
Abstract
Background
Despite extensive research, the identification of effective biomarkers for early prediction of preterm birth (PTB) continues to be a challenging endeavor. This study aims to identify amniotic fluid (AF) protein biomarkers useful for the early diagnosis of PTB.
Methods
We initially identified the protein expression profiles in the AF of women with PTB (n = 22) and full-term birth (FTB, n = 22), from the First People’s Hospital of Yunnan Province who underwent amniocentesis from November 2019 to February 2020, using mass spectrometry employing the data-independent acquisition (DIA) technique, and then analyzed differentially expressed proteins (DEPs). Subsequently, the least absolute shrinkage and selection operator (LASSO) and random forest analysis were employed to further screen the key proteins for PTB biomarker identification. The receiver operating characteristic (ROC) analysis, calibration plots, and decision curve analyses (DCA) were utilized to assess the discrimination and calibration of the key biomarkers.
Results
A total of 25 DEPs were identified between the PTB and FTB groups, comprising 13 up-regulated and 12 down-regulated proteins. Three key protein biomarkers for early PTB diagnosis were identified: IL1RL1 (interleukin-1 receptor-like 1), APOE (apolipoprotein E), and NECTIN4 (nectin cell adhesion molecule 4). The results of the ROC analysis showed that the area under the curve (AUC) of the three proteins combined as a biomarker for early diagnosis of PTB was 0.913 (95% CI: 0.823-1.000), with a sensitivity of 0.864 and a specificity of 0.955, both superior to those of the individual biomarkers. Bootstrap internal validation revealed a concordance index (C-index) of 0.878, with a sensitivity of 0.812 and a specificity of 0.773, indicating the robust predictive performance of these biomarkers.
Conclusions
We identified three previously unexplored yet potentially useful protein biomarkers in AF for early PTB diagnosis: IL1RL1, APOE, and NECTIN4.
Similar content being viewed by others


Background
Preterm birth (PTB), defined as delivery occurring before 37 weeks of gestation, represents the leading cause of death in newborns and children under 5 years of age [1, 2]. Worldwide, approximately 15 million newborns are delivered prematurely each year, resulting in roughly 1 million fatalities [3]. PTB is considered to be potentially associated with genetics, infections, immune imbalances, hormonal disorders, and psychological stress [4]. In addition to an increased risk of death, preterm infants are afflicted with a variety of complications and long-term adverse effects, such as a high prevalence of respiratory distress syndrome, bronchopulmonary dysplasia, sepsis, and cerebral palsy [5], which is a major global health problem.
The early prediction of PTB is essential for aiding clinicians in making more informed decisions. Over the past decades, numerous efforts have focused on identifying novel biomarkers capable of predicting a higher risk of PTB, including fetal fibronectin, placental α-microglobulin-1, phosphorylated insulin-like growth factor-binding protein 1, and C-reactive protein, among others. However, these predictors of PTB are frequently inaccessible in resource-limited settings and are characterized by suboptimal accuracy [6,7,8,9,10,11,12], highlighting the absence of a reliable method for predicting PTB. Amniotic fluid (AF), serving as the environment for fetal growth and development, contains substances originating from the placenta and fetus, and can avoid the occurrence of false positive results caused by contamination of the sample by maternal blood, representing a significant potential source of biomarkers for identifying fetal pathology [13,14,15]. However, studies based on AF for PTB are relatively scarce due to its challenging accessibility. Meanwhile, owing to the limitations of traditional proteomics methods, the range of identified proteins remains narrow. Given PTB’s status as a multifactorial dysfunctional syndrome mediated by complex molecular mechanisms, its precise molecular mechanisms are still not well understood [16], making the prevention and treatment of PTB a long-standing challenge.
Our objective was to identify biomarkers for the early diagnosis of PTB by analyzing the AF of pregnant women utilizing quantitative proteomics data-independent acquisition(DIA) technology [17], a method offering the advantages of high-throughput and comprehensive proteomic quantification. The potential implications of this research include improved maternal management strategies and enhanced understanding of the biological mechanisms influencing PTB, along with the identification of novel biomarkers for PTB.
