
Abstract
Homo sapiens chromosome 2 clone RP11-339H12 (AC010883.5) is a dysregulated long non-coding RNA (lncRNA) that has never been investigated in cervical cancer (CC). Thus, the potential function and molecular mechanism remain unclear. Our study explored the biological function of AC010883.5 to determine the underlying mechanisms in CC and provide potential therapeutic targets for improving the clinical treatment strategy. We used quantitative real-time polymerase chain reaction to measure mitochondrial RNA levels and western blot to measure the protein levels of target genes. Further, we used Cell Counting Kit-8 and 5‐Bromo-2'-deoxyuridine incorporation assays to evaluate cell proliferation in vitro. Cell apoptosis was analyzed by flow cytometry. Cell invasion was analyzed by wound healing and Transwell migration assays was ued to analyze cell migration. Finally, the biological function and mechanism of AC010883.5 in CC growth were evaluated by in vivo xenograft assay. AC010883.5 was enhanced in CC tissues and cell lines, and enhanced AC010883.5 expression accelerated CC cell proliferation, migration, and invasion and induced epithelial–mesenchymal transition in vitro and in vivo. AC010883.5 also activated the mitogen-activated protein kinase (MAPK) signaling pathway by promoting phosphorylation of extracellular signal-regulated kinase 1/2 (i.e., ERK1/2) and MAPK kinase 1/2 (i.e., MEK1/2). Blocking the MAPK signaling pathway could counteract the pro-proliferative, pro-migrative, and pro-invasive effects of AC010883.5 over-expression. We found that the lncRNA, AC010883.5, is an oncogenic molecule involved in CC tumor progression via dysregulation of the MAPK signaling pathway, implying that AC010883.5 could be a tumor progression and therapeutic response biomarker.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cervical cancer (CC) is the second most common malignancy with the highest mortality in females worldwide, ranking just after breast cancer. Globally, there are almost 527 000 new cancer cases and 265 000 patient deaths per year [1]. Despite worldwide screening and advances in early diagnosis and targeted treatments, the overall prognosis of patients with advanced CC remains poor. Therefore, it is imperative to elucidate the pathogenesis underpinning carcinogenesis and progression.
Long non-coding RNAs (lncRNAs) are RNA transcripts longer than 200 nucleotides, often polyadenylated, and devoid of evident open reading frames [2]. Accumulating evidence suggests that lncRNAs participate in CC progression. For example, HOX transcript antisense RNA (i.e., HOTAIR) gene expression is strictly controlled by the human papillomavirus (HPV) E7 protein [3], acting as a sponge to alter the miR-143-3p/BCL2 axis favoring cancer cell growth [4] and the miR-129-5p/RPL14 axis promoting cell proliferation and metastasis [5]. Further, miR-145 sponging by metastasis-associated lung adenocarcinoma transcript 1 (i.e., MALAT1) was involved in radio-resistance in radiotherapy [6] and identified as a potential therapeutic target and prognostic biomarker [7].
Our research group screened out five lncRNAs most related to the biological functions of cervical cancer through bioinformatics analysis, and determined that AC010883.5 has the strongest ability to regulate cell growth compared with other lncRNAs through cell experiments. To our knowledge, the lncRNA, homo sapiens chromosome 2 clone RP11-339H12 (AC010883.5), located on human chromosome 2p, has never been studied in CC. Our previous research on AC010883.5 was limited to cell proliferation assays and the correlation between its expression and clinical pathology. Thus, the potential function and molecular mechanism remain unclear. Our study explored the biological function of AC010883.5 to determine the underlying mechanisms in CC and provide potential therapeutic targets for improving the clinical treatment strategy.
Materials and methods
Clinical samples
We collected ten CC and paracancerous tissue pairs from patients with CC from the East Hospital affiliated with the Tongji University of Medicine between 2019 and 2020. Patients pathologically diagnosed with CC with no history of radiochemotherapy or other adjuvant therapies were included. Patients with distant metastasis or other malignant tumors or who were pregnant or lactating were excluded. The Ethics Committee of the Affiliated Tumor Hospital of Xinjiang Medical University approved the present study. All patients have signed the written informed consent forms before surgical resection. Following collection, tissue samples were snap-frozen and stored at − 80 °C before use.
Cell culture and reagents
CC-derived cell lines (SiHa, Hela, Caski, and C-33A) and a normal epithelial-derived cell line (HcerEpic) were obtained from the Institute of Cell Research, Chinese Academy of Sciences (Shanghai, China) for functional analyses. Hela and SiHa were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Procell, Wuhan, China), Caski in Roswell Park Memorial Institute medium (RPMI; Invitrogen, Carlsbad, USA), and C-33A in minimal essential medium (MEM; Invitrogen, Carlsbad, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (HyClone, Logan, UT, USA) with 5% carbon dioxide (CO2) at 37 °C. When appropriate, the cells were treated with 10 μM of PD98059 (MCE; Beverly, MA, USA).
