. Front Neurosci. 2022;16:815556. https://doi.org/10.3389/fnins.2022.815556.
Alharbi N, Shosha E, Murad H, et al. Clinical and genetic features of Calpainopathies in Saudi Arabia - a descriptive cross-sectional study. Eur Rev Med Pharmacol Sci. 2021;25(15):4941–52. https://doi.org/10.26355/eurrev_202108_26451.
Bohlega S, Monies DM, Abulaban AA, Murad HN, Alhindi HN, Meyer BF. Clinical and genetic features of anoctaminopathy in Saudi Arabia. Neurosciences (Riyadh). 2015;20(2):173–7. https://doi.org/10.17712/nsj.2015.2.20140547.
Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574–87. https://doi.org/10.1002/acn3.649.
Ghaoui R, Cooper ST, Lek M, et al. Use of whole-exome sequencing for diagnosis of limb-girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. 2015;72(12):1424–32. https://doi.org/10.1001/jamaneurol.2015.2274.
Yu M, Zheng Y, Jin S, et al. Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE. 2017;12(4):e0175343. https://doi.org/10.1371/journal.pone.0175343.
Bugiardini E, Khan AM, Phadke R, et al. Genetic and phenotypic characterisation of inherited myopathies in a tertiary neuromuscular centre. Neuromuscul Disord. 2019;29(10):747–57. https://doi.org/10.1016/j.nmd.2019.08.003.
Gonzalez-Quereda L, Rodriguez MJ, Diaz-Manera J, et al. Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes (Basel). 2020;11(5):539. https://doi.org/10.3390/genes11050539.
Çavdarlı B, Köken ÖY, Satılmış SBA, et al. High diagnostic yield of targeted next-generation sequencing panel as a first-tier molecular test for the patients with myopathy or muscular dystrophy. Ann Hum Genet. 2023;87(3):104–14. https://doi.org/10.1111/ahg.12492.
Vasli N, Böhm J, Le Gras S, et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012;124(2):273–83. https://doi.org/10.1007/s00401-012-0982-8.
Evilä A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord. 2016;26(1):7–15. https://doi.org/10.1016/j.nmd.2015.10.003.
Palmio J, Leonard-Louis S, Sacconi S, et al. Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol. 2019;266(3):680–90. https://doi.org/10.1007/s00415-019-09187-2.
Bucher RM, Svergun DI, Muhle-Goll C, Mayans O. The structure of the FnIII Tandem A77-A78 points to a periodically conserved Architecture in the myosin-binding region of Titin. J Mol Biol. 2010;401(5):843–53. https://doi.org/10.1016/j.jmb.2010.06.011.
Mul K. Facioscapulohumeral muscular dystrophy. Continuum (Minneap Minn). 2022;28(6):1735–51. https://doi.org/10.1212/CON.0000000000001155.
Mohassel P, Landon-Cardinal O, Foley AR, et al. Anti-HMGCR myopathy may resemble limb-girdle muscular dystrophy. Neurol Neuroimmunol Neuroinflamm. 2019;6(1):e523. https://doi.org/10.1212/NXI.0000000000000523.
Benveniste O, Romero NB. Myositis or dystrophy? Traps and pitfalls. Presse Med. 2011;40(4 Pt 2):e249–255. https://doi.org/10.1016/j.lpm.2010.11.023.
Tard C, Tiffreau V, Jaillette E, et al. Anti-HMGCR antibody-related necrotizing autoimmune myopathy mimicking muscular dystrophy. Neuropediatrics. 2017;48(6):473–6. https://doi.org/10.1055/s-0037-1604402.
Ikeda K, Mori-Yoshimura M, Yamamoto T, et al. Chronic Myopathy Associated with Anti-signal Recognition particle antibodies can be misdiagnosed as Facioscapulohumeral muscular dystrophy. J Clin Neuromuscul Dis. 2016;17(4):197–206. https://doi.org/10.1097/CND.0000000000000115.
Acknowledgements
We thank the Investigator support Unit at Naif Health Research Center, for the language editing service provided.
Funding
No funding was received for this work.
Author information
Authors and Affiliations
Contributions
Study conceptualization, data collection and analysis, manuscript writing: MHA, RMA, MLA, SSA. Data collection and analysis: HMA. All authors reviewed the manuscript. RMA and MLA contributed as joint first authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by institutional review board Sub-Committee at King Saud University–College of Medicine, approval number: 22/0232/IRB and is an accordance with equator network guidelines for observational studies. This study has been granted an exemption from requiring written informed consent by institutional review board Sub-Committee at King Saud University–College of Medicine, Ref.No.22/0232/IRB.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Human ethics and consent to participate
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12883_2024_3838_MOESM1_ESM.docx
Supplementary Material 1: Supplementary Table 1: Cardiac and pulmonary function testing in patients with hereditary myopathy. Supplementary Table 2: Characteristics of pathogenic or likely pathogenic variants in hereditary myopathy. Supplementary Table 3: Clinical phenotypes, electromyographic and muscle histopathologic findings in patients with indeterminate molecular genetic diagnoses. Supplementary Table 4: Characteristics of variants of uncertain significance (VUS) in 8 patients with suspected hereditary myopathy. Supplementary Table 5: Distribution of clinical diagnoses among 8 families with 2 or more members suspected to have hereditary myopathy. Supplementary Table 6: Number of repeat expansions in 3 patients with Myotonic dystrophy type 1. Appendix 1- Molecular Testing methods
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alhammad, R.M., Alrehaili, M.L., Albulaihe, H.M. et al. Clinical and genetic evaluation of hereditary myopathies in an adult Saudi cohort. BMC Neurol 24, 312 (2024). https://doi.org/10.1186/s12883-024-03838-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-024-03838-2