Methods
Participants and sample collection
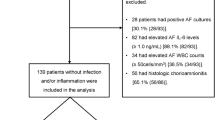
This is a nested case-control study. The study design is shown in Fig. 1. The study received approval from the Medical Ethics Committee of Kunming Medical University (approval number: KMMU2020MEC056). Additionally, informed consent forms were duly signed by all participants.Participants were recruited between November 2019 and February 2020 from the First People’s Hospital of Yunnan Province. Participants were enrolled without prior consideration to their gestational age. Post-delivery, women who gave birth before 37 weeks were categorized into the PTB group, while those delivering after 37 weeks were placed in the FTB group. The case and control groups were matched on a 1:1 basis according to age, ethnicity, and gestational time at amniocentesis. Inclusion criteria included: (1) live fetuses; (2) available AF samples for analysis; (3) no preterm premature rupture of membranes; (4) no major congenital anomalies. Participants with multiple pregnancies or with pregnancy complications were excluded from this study. We used the last menstrual period to determine gestational age. General maternal characteristics and delivery information were obtained through hospital medical records and telephone follow-up.

A flow chart for study population selection
Amniocentesis was employed to detect chromosomal abnormalities and various genetic disorders. AF samples (10 mL) were collected from patients who underwent amniocentesis at our hospital. Amniotic cells were separated by centrifugation, thereby enriching the cell-free supernatant. Subsequently, the AF samples were stored at -80 °C for future analysis.
Data acquisition and LC-MS/MS analysis
Samples were pooled for spectral library construction and digested with trypsin according to internal standard operating procedures. Dithiothreitol was added to a final concentration of 20 mM and incubated at 30 °C for 2 h. After cooling, iodoacetamide was added to 25 mM, agitated, and left in the dark for 30 min. Urea concentration was diluted with 50 mM ammonium bicarbonate (NH4HCO3) buffer. Lys-C (2 µg) in NH4HCO3 was added, followed by incubation at 37 °C for 4 h. Trypsin (2 µg) was then added, and samples were incubated at 37 °C for 16 h. Samples were desalted, lyophilized, and reconstituted in 0.1% formic acid. Peptide concentration was measured by OD280. High-abundance peptides were isolated using High-Performance Reversed-Phase chromatography. Post-lyophilization, peptides were reconstituted in 10 µL of 0.1% formic acid, and concentrations determined again by OD280. Finally, 2 µg of peptides were combined with indexed retention time (iRT) standards for data-dependent acquisition (DDA) mass spectrometry.
DDA and DIA analyses were performed on a Q-Exactive HFX mass spectrometer with an Easy nLC-1200 system using 0.1% formic acid in water (Liquid A) and 84% acetonitrile (Liquid B). Samples were separated on a 25 cm tip-column at 300 nl/min with an 8–30% Liquid B gradient over 40 min, 30–100% over 10 min, and 100% for 15 min. DDA MS scan range was 300–1800 m/z, resolution 60,000, AGC target 3e6, IT 50 ms, with MS2 scans at 30,000 resolution, AGC target 3e6, IT 120 ms, and 27 normalized collision energy. DIA MS scan range was 350–1650 m/z, with MS2 using 30 DIA windows, 30,000 resolution, AGC target 3e6, auto IT, and 30 normalized collision energy.
DDA and DIA data were processed using Spectronaut (Version 14.4.200727.47784) with trypsin digestion (one missed cleavage), fixed carbamidomethylation, variable oxidation, and acetylation. Proteins were identified with FDR < 1% using dynamic iRT, MS2 interference correction, cross-run normalization, and a Q value cutoff of 0.01.An additional file shows this in more detail [see Additional file 1]
Identification of differentially expressed proteins (DEPs)
DEPs in AF samples from the PTB and FTB groups were identified using the R “limma” package, applying the criteria of fold change (FC) ≥ 1.23 or FC ≤ 0.81, and a P-value < 0.05. Volcano plots were subsequently generated to illustrate these DEPs.