AC010883.5 knockdown and overexpression
For in vitro experiments, to knock down AC010883.5 expression in SiHa cells, small interfering RNAs (siRNAs) targeting AC010883.5 (siRNA1-siRNA4) and a negative control (siNC) were purchased from GenePharma (Shanghai, China). The siRNA sequences were as follows: siRNA1, CUACUACCCUUCCAAAGCU; siRNA2, CUCAAGAUAACUAUUACCA; siRNA3, CAAGACACAUCCUAGAGCA; siRNA4, GAUAACUAUUACCAUCCCA. To upregulate AC010883.5 expression in C-33A cells, the AC010883.5 sequence was inserted into pcDNA3.1 vectors to construct AC010883.5 expression vectors (oeAC010883.5). Empty plasmid pcDNA3.1 was used as a negative control. For subsequent experiments, SiHa and C-33A were harvested 24 h after siRNA or plasmid transfection. AC010883.5 knockdown and overexpression efficiencies were detected by quantitative real-time polymerase chain reaction (qRT-PCR). The two most effective siRNAs were used in the functional experiments.
For in vivo experiments, short hairpin RNA (shRNA) targeting AC010883.5 generated from siRNA2 and a scrambled shRNA were cloned into pLKO.1-puro (Addgen, MA, USA). Lentiviral particles were generated in 293 T cells by co-transfecting a lentiviral plasmid and packaging plasmid (psPAX and pMD2.G, Addgene, MA, USA) using Lipofectamine 2000 (Thermo Fisher Scientific, NY, USA). After 48 h, the transfected lentiviral supernatants were filtered for further infection of SiHa cells and prepared for tumor formation in a nude mice experiment.
Cell proliferation assay
The proliferative capacity was evaluated by Cell Counting Kit-8 (CCK-8) assay (SAB, MA, USA, CP002) at 0, 24, 48, and 72 h. Cells (3 × 104/well) were seeded into 96-well plates and incubated at 37 °C in 5% CO2 and treated with CCK-8 every 24 h. The absorbance was measured at 450 nm, and a proliferation curve was plotted based on optical density values detected by a microplate reader (Perlove Medical; DNM-9602).
A 5‐Bromo-2'-deoxyuridine (BrdU) incorporation assay was performed with the Cell-Light™ BrdU Apollo®643 In Vitro Imaging Kit (RiboBio, Guangzhou, China). Cells (5 × 104/well) were harvested and treated with BrdU, fixed for 30 min and incubated using peroxidase-coupled anti-BrdU-antibody (Sigma Aldrich) for 1 h. The samples were analyzed using a Zeiss fluorescence photomicroscope (Carl Zeiss, Oberkochen, Germany) by randomly counting ten fields.
qRT-PCR
The gene mRNA expression levels were estimated by qRT-PCR. Total RNA was extracted using TRIzol reagent (1596–026, Invitrogen, Carlsbad, USA). First-strand complementary DNA was synthesized from total RNA using the SuperScript II first-strand synthesis system (Invitrogen Carlsbad, USA). The target gene primer sequences were: glyceraldehyde 3-phosphate dehydrogenase (GAPDH): F 5′-TGCACCACCAACTGCTTAGC-3′, R 5′-GGCATGGACTGTGGTCATGA-3′; AC010883.5: F 5’-AAGGCAAACTCTGCAAAGACAG-3’, R 5′-TCCAAGGAAAGGGCAAACC-3’. The relative mRNA levels were quantified using the 2−ΔΔCT method with GAPDH as an internal control.
Cell apoptosis
Annexin V and propidium iodide (PI) staining were applied using an Annexin V-FITC Apoptosis Detection kit (Beyotime Biotechnology, Shanghai, CHN, C1062). Cells (3 × 104/well) were harvested and resuspended in staining buffer then stained with 10 μL of Annexin V and 5 μL of PI solution in the dark for 15 min. Apoptotic cells were analyzed via a BD FACSCanto™ II flow cytometer (BD Biosciences, USA), after which FlowJo Software (v10.4; FlowJo LLC, Ashland, OR, USA) was used for data analysis.
Wound-healing assay
For the wound healing assay, 8 × 105 cells were seeded into 6—well plates until reaching to a density of 60%. Cells were wounded using a pipette tip and incubated in serum-free medium. Cell images were captured at 12-h intervals and photographed with an inverted microscope. Then, the wound closure percentage was measured. Scratch areas were measured using the Image J software. Wound healing percentage (%) was quantified using the percentage change in the normalized measurement area divided by the original open area according to the formula: Wound healing percentage (%) = [A (0)—A (t)/A (0)] × 100 where the area (A) at time zero (0) and the area after incubation time (t) were used to calculate the percent wound healing [8].
Transwell assay
For the migration assay, cells were suspended in a serum-free medium, then 5 × 104 cells in 200 μL of serum-free medium were seeded into the upper chamber coated with or without Matrigel. The lower chamber received 700 μL of complete medium supplemented with 10% FBS. After 24 h, the invaded or migrated cells were fixed with 50% methanol, then stained with 0.5% crystal violet solution for 30 min at room temperature. The numbers of invaded and migrated cells were evaluated via microscopy (Olympus Corporation) by randomly selecting three fields.
