
Abstract
Background
Oral microbiota comprises polymicrobial communities shaped by mutualistic coevolution with the host, contributing to homeostasis and regulating immune function. Nevertheless, dysbiosis of oral bacterial communities is associated with a number of clinical symptoms that ranges from infections to oral cancer. Peri-implant diseases are biofilm-associated inflammatory conditions affecting the soft and hard tissues around dental implants. Characterization and identification of the biofilm community are essential for the understanding of the pathophysiology of such diseases. For that sampling methods should be representative of the biofilm communities Therefore, there is a need to know the effect of different sampling strategies on the biofilm characterization by next generation sequencing.
Methods
With the aim of selecting an appropriate microbiome sampling procedure for periimplant biofilms, next generation sequencing was used for characterizing the bacterial communities obtained by three different sampling strategies two months after transepithelial abutment placement: adjacent periodontal crevicular fluid (ToCF), crevicular fluid from transepithelial abutment (TACF) and transepithelial abutment (TA).
Results
Significant differences in multiple alpha diversity indices were detected at both the OTU and the genus level between different sampling procedures. Differentially abundant taxa were detected between sample collection strategies, including peri-implant health and disease related taxa. At the community level significant differences were also detected between TACF and TA and also between TA and ToCF. Moreover, differential network properties and association patterns were identified.
Conclusions
The selection of sample collection strategy can significantly affect the community composition and structure.
Trial registration
This research is part of a randomized clinical trial that was designed to assess the effect of transepithelial abutment surface on the biofilm formation. The trial was registered at Trial Registration ClinicalTrials.gov under the number NCT03554876.
Similar content being viewed by others


Background
Peri-implant health is defined by the absence of erythema, redness, bleeding on probing, swelling and suppuration around dental implants. Despite high implant survival rates, different biological complications can affect osseointegrated implants, including peri-implant mucositis and peri-implantitis [58]. Peri-implantitis is an oral inflammatory process that affects the surrounding tissues of osseointegrated implants and results in the loss of supporting bone and destruction of soft tissues [99]. According to Daubert et al. [29] and Konstantinidis et al. [59], the prevalence of peri-implant disease range between 13 and 25%, leading to a significant increase of patient morbidity, economic burden and eventual implant loss [5, 32]. As is the case for periodontal diseases [67], the primary etiological factor in the development of peri-implant diseases is the biofilm, which is a complex synthropic microbial community consisting of adherent cells embedded within a matrix composed of extracellular polymeric substances [46]. However, some discrepancies have been identified in the biofilms from peri-implantitis and periodontitis sites [8, 79].
Approximately 700 species of Prokaryota have been identified in the oral cavity, predominantly ascribed to 12 phyla: Firmicutes, Fusobacteria, Proteobacteria, Actinobacteria, Bacteroidetes, Chlamydiae, Chloroflexi, Spirochaetes, SR1, Synergistetes, Saccharibacteria (TM7) and Gracilibacteria [93]. Specifically, the mouth contains distinct niches with dynamic microbial communities, including saliva, gingiva, the hard and soft tissues of teeth, the tonsil, the gingival sulcus, the throat, the hard and soft palates and the buccal mucosa [31], and eventually dental implants [23]. These surfaces—whose differing chemistry