Functional and pathway enrichment analyses
The DEPs were subjected to gene ontology (GO) and kyoto encyclopedia of genes and genomes (KEGG) functional enrichment analysis using DAVID (https://david.ncifcrf.gov/). The GO analysis encompassed three categories: biological process (BP), cellular component (CC), and molecular function (MF). Additionally, KEGG analysis facilitated the exploration of potential signaling pathways.
Identification of key proteins
Initially, candidate proteins were screened using the aforementioned method. Subsequently, two machine learning algorithms, the least absolute shrinkage and selection operator (LASSO) and random forest, were utilized to further screen for key proteins. For LASSO analysis, the R package “glmnet” was employed. Additionally, the R package “randomForest” was applied to ascertain the importance sores of the candidate proteins. The top 10 proteins, ranked by importance value, were identified and overlaid with proteins from the LASSO analysis, then combined with the P-value to ascertain the final key proteins.
Statistical analysis
Statistical analysis was conducted utilizing logistic regression to construct a nomogram model depicting PTB occurrence. Receiver operating characteristic (ROC) and calibration curves were assessed to evaluate the predictive accuracy and compliance of the protein. Decision curve analyses (DCA) were utilized to reflect the net benefit of the model to the patient. The model’s internal validation was conducted using the bootstrap method. All statistical analyses utilized the “limma, glmnet, randomForest, rms, ggplot2, and pROC” packages in the R language (Version 4.3.0). Measures with normal distribution were expressed as the mean (standard deviation, SD), with two independent samples t-tests employed for between-group comparisons. For skewed measures, the median (inter-quartile range, IQR) was reported, and non-parametric tests were utilized for between-group comparisons. Constituent ratios (%) representing categorical variables were analyzed using the χ2 test or Fisher’s exact probability method. Protein concentrations underwent logarithmic (base 2) transformation for analysis. Following the removal of proteins missing in over 50% of samples, the remaining missing values were imputed using the K-nearest neighbor (KNN) method. All statistical tests were two-sided, and a P value < 0.05 indicated statistical significance.
Results
Characteristics of participants
A total of 44 pregnant women were included in this study, with 22 cases of PTB and 22 cases of FTB. The participants’ mean (± SD) age was 33.30(± 5.30) years. As expected, There were no significant differences between the two groups in age, ethnic group, residence, pregnancy time at amniocentesis, mode of delivery, and neonatal sex (all P > 0.05);Among the PTB group, The participants’ mean (± SD) gestational age was 34.96(± 1.40) weeks, and neonatal weight was 2.20(± 4.74) kg. In the FTB group, The participants’ mean (± SD) gestational age was 39.49(± 0.82) weeks, and neonatal weight was 3.34(± 3.40)kg, There were significant differences in gestational age and neonatal weight between the PTB and FTB groups (P < 0.05) (Table 1).
Identification of DEPs
We averaged the same proteins and deleted proteins that were missing in more than 50% of the samples, and finally retained the expression profiles of 611 proteins in 44 samples. A total of 25 DEPs were identified, comprising 13 up-regulated proteins: CTSL, C8A, AXL, NENF, ISLR, HMCN1, ALCAM, F10, C8B, ITIH2, IL1RL1, CST2, and MYH9 and 12 down-regulated proteins: NECTIN4, HBA2, CLPS, ANXA13, PLEC, SLIT1, GOLPH2, MRC2, APOE, CEACAM5, OLFML3, and ARHGDIA (Fig. 2A).

Differential protein expression and functional enrichment analysis of DEPs. (A) Volcano plot illustrating DEPs between PTB and FTB groups. Red dots represent upregulated proteins, blue dots represent downregulated proteins, and gray dots represent proteins with no significant change; (B) Functional enrichment analysis of DEPs in BP, CC, and MF; (C) KEGG pathway analysis of DEPs, showing the primary signaling pathways these proteins are involved in
GO and KEGG enrichment analysis
The GO analysis comprised three categories: BP, CC, and MF (Fig. 2B). In BP analysis, the three most prominent processes identified were immune response, heterophilic cell-cell adhesion via plasma membrane cell adhesion molecules, and virus entry into host cells. In CC analysis, the top three components were extracellular exosome, extracellular region, and extracellular space. Additionally, protein binding and macromolecular complex binding were identified as important roles in MF. KEGG analysis revealed that the primary enrichment pathways were “complement and coagulation cascades” and “regulation of actin cytoskeleton” (Fig. 2C).