Western blotting
Radio-Immunoprecipitation Assay (i.e., RIPA) buffer (Beyotime Biotechnology, CA, China) supplemented with 1 nM of benzylsulfinyl fluoride was added to extract total proteins from the cells or tumor samples, followed by a bicinchoninic acid (i.e., BCA) assay (Beyotime) to quantify the protein concentrations. The proteins (30 μg) were separated using a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (i.e., SDS-PAGE) gel, then transferred to polyvinylidene fluoride membrane (EMD Millipore, Burlington, MA, USA). The membrane was blocked for 2 h with non-fat milk followed by primary antibody incubation, including E-cadherin (1:1000; Abcam; No. ab231303), N-cadherin (1:1000; Abcam; No. ab18203), ERK1/2 (1:1000; Abcam; No. ab184699), p-ERK1/2 (1:1000; Abcam; No. ab223500), MEK1/2 (1:500; Affinity; No. AF6385), p-MEK1/2 (1:500; Affinity; No. AF8035), and GAPDH (1:1000; Proteintech; #60,004–1-1G) at 4 °C overnight. The blots were washed with TBS-T and treated with horseradish peroxidase-conjugated polyclonal goat anti-rabbit immunoglobulin G (IgG; 1:1000; No. A0208) or polyclonal goat anti-mouse IgG (1:1000; Beyotime; No. A0216) for 1 h at ambient temperatures. An enhanced chemiluminescence kit (Beyotime; WBKLS0100) was used to assess the protein bands.
Mouse tumor xenograft model
Female BALB/c nude mice (5–6 weeks old, 15–20 g) from Shanghai Slark Experimental Animal Company were housed in specific pathogen-free conditions. Animals were randomized into two groups (six mice per group). SiHa cells were suspended in phosphate-buffered saline at 2 × 107/mL, then 100 μL were subcutaneously injected to the right flank of each mouse. The tumor volume was measured every three days and calculated as follows: tumor volume [mm3] = length × width [2] × 0.5. Mice were sacrificed 33 days after implantation, and the specimen was collected for weight measurements and in vitro assays. All experiments involving animals were approved by the Animal Care and Use Committee at Tongji University and followed by Guidelines for the ethical review of laboratory animal welfare People’s Republic of China National Standard GB/T 35,892–2018.
Statistical analyses
All data analysis was performed using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, USA) and SPSS version 22 (IBM Corp., Armonk, NY, USA). The data were expressed as means ± standard deviations. Continuous data were analyzed using Student’s t-test or one-way analysis of variance. p < 0.05 was considered statistically significant.
Results
AC010883.5 expression in CC tissues and cell lines
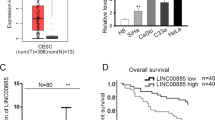
AC010883.5 expression was significantly enhanced in the cancer tissues compared with the adjacent tissues (Fig. 1A). Generally, AC010883.5 was expressed more in the CC cell lines than in the HcerEpic cell line (Fig. 1B). The function of lncRNAs is strongly linked to subcellular distribution in cells. Therefore, qRT-PCR was performed to distinguish the subcellular distribution of AC010883.5 in the SiHa and C-33A cell lines using GAPDH and U6 as cytoplasmic and nuclear references, respectively. AC010883.5 was substantially enriched in the cytoplasm (Fig. 1C). Further, AC010883.5 knockdown with siRNAs (siAC010883.5–1, siAC010883.5–2, siAC010883.5–3, siAC010883.5–4) dramatically decreased AC010883.5 expression in SiHa cells (Fig. 1D). The CCK-8 analysis also demonstrated that viable SiHa cells were remarkably decreased after silencing AC010883.5 (Fig. 1E).

The relative expression and subcellular location of AC010883.5. A. qRT-PCR analysis of AC010883.5 expression in CC and adjacent cervical tissues from ten patients. B. qRT-PCR analyses of AC010883.5 expression in four CC cell lines and one normal cell line. C. An investigation of the subcellular location of AC010883.5 in SiHa and C-33A cells by qPCR. The original magnification was 400 × . D. AC010883.5 expression in SiHa cells transfected with si-NC or one of four experimental siRNAs. E. CCK-8 analysis of AC010883.5 expression in SiHa cells transfected with si-NC or siRNA 1 to 4. The error bars represent the mean ± SD of at least three experiments. N = 3. All qPCR results were normalized to GAPDH. *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: qRT-PCR, quantitative real-time polymerase chain reaction; CC, cervical cancer; si-NC, negative control siRNA; siRNA, small interfering RNA; CCK-8, Cell Counting Kit‐8; DAPI, 4′,6‐diamidino‐2‐phenylindole
Silenced AC010883.5 suppressed SiHa cell proliferation, induced apoptosis, and repressed cell migration and invasion
To discriminate the effects of AC010883.5 on cervical cells, we first generated cells with stably knocked down AC010883.5 (transfection with siAC010883.5–2 and siAC010883.5–3) and SiHa control cells (transfection with siNC, an empty control); we evaluated the efficiency by qRT-PCR (Fig. 2A).