Identification of key proteins based on LASSO and Random Forest algorithm
LASSO analysis resulted in the selection of 8 proteins (Fig. 3A and B), while random forest analysis identified the top 10 proteins based on importance values (Fig. 3C and D). Upon overlapping these two algorithms and to prevent overfitting, the top 3 proteins with the smallest P-values were chosen as key predictors for PTB, namely IL1RL1, APOE, and NECTIN4 (Fig. 3E). The identified proteins exhibited differential expression between the PTB and FTB groups, indicating their potential role in PTB among pregnant women (Fig. 3F&G&H).

The machine algorithms and expression for key proteins. (A) Penalty plot of the LASSO model with error bars denoting standard errors.This plot demonstrates the impact of different penalty values on model error; (B) LASSO plot showing the variations in the size of coefficients for parameters as the penalty value (λ) increases, indicating the process of variable selection and regularization; (C) Error rate confidence intervals for the random forest model, illustrating the variability in model error; (D) Relative importance of proteins in the random forest model, with longer bars indicating higher importance of the proteins; (E) Interaction analysis between the LASSO and random forest algorithms, highlighting the combined effect of both methods; (F-H) Expression levels of key proteins between PTB and FTB groups, presented as bar charts showing differential expression
Evaluation of key protein biomarkers: IL1RL1, APOE, and NECTIN4
A logistic regression model was constructed based on the above 3 factors and then integrated into the nomogram (Fig. 4A). In this model, a higher total score for each patient indicates an increased risk of maternal PTB.

Development and validation of a nomogram for PTB prediction.(A) Nomogram for predicting the rate of PTB, incorporating different variables to estimate the risk of PTB; (B) ROC curve analysis of key proteins, demonstrating the diagnostic performance of each biomarker with AUC values. Higher AUC values indicate better diagnostic accuracy; (C) Calibration plot of the predictive model, comparing predicted probabilities with observed outcomes to assess the model’s accuracy; (D) DCA of the predictive model, evaluating the clinical usefulness of the model across different threshold probabilities
The combined AUC of the three proteins as a biomarker for early PTB diagnosis was 0.913 (95% CI: 0.823-1.000), which was greater than that of the individual biomarkers IL1RL1:0.731 (95%CI: 0.581–0.882), APOE:0.713 (95%CI: 0.557–0.868), and NECTIN4:0.731 (95% CI: 0.571–0.891) (Fig. 4B), Furthermore, the calibration curves closely approximated the ideal diagonal (Fig. 4C), and the DCA demonstrated a significantly better net gain in the prediction model (Fig. 4D).In addition, internal validation using bootstrap self-sampling method (1000 times) showed that the C-index of the combined marker prediction model was 0.878, sensitivity was 0.812, and specificity was 0.773. These results indicate that the protein biomarkers exhibit superior performance.
Discussion
We identified 25 proteins that were significantly different between the AF samples of PTB and FTB groups. By overlapping two machine learning algorithms - LASSO and random forest analysis and integrating the P-values, we further identified three key biomarkers for PTB: IL1RL1, APOE, and NECTIN4; together these three biomarkers demonstrated a high-performance prediction of PTB with an AUC of 0.913.
In our study, IL1RL1 levels were higher in PTB AF samples than in FTB. Similar to our study, IL1RL1 mRNA was also found to be detected more frequently in prenatal maternal plasma samples collected from women with spontaneous PTB in a previous plasma/serum-based study [18]. Furthermore, in the research conducted by Kaitu’u-Lino et al [19], it was observed that pregnancy failure occurred in women who exhibited dysregulated serum sST2 levels at 6 weeks of gestation. This evidence suggests that the IL-33/ST2 pathway might have a significant role in the pathogenesis of PTB, thereby reinforcing our findings. IL1RL1 (Interleukin-1 receptor-like 1), comprising two different variants, ST2 (ST2L) and sST2, forms part of the IL-1R family of proteins and serves as an IL-33R, notably expressed in the placenta, kidney, and other tissues. This protein is crucial in modulating the immune system and inflammatory response. Additionally, mounting evidence suggests that elevated serum or tissue levels of IL1RL1 may serve as a clinical biomarker for inflammatory diseases [20,21,22,23]. In the study by Chen S et al. [24], the expression of IL1RL1 in amniotic fluid cells was detected by PCR after PROZ intervention. The results showed that IL1RL1 expression was significantly upregulated following PROZ intervention. This suggests that PROZ may regulate the secretion of collagen I by influencing the expression of IL1RL1, thereby affecting the function of AF cells and potentially playing an important role in PTB and membrane rupture. IL1RL1 could potentially function as a biomarker for the early diagnosis of PTB; however, the underlying biological mechanisms of its presence in the AF warrant further investigation.
To the best of our knowledge, this is the first study to observe in AF that the expression levels of APOE (apolipoprotein E) and NECTIN4 (nectin cell adhesion molecule 4) are significantly lower in PTB women than in the FTB group. It is well known that APOE4 represents the strongest genetic risk factor for Alzheimer’s disease [25, 26], however, the literature is sparse regarding the association of APOE with PTB. APOE, as a lipid-binding protein, forms lipoprotein complexes that bind to lipid molecules. These complexes facilitate the transport of cholesterol, triglycerides, and other lipids, which are vital for maintaining cell membrane integrity, energy metabolism, and lipid homeostasis [27, 28]. Additionally, APOE plays a significant role in regulating inflammatory responses and is posited to have a neuroprotective function in the central nervous system [29,30,31]. We propose that a deficiency in APOE may impede the realization of its critical functions, suggesting that APOE could serve as a novel biomarker and a significant indicator for the early diagnosis of PTB. Although NECTIN4 shows high expression in a variety of cancers [32,33,34,35], differently, in our current study, low expression of NECTIN4 was observed in the AF of patients with PTB.NECTIN4, also referred to as LNIR, PRR4, PVRL4, functions as an intercellular adhesion protein within the immunoglobulin-like superfamily, playing a pivotal role in cell-to-cell adhesion, as well as facilitating intercellular migration and proliferation [36, 37]. We hypothesize that its involvement in the incidence of PTB may be associated with the diminished execution of NECTIN4’s adhesive role, a factor crucial for maintaining cellular organization, structure formation, and the regulation of cellular interactions and signaling. Although its function and mechanism of action in PTB still require further in-depth study, the prospect for the use of NECTIN4 as a predictor of PTB is compelling.
The principal strength of our study lies in the amalgamation of a nested case-control design with DIA proteomics. This approach, employed in identifying biomarkers associated with PTB within a cohort of pregnant women, meticulously balanced study feasibility and economic considerations. Moreover, the amalgamation of two distinct machine-learning algorithms significantly augmented the interpretability of our findings. Notably, three out of the 25 proteins identified in association with PTB emerged as potential biomarkers for PTB. These biomarkers could potentially serve as a straightforward and valuable tool in clinical practice, aiding in the formulation of more precise treatment strategies for patients experiencing PTB.
Despite the high predictive performance of the three key proteins identified for PTB in this study, several limitations must be acknowledged. Firstly, the diagnostic utility of these potential biomarkers requires validation through alternative methodologies, such as enzyme-linked immunosorbent assay (ELISA). Secondly, the predictive model constructed in this study was exclusively focused on protein analysis and did not incorporate other contributory factors. Furthermore, its performance was not evaluated in an independent cohort, and being a single-center study, it limits the generalizability of our findings. Additionally, the study was constrained by the relatively small cohort of samples and the random selection of a subset from the PTB group. This selection was necessary due to the limited availability of amniotic fluid samples, which we recognize as a significant limitation. We also acknowledge that studying only one ethnicity further restricts the generalizability of our findings. To address these limitations and broaden the scope and applicability of our results, future studies should include multi-center cohorts with larger sample sizes and more diverse populations. Moreover, it is crucial to classify patient series samples into groups based on spontaneous and iatrogenic PTB. This classification would facilitate a more focused search for suitable biomarkers, thereby distinguishing between different types of PTB and enhancing the management and treatment of these cases. By incorporating these considerations, we aim to refine our understanding and improve the diagnostic utility of the identified biomarkers.