Silencing AC010883.5 suppressed proliferation and progression in SiHa cells. A. qRT-PCR analysis of AC010883.5 expression in SiHa cells transfected with shRNA-2 and shRNA-3. B. CCK-8 analysis of viable SiHa cells under AC010883.5 treatment (silenced vs. control). C. BrdU incorporation analysis of proliferative SiHa cells under AC010883.5 treatment (silenced vs. control). D. Cell cycle quantification of SiHa cells using flow cytometry. E. Flow cytometry analysis of apoptotic SiHa cells under AC010883.5 treatment (silenced vs. control). F. Transwell migration analysis of migrative SiHa cells under AC010883.5 treatment (silenced vs. control). G. Transwell invasion analysis of invasive SiHa cells under AC010883.5 treatment (silenced vs. control); H. Wound healing analysis of migrative SiHa cells under AC010883.5 treatment (silenced vs. control). I. Western blots of E-cadherin and N-cadherin in SiHa cells under AC010883.5 treatment (silenced vs. control). N = 3; Abbreviations: qRT-PCR, quantitative real-time polymerase chain reaction; shRNA, short hairpin RNA; CCK-8, Cell Counting Kit‐8; BrdU, 5‐Bromo-2'-deoxyuridine. *P < 0.05, **P < 0.01, and ***P < 0.001
In SiHa cells, silencing AC010883.5 suppressed cell survival (CCK-8 assay; Fig. 2B) and significantly restrained cell proliferation (BrdU incorporation assay; Fig. 2C). Silencing AC010883.5 also arrested the cell cycle at the G1 phase (Fig. 2D) and induced apoptosis (flow cytometry, Fig. 2E). Furthermore, silencing AC010883.5 greatly inhibited cells migration (Transwell migration and wound healing assays; Fig. 2F, H) and remarkably slowed cell invasion (Transwell invasion assay; Fig. 2G).
Epithelial–mesenchymal transition (EMT) is one of the most important indicators of cancer metastasis. Thus, we explored the correlation between AC010883.5 and EMT. Silencing AC010883.5 decreased the N-cadherin protein level and enhanced the E-cadherin protein level in SiHa cells compared to control cells (Fig. 2I), indicating that AC010883.5 could be involved in EMT.
Overexpressing AC010883.5 accelerated C-33A cell proliferation, inhibited apoptosis, and promoted cell migration and invasion
We also constructed stably overexpressed AC010883.5 cells (transfection with an AC010883.5 overexpression vector) and control C-33A cells (transfection with an empty vector); we evaluated the efficiency by qRT-PCR (Fig. 3A).

Overexpressed AC010883.5 promoted proliferation and progression in C-33A cells. A. qRT-PCR analyses of AC010883.5 expression in C-33A cells transfected with the oeAC010883.5 vector. B. CCK-8 analysis of viable C-33A cells under AC010883.5 treatment (overexpressed vs. control). C. BrdU incorporation analysis of proliferative C-33A cells under AC010883.5 treatment (overexpressed vs. control). D. Quantification of flow cytometry results in C33-A cells under AC010883.5 treatment (overexpressed vs. control). E. Flow cytometry analysis of apoptotic C-33A cells under AC010883.5 treatment (overexpressed vs. control). F. Transwell migration analysis of migrative C-33A cells under AC010883.5 treatment (overexpressed vs. control). G. Transwell invasion analysis of invasive C-33A cells under AC010883.5 treatment (overexpressed vs. control). H. Wound healing analysis of migrative C-33A cells under AC010883.5 treatment (overexpressed vs. control). I. Western blots of E-cadherin and N-cadherin in C-33A cells under AC010883.5 treatment (overexpressed vs. control). Abbreviations: qRT-PCR, quantitative real-time polymerase chain reaction; CCK-8, Cell Counting Kit‐8; BrdU, 5‐Bromo-2'-deoxyuridine
In C-33A cells, overexpressing AC010883.5 promoted cell survival (CCK-8 assay; Fig. 3B) and significantly improved cell proliferation (BrdU incorporation assay; Fig. 3C). Overexpressing AC010883.5 also induced cell cycle transitions; the ratio of cells in the G1 phase decreased while the ratio in the S phase increased (Fig. 3D). Overexpressed AC010883.5 repressed cell apoptosis (flow cytometry, Fig. 3E), increased cell migration (wound healing assay; Fig. 3F, H), and markedly accelerated cell invasion (Transwell migration assay; Fig. 3G). Regarding the EMT analysis, overexpressing AC010883.5 enhanced the N-cadherin protein level and decreased the E-cadherin protein level in C-33A cells (Fig. 3I).
AC010883.5 activates the mitogen-activated protein kinase (MAPK) signaling pathway
The MAPK signaling pathway is of known importance in CC progression [9]. Therefore, we investigated the role of this pathway in cell phenotype adjustments under the influence of AC010883.5. We evaluated the phosphorylation levels of MAPK kinase (MEK; p-MEK) and extracellular signal-regulated kinase (ERK) 1/2 (p-ERK1/2), key biomarkers of MAPK signaling pathway activation, by western blot. p-MEK1/2 and p-ERK1/2 significantly increased in AC010883.5 overexpressing cells and decreased in AC010883.5 knockdown cells (Fig. 4A). Therefore, we hypothesized that the MAPK pathway was involved in AC010883.5 function in CC cells.