Conclusions
In conclusion, our study indicates that analyzing AF proteomic biomarkers may offer a dependable approach for assessing the risk of PTB. This research has, identified a set of AF proteomic biomarkers strongly associated with PTB, including IL1RL1, APOE, and NECTIN4. These findings pave the way for a novel strategy in the early detection of PTB, potentially contributing to improved outcomes through timely intervention and management.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- PTB:
-
Preterm birth
- AF:
-
Amniotic fluid
- FTB:
-
Full-term birth
- DIA:
-
Data-independent acquisition
- DEPs:
-
Differentially expressed proteins
- LASSO:
-
Least absolute shrinkage and selection operator
- ROC:
-
Receiver operating characteristic analysis
- DCA:
-
Decision curve analyses
- IL1RL1:
-
Interleukin-1 receptor-like 1
- APOE:
-
Apolipoprotein E
- NECTIN4:
-
Nectin cell adhesion molecule 4
- AUC:
-
Area under the curve
- CI:
-
Confidence interval
- C-Index:
-
Concordance index
- FC:
-
Fold change
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- BP:
-
Biological process
- CC:
-
Cellular component
- MF:
-
Molecular function
- SD:
-
Standard deviation
- IQR:
-
Inter-quartile range
- KNN:
-
K-nearest neighbor
- ELISA:
-
Enzyme-linked immunosorbent assay
References
Yang R, Li X, Ying Z, Zhao Z, Wang Y, Wang Q, Shen B, Peng W. Prematurely delivering mothers show reductions of lachnospiraceae in their gut microbiomes. BMC Microbiol. 2023;23(1):169.
Perin J, Mulick A, Yeung D, Villavicencio F, Lopez G, Strong KL, Prieto-Merino D, Cousens S, Black RE, Liu L. Global, regional, and national causes of under-5 mortality in 2000-19: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet Child Adolesc Health. 2022;6(2):106–15.
Harrison MS, Goldenberg RL. Global burden of prematurity. Semin Fetal Neonatal Med. 2016;21(2):74–9.
Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345(6198):760–5.
Chawanpaiboon S, Vogel JP, Moller AB, Lumbiganon P, Petzold M, Hogan D, Landoulsi S, Jampathong N, Kongwattanakul K, Laopaiboon M, et al. Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob Health. 2019;7(1):e37–46.
Berghella V, Saccone G. Fetal fibronectin testing for reducing the risk of preterm birth. Cochrane Database Syst Rev. 2019;7(null):Cd006843.
Kuhrt K, Watson H, Seed P, Shennan A. Placental alpha Microglobulin-1 compared with fetal fibronectin to Predict Preterm Delivery in Symptomatic Women. Obstet Gynecol. 2018;131(4):743.
Nikolova T, Uotila J, Nikolova N, Bolotskikh VM, Borisova VY, Di Renzo GC. Prediction of spontaneous preterm delivery in women presenting with premature labor: a comparison of placenta alpha microglobulin-1, phosphorylated insulin-like growth factor binding protein-1, and cervical length. American journal of obstetrics and gynecology 2018, 219(6):610.e611-610.e619.
Hsu TY, Cheng HH, Lan KC, Hung HN, Lai YJ, Tsai CC, Fan WL, Li SC. The abundances of LTF and SOD2 in amniotic fluid are potential biomarkers of gestational age and preterm birth. Sci Rep. 2023;13(1):4903.
Pons-Duran C, Wilder B, Hunegnaw BM, Haneuse S, Goddard FG, Bekele D, Chan GJ. Development of risk prediction models for preterm delivery in a rural setting in Ethiopia. J Glob Health. 2023;13:04051.
Lucaroni F, Morciano L, Rizzo G, D’ Antonio F, Buonuomo E, Palombi L, Arduini D. Biomarkers for predicting spontaneous preterm birth: an umbrella systematic review. J Maternal-Fetal Neonatal Med. 2018;31(6):726–34.