Overexpressed AC010883.5 activates the MAPK signaling pathway and promoted C33A cell progression. A. Western blots of ERK1/2, p-ERK1/2, MEK1/2, and p-MEK 1/2 protein in SiHa cells under AC010883.5 treatment (silenced vs. control) and C-33A cells under AC010883.5 (overexpressed vs. control). B. CCK-8 analysis of viable C-33A cells in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). C. BrdU incorporation analysis of proliferative C-33A cells in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). D. Quantification of flow cytometry results in C33-A cells under AC010883.5 treatment (overexpressed vs. control). E. Flow cytometry analysis apoptotic C-33A cell in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). F. Transwell migration analysis of migrative C-33A cell in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). G. Transwell invasive analysis of invasive C-33A cell in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). H. Wound healing analysis of migrative C-33A cells in vehicle or PD98059-treated C-33A cells in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). I. Western blots of MAPK signal molecules in a vehicle or PD98059-treated C-33A cells under AC010883.5 treatment (overexpressed vs. control). N = 3; *P < 0.05, **P < 0.01, and ***P < 0.001. a: Vector; b: oeAC010883.5; c: PD98059 + Vector; d: PD98059 + oeAC010883.5; Abbreviations: MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1/2; p-ERK1/2, phosphorylated ERK1/2; MEK1/2, MAPK kinase 1/2; p-MEK1/2, phosphorylated MEK1/2; CCK-8, Cell Counting Kit‐8; BrdU, 5‐Bromo-2'-deoxyuridine
The MAPK pathway inhibitor, PD98059, was used to block the activation signal in AC010883.5 overexpressed C-33A cells to investigate whether AC010883.5 promotes CC progression through the MAPK signaling pathway. Proliferation and cell cycle analyses revealed that PD98059 suppressed AC010883.5-mediated cell proliferation and cell cycle transition and reversed apoptosis inhibition in C-33A cells (Fig. 4B–E). Consistently, PD98059 suppressed AC010883.5-mediated cell migration and invasion in C-33A cells (wound healing and Transwell migration assays; Fig. 4F–H). Moreover, PD98059 reversed the effects of AC010883.5 overexpression on the ERK1/2 and MEK1/2 phosphorylation (western blot; Fig. 4I). These results suggest that AC010883.5 promotes proliferation, invasion, and migration of CC cells by activating the MAPK pathway.
AC010883.5 knockdown inhibited CC tumor growth in vivo
To explore the function of AC010883.5 in vivo, nude mice were used to establish a xenograft mouse model by subcutaneously injecting control or stably knocking down AC010883.5 SiHa cells. The tumor sizes and weights of the shAC010883.5 mice were significantly less than the control mice (Fig. 5A–C). As expected, the shAC010883.5 mice had lower AC010883.5 expression than the control mice (Fig. 5D).

Silencing AC010883.5 inhibited tumor cell proliferation and progression by inactivating the MAPK signaling pathway. A. Tumor xenografts images on the day of sacrifice (day 33; N = 6). B. Dissected tumor tissue growth with AC010883.5 pLVX-shRNAs-puro SiHa cells. C. Dissected tumor tissue weight with AC010883.5 pLVX-shRNAs-puro SiHa cells. D. AC010883.5 expression in SiHa cells transfected with pLVX-shRNAs-puro in tumor tissue. E–G. PCNA and Ki67 expression in C-33A cells were detected by immunohistochemistry staining of the tumor tissue. H. ERK1/2, p-ERK, MEK1/2, and p-MEK protein levels with pLVX-shRNAs-puro SiHa cells by western blot; N = 6; *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: MAPK, mitogen-activated protein kinase; PCNA, proliferating cell nuclear antigen; ERK1/2, extracellular signal-regulated kinase 1/2; p-ERK1/2, phosphorylated ERK1/2; MEK1/2, MAPK kinase 1/2; p-MEK1/2, phosphorylated MEK1/2
Next, we evaluated proliferation biomarkers in the xenograft tumors. Proliferating cell nuclear antigen (PCNA) and Ki67 expression were significantly down-regulated in the shAC010883.5 mice compared with the control mice (Fig. 5E-G), indicating that AC010883.5 downregulation suppressed tumor growth in vivo, consistent with in vitro.
More importantly, AC010883.5 knockdown downregulated the ERK1/2 and MEK1/2 phosphorylation levels in xenograft tumors (Fig. 5H). Collectively, these data demonstrate that AC010883.5 modulates CC growth via the MAPK signaling pathway.
Discussion
CC is a lethal malignancy posing a serious threat to the lives of females [10]. Despite advances in treatment and screening, the resistance to therapy and high recurrence rates pose challenges to physicians and patients [11, 12]. Therefore, finding new biomarkers to CC improve treatment strategies and prognosis predictions is crucial.