Gunko VO, Pogorelova TN, Linde VA. Proteomic profiling of the blood serum for prediction of premature delivery. Bull Exp Biol Med. 2016;161(6):829–32.
Jha P, Raghu P, Kennedy AM, Sugi M, Morgan TA, Feldstein V, Pōder L, Penna R. Assessment of amniotic fluid volume in pregnancy. Radiographics. 2023;43(6):e220146.
Liu X, Song Y, Guo Z, Sun W, Liu J. A comprehensive profile and inter-individual variations analysis of the human normal amniotic fluid proteome. J Proteom. 2019;192(null):1–9.
Vuadens F, Benay C, Crettaz D, Gallot D, Sapin V, Schneider P, Bienvenut WV, Lémery D, Quadroni M, Dastugue B, et al. Identification of biologic markers of the premature rupture of fetal membranes: proteomic approach. Proteomics. 2003;3(8):1521–5.
Camunas-Soler J, Gee EPS, Reddy M, Mi JD, Thao M, Brundage T, Siddiqui F, Hezelgrave NL, Shennan AH, Namsaraev E et al. Predictive RNA profiles for early and very early spontaneous preterm birth. American journal of obstetrics and gynecology 2022, 227(1):72.e71-72.e16.
Meyer JG, Schilling B. Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Rev Proteomics. 2017;14(5):419–29.
Chim SS, Lee WS, Ting YH, Chan OK, Lee SW, Leung TY. Systematic identification of spontaneous preterm birth-associated RNA transcripts in maternal plasma. PLoS ONE. 2012;7(4):e34328.
Kaitu’u-Lino TJ, Tuohey L, Tong S. Maternal serum interleukin-33 and soluble ST2 across early pregnancy, and their association with miscarriage. J Reprod Immunol. 2012;95(1–2):46–9.
Gungor O, Unal HU, Guclu A, Gezer M, Eyileten T, Guzel FB, Altunoren O, Erken E, Oguz Y, Kocyigit I, et al. IL-33 and ST2 levels in chronic kidney disease: associations with inflammation, vascular abnormalities, cardiovascular events, and survival. PLoS ONE. 2017;12(6):e0178939.
Cui G, Yuan A, Li Z, Goll R, Florholmen J. ST2 and regulatory T cells in the colorectal adenoma/carcinoma microenvironment: implications for diseases progression and prognosis. Sci Rep. 2020;10(1):5892.
Sun Z, Chang B, Huang A, Hao S, Gao M, Sun Y, Shi M, Jin L, Zhang W, Zhao J, et al. Plasma levels of soluble ST2, but not IL-33, correlate with the severity of alcoholic liver disease. J Cell Mol Med. 2019;23(2):887–97.
Lu DP, Zhou XY, Yao LT, Liu CG, Ma W, Jin F, Wu YF. Serum soluble ST2 is associated with ER-positive breast cancer. BMC Cancer. 2014;14:198.
Chen S, Wang W, Yan G, Liu M, Li M, Chen P, Ma Q, Zhang J, Tang Y, Zhou L, et al. Amniotic Fluid Proteomics Analysis and in Vitro Validation to identify potential biomarkers of Preterm Birth. Reproductive Sci. 2024;31(7):2032–42.
Huggins LKL, Min SH, Kaplan S, Wei J, Welsh-Bohmer K, Xu H. Meta-analysis of variations in Association between APOE ɛ4 and Alzheimer’s Disease and related dementias across hispanic regions of Origin. J Alzheimer’s Disease: JAD. 2023;93(3):1095–109.
Zheng L, Eramudugolla R, Cherbuin N, Drouin SM, Dixon RA, Anstey KJ. Gender specific factors contributing to cognitive resilience in APOE varepsilon4 positive older adults in a population-based sample. Sci Rep. 2023;13(1):8037.
Frieden C, Garai K. Concerning the structure of apoE. Protein Sci. 2013;22(12):1820–5.
Lozano S, Padilla V, Avila ML, Gil M, Maestre G, Wang K, Xu C. APOE Gene Associated with cholesterol-related traits in the Hispanic Population. Genes. 2021;12(11):null.
Hansen SB, Wang H. The shared role of cholesterol in neuronal and peripheral inflammation. Pharmacology & therapeutics 2023, 249(null):108486.
Ma J, Guo Q, Shen MQ, Li W, Zhong QX, Qian ZM. Apolipoprotein E is required for brain iron homeostasis in mice. Redox Biol. 2023;64(null):102779.
Jiang L, Zhong J, Dou X, Cheng C, Huang Z, Sun X. Effects of ApoE on intracellular calcium levels and apoptosis of neurons after mechanical injury. Neuroscience. 2015;301(null):375–83.
Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, Raitano A, Nadell R, Liu W, Lortie DR, et al. Enfortumab Vedotin antibody-drug Conjugate Targeting Nectin-4 is a highly potent Therapeutic Agent in multiple preclinical Cancer models. Cancer Res. 2016;76(10):3003–13.
Hashimoto M, Fujita K, Tomiyama E, Fujimoto S, Adomi S, Banno E, Minami T, Takao T, Nozawa M, Fushimi H, et al. Immunohistochemical analysis of HER2, EGFR, and Nectin-4 expression in Upper urinary tract Urothelial Carcinoma. Anticancer Res. 2023;43(1):167–74.
Murata M, Ito T, Tanaka Y, Kaku-Ito Y, Furue M. NECTIN4 expression in Extramammary Paget’s Disease: implication of a New Therapeutic Target. Int J Mol Sci. 2020;21(16):null.
Tanaka Y, Murata M, Oda Y, Furue M, Ito T. Nectin Cell Adhesion Molecule 4 (NECTIN4) expression in cutaneous squamous cell carcinoma: a New Therapeutic Target? Biomedicines 2021, 9(4):null.
Boylan KL, Buchanan PC, Manion RD, Shukla DM, Braumberger K, Bruggemeyer C, Skubitz AP. The expression of Nectin-4 on the surface of ovarian cancer cells alters their ability to adhere, migrate, aggregate, and proliferate. Oncotarget. 2017;8(6):9717–38.
Bedir R, Sehitoglu I, Balik G, Kagitci M, Gucer H, Yurdakul C, Bagci P. The role of the adhesion molecule Nectin-4 in the pathogenesis of endometriosis. Clin Exp Obstet Gynecol. 2016;43(3):463–6.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (grant numbers 82073569, 81960592), the Outstanding Youth Science Foundation of Yunnan Basic Research Project (202001AW070021), the Reserve Talent Project for Young and Middle-aged Academic and Technical Leaders (2012005AC160023), the Key Science Foundation of Yunnan Basic Research (202101AS070040), and the science and technology plan project of Yunnan Province(202101AY070001-225).
Author information
Authors and Affiliations
Contributions
Min Li: Conceptualization, Methodology, Investigation, Formal Analysis, Writing - Original Draft, Writing Review Editing; Mengmei Liu and Ping Chen: Investigation, Visualization, Formal Analysis, Writing - Original Draft; Sifan Duan, Yani Li, and Qingyan Ma: Investigation, Data Collection; Guanghong Yan: Data Collection, Visualization; Hongying Li, Jinman Zhang, and Dingyun You: Conceptualization, Funding Acquisition, Resources, Supervision, Writing - Review & Editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study has been approved by the Medical Ethics Committee of Kunming Medical University and meets the relevant ethical standards (KMMU2020MEC056). All participants voluntarily participated in the study and signed an informed consent form, and patient information was kept confidential.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12884_2024_6734_MOESM1_ESM.docx
Additional file 1: DIA proteomic analysis. Quantitative proteomic analysis by DIA was performed on each AF sample. This comprehensive process encompassed protein extraction, peptide digestion, mass spectrometry analysis, and data analysis of the mass spectrometry results.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, M., Liu, M., Chen, P. et al. Amniotic fluid proteomic analysis identifies IL1RL1, APOE, and NECTIN4 as new biomarkers for preterm birth. BMC Pregnancy Childbirth 24, 530 (2024). https://doi.org/10.1186/s12884-024-06734-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-024-06734-2