In recent decades, the biological functions of lncRNAs have been widely investigated [13, 14]. Abnormal expression of lincRNAs are considered to be key factors in the tumorigenesis of cancers [15, 16]. Additionally, an increasing number of studies implicate lncRNAs in CC. Several lncRNAs, including HOTAIR [17], H19 [18], mucosa-associated lymphoid tissue 1 (i.e., MALT1) [19], growth arrest-specific 5 (i.e., GAS5) [20], cervical carcinoma high expressed 1 (i.e., CCHE1) [17], and Pvt1 oncogene (i.e., PVT1) [21], are crucial in tumorigenesis, invasion, metastasis, and radio-resistance [22,23,24]. HOTAIR is significantly associated with CC progression, increasing proliferation, migration, invasion, and EMT by increasing enhancer of zeste homolog 2 (i.e., EZH2) and trimethylation of histone H3 at lysine 27 (i.e., H3K27me3) binding of the antagonist naked cuticle homolog 1 (i.e., NKD1) [3]. Differentiation antagonizing non-protein coding RNA (i.e., DANCR) has also been significantly implicated in CC development, augmenting cell adhesion, viability, invasiveness, and migration by down-regulating p‐GSK3β and β‐catenin expression and suppressing Wnt/β‐catenin pathway activation [25]. Further, H19, controlled by the HPV16 E6 protein, remarkably correlated with CC progression by increasing cell migration, invasion, and proliferation through working as a molecular sponge for miR-138-5p in epithelial cells [26]. These results highlight that lncRNA deregulation occurs in carcinogenesis and progression and exerts both oncogenic and tumor inhibition functions related to CC. AC010883.5 is located at chromosome 2:43.46, is 216626 bp long [27], and is involved in inflammatory bowel disease [28]. In this study, we reported the function and localization of AC010883.5 in cells for the first time. Our study showed that AC010883.5 expression was upregulated in CC tissues and cell lines. We examined the expression of AC010883.5 in 4 different cervical cancer cell lines, all of which were elevated compared with cervical epithelial cells (HcerEpic). However, the level of AC010883.5 was higher in SiHa and Caski cells than in HeLa and C33A, which was caused that cell lines derived from different individuals have different genetic backgrounds. In addition, we discovered that AC010883.5 was substantially enriched in the cytoplasm. AC010883.5 depletion suppressed CC cell proliferation, migration, invasion, and epithelial-to-mesenchymal transition (EMT).
Furthermore, we explored the molecular mechanisms by which knockdown of AC010883.5 inhibited CC cell growth in vitro. The MAPK signaling pathway modulates the cell’s response to proliferation, migration, apoptosis, and stress. Further, ERK1/2 and MEK1/2 expression and hyperactivation play an essential role in the survival and development of tumor cell proliferation, migration, invasion, and EMT [29, 30]. An aberrant MAPK signaling pathway is a critical regulator of carcinogenesis and the development of multiple tumor types [31]. It has been recognized that lncRNAs can influence cancer occurrence and development via the MAPK pathway [32, 33]. Liao et al. discovered that lncRNA-CCHE1 promoted the proliferation, migration, and invasion in non-small cell lung cancer (NSCLC) cell line through the ERK/MAPK pathway [34]. Zhang et al. found that GINS2 also inhibits cell activity by interfering with the MAPK/ERK pathway and induces cell cycle arrest, thus promoting apoptosis of pancreatic cancer cells [35]. Moreover, Du et al. demonstrated that inhibition of SLC25A22 by the MAPK/ERK pathway could promote mitochondrial apoptosis, thus inhibiting the growth and proliferation gallbladder carcinoma cells [36]. Therefore, we examined the effects of AC010883.5 knockdown on the ERK/MAPK pathway. In the present study, we demonstrated that upregulation of AC010883.5 promote the growth and invasion of tumor by activating this signaling cascade. It was observed that overexpression of AC010883.5 significantly increased the expression of phosphorylated ERK1/2. However, no detectable changes in the expression of total ERK1/2 protein were observed. To further verify the role of signaling activation and inactivation in AC010883.5-aberrant expressed CC, the addition of PD98059 was used to pretreat cells. Results showed that inhibition of MAPK signaling suppressed AC010883.5 high expression-induced levels of phosphorylated ERK1/2 and MEK1/2. Thus, we concluded that AC010883.5 promoted cell proliferation, invasion, migration, and epithelial-to-mesenchymal transition in cervical cancer by activating the MAPK signaling pathway.
This study had several limitations, including the low number of clinical samples, the lack of a reliable clinical characteristics analysis, and insufficient research on the regulatory mechanism. These limitations might contribute to unilateral results. Future research should include a larger sample size and involve more complex regulation processing to validate our results.
Conclusions
In summary, in this study, we first found that AC010883.5 is markedly enhanced in CC tissues and cell lines. Further, it was involved in cell proliferation, invasiveness, and migration and reduced apoptosis, accelerating tumor growth and EMT in vitro and in vivo. We also found that enhanced AC010883.5 expression induced an ERK1/2 and MEK1/2 phosphorylation increase and activated the MAPK signaling pathway to promote tumor progression. The results demonstrate that AC010883.5 is a possible CC oncogene, and therapies targeting AC010883.5 may be of interest.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Canfell K, Kim JJ, Brisson M, Keane A, Simms KT, Caruana M, et al. Mortality impact of achieving WHO cervical cancer elimination targets: a comparative modelling analysis in 78 low-income and lower-middle-income countries. Lancet. 2020;395(10224):591–603.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Sharma S, Mandal P, Sadhukhan T, Roy Chowdhury R, RanjanMondal N, Chakravarty B, et al. Bridging Links between long noncoding RNA HOTAIR and HPV oncoprotein E7 in cervical cancer pathogenesis. Sci Rep. 2015;5:11724.
Liu M, Jia J, Wang X, Liu Y, Wang C, Fan R. Long non-coding RNA HOTAIR promotes cervical cancer progression through regulating BCL2 via targeting miR-143-3p. Cancer Biol Ther. 2018;19(5):391–9.
Sun N, Zhang W, Liu J, Yang X, Chu Q. Propofol Inhibits the Progression of Cervical Cancer by Regulating HOTAIR/miR-129-5p/RPL14 Axis. Onco Targets Ther. 2021;14:551–64.
Lu H, He Y, Lin L, Qi Z, Ma L, Li L, et al. Long non-coding RNA MALAT1 modulates radiosensitivity of HR-HPV+ cervical cancer via sponging miR-145. Tumour Biol. 2016;37(2):1683–91.
Chen Q, Hu L, Huang D, Chen K, Qiu X, Qiu B. Six-lncRNA immune prognostic signature for cervical cancer. Front Genet. 2020;11:533628.
Treloar KK, Simpson MJ. Sensitivity of edge detection methods for quantifying cell migration assays. PLoS One. 2013;8(6):e67389.
Sun Q, Liang Y, Zhang T, Wang K, Yang X. ER-alpha36 mediates estrogen-stimulated MAPK/ERK activation and regulates migration, invasion, proliferation in cervical cancer cells. Biochem Biophys Res Commun. 2017;487(3):625–32.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Adiga D, Eswaran S, Pandey D, Sharan K, Kabekkodu SP. Molecular landscape of recurrent cervical cancer. Crit Rev Oncol Hematol. 2021;157:103178.
Lima J, Ali Z, Banerjee S. Immunotherapy and systemic therapy in metastatic/recurrent endometrial and cervical cancers. Clin Oncol (R Coll Radiol). 2021;33(9):608–15.
Jiang L, Wang R, Fang L, Ge X, Chen L, Zhou M, et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics. 2019;9(9):2460–74.
Qin G, Tu X, Li H, Cao P, Chen X, Song J, et al. Long Noncoding RNA p53-stabilizing and activating RNA promotes p53 signaling by inhibiting heterogeneous nuclear ribonucleoprotein K deSUMOylation and suppresses hepatocellular carcinoma. Hepatology. 2020;71(1):112–29.
He F, Song Z, Chen H, Chen Z, Yang P, Li W, et al. Long noncoding RNA PVT1–214 promotes proliferation and invasion of colorectal cancer by stabilizing Lin28 and interacting with miR-128. Oncogene. 2019;38(2):164–79.
Kong F, Deng X, Kong X, Du Y, Li L, Zhu H, et al. ZFPM2-AS1, a novel lncRNA, attenuates the p53 pathway and promotes gastric carcinogenesis by stabilizing MIF. Oncogene. 2018;37(45):5982–96.
Ghafouri-Fard S, Azimi T, Taheri M. Cervical carcinoma high expressed 1 (CCHE1): An oncogenic lncRNA in diverse neoplasms. Biomed Pharmacother. 2021;142:112003.
Roychowdhury A, Samadder S, Das P, Mazumder DI, Chatterjee A, Addya S, et al. Deregulation of H19 is associated with cervical carcinoma. Genomics. 2020;112(1):961–70.
Chiarini A, Marconi M, Pacchiana R, Dal Pra I, Wu J, Armato U. Role-shifting PKCzeta fosters its own proapoptotic destruction by complexing with Bcl10 at the nuclear envelope of human cervical carcinoma cells: a proteomic and biochemical study. J Proteome Res. 2012;11(8):3996–4012.
Lambrou GI, Hatziagapiou K, Zaravinos A. The Non-Coding RNA GAS5 and Its Role in Tumor Therapy-Induced Resistance. Int J Mol Sci. 2020;21(20):7633.
Wang H, Wei M, Kang Y, Xing J, Zhao Y. Circular RNA circ_PVT1 induces epithelial-mesenchymal transition to promote metastasis of cervical cancer. Aging (Albany NY). 2020;12(20):20139–51.
Dong J, Su M, Chang W, Zhang K, Wu S, Xu T. Long non-coding RNAs on the stage of cervical cancer (Review). Oncol Rep. 2017;38(4):1923–31.
Tornesello ML, Faraonio R, Buonaguro L, Annunziata C, Starita N, Cerasuolo A, et al. The role of microRNAs, long non-coding RNAs, and circular RNAs in cervical cancer. Front Oncol. 2020;10:150.
Caceres-Duran MA, Ribeiro-Dos-Santos A, Vidal AF. Roles and mechanisms of the long noncoding RNAs in cervical cancer. Int J Mol Sci. 2020;21(24):9742.
Tian W, Lei N, Guo R, Yuan Z, Chang L. Long non-coding RNA DANCR promotes cervical cancer growth via activation of the Wnt/beta-catenin signaling pathway. Cancer Cell Int. 2020;20:61.
Ou L, Wang D, Zhang H, Yu Q, Hua F. Decreased expression of miR-138-5p by lncRNA H19 in cervical cancer promotes tumor proliferation. Oncol Res. 2018;26(3):401–10.
Chang SW, McDonough CW, Gong Y, Johnson TA, Tsunoda T, Gamazon ER, et al. Genome-wide association study identifies pharmacogenomic loci linked with specific antihypertensive drug treatment and new-onset diabetes. Pharmacogenomics J. 2018;18(1):106–12.
Zhang Y, Tian L, Sleiman P, Ghosh S, Hakonarson H, International IBDGC. Bayesian analysis of genome-wide inflammatory bowel disease data sets reveals new risk loci. Eur J Hum Genet. 2018;26(2):265–74.
Olea-Flores M, Zuniga-Eulogio MD, Mendoza-Catalan MA, Rodriguez-Ruiz HA, Castaneda-Saucedo E, Ortuno-Pineda C, et al. Extracellular-signal regulated kinase: a central molecule driving epithelial-mesenchymal transition in cancer. Int J Mol Sci. 2019;20(12):2885.
Asl ER, Amini M, Najafi S, Mansoori B, Mokhtarzadeh A, Mohammadi A, et al. Interplay between MAPK/ERK signaling pathway and MicroRNAs: A crucial mechanism regulating cancer cell metabolism and tumor progression. Life Sci. 2021;278:119499.
Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
Peng W, Fan H. Long noncoding RNA CCHE1 indicates a poor prognosis of hepatocellular carcinoma and promotes carcinogenesis via activation of the ERK/MAPK pathway. Biomed Pharmacother. 2016;83:450–5.
Han Y, Wu Z, Wu T, Huang Y, Cheng Z, Li X, et al. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016;7(3):e2123.
Liao Y, Cheng S, Xiang J, Luo C. lncRNA CCHE1 increased proliferation, metastasis and invasion of non-small lung cancer cells and predicted poor survival in non-small lung cancer patients. Eur Rev Med Pharmacol Sci. 2018;22(6):1686–92.
Zhang M, He S, Ma X, Ye Y, Wang G, Zhuang J, et al. GINS2 affects cell viability, cell apoptosis, and cell cycle progression of pancreatic cancer cells via MAPK/ERK pathway. J Cancer. 2020;11(16):4662–70.
Du PA-O, Liang H, Fu X, Wu P, Wang C, Chen H, et al. SLC25A22 promotes proliferation and metastasis by activating MAPK/ERK pathway in gallbladder cancer. Cancer Cell Int. 2019;19:33.
Acknowledgements
No applicable.
Funding
This study was funded by the National Natural Science Foundation of China (grant number 82260587), the Natural Science Foundation of Jiangxi Province (grant number 20202ACB206006), Science Foundation of Shanghai Municipal Health Commission (grant number 201840166) and The Top-level Clinical Discipline Project of Shanghai Pudong (grant number PWYgf2021-07).
Author information
Authors and Affiliations
Contributions
Qiyu Gan, Xia Huang and Wenrong Zhao conceived and designed the experiments, authored, approved the final manuscript. Yan Xu and Xia Huang analyzed the data and prepared figures. Hui Liu provided suggestions on the study design and analyzed the data. Xiaohua Zhang reviewed the manuscript, Jingxin Cheng and Rui Chen designed and supervised the research, Jingxin Cheng obtained funding. Rui Chen gave intelligent support. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study procedures for human participates were in accordance with the ethical standards of the Ethics Committee of the Affiliated Shanghai East Hospital of Tongji University School of Medicine. All patients understood the aims of specimen collection and signed written form informed consent. The study was reviewed and approved by the ethics committee of Ji an Hospital East Hospital Affiliated to Tongji University of Medicine and the Ethics Committee of the Affiliated Shanghai East Hospital of Tongji University School of Medicine. The animal studies were reviewed and approved by the Tongji University Animal Ethics Committee. The study was carried out in compliance with the ARRIVE guidelines, and all procedures for animal experiments followed the ethical standards. All experiments involving animals were approved by the Animal Care and Use Committee at Tongji University and followed by Guidelines for the ethical review of laboratory animal welfare People’s Republic of China National Standard GB/T 35892–2018.
Consent for publication
No applicable.
Competing interests
The authors have no competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gan, Q., Huang, X., Zhao, W. et al. AC010883.5 promotes cell proliferation, invasion, migration, and epithelial-to-mesenchymal transition in cervical cancer by modulating the MAPK signaling pathway. BMC Cancer 23, 364 (2023). https://doi.org/10.1186/s12885-023-10825-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-023-10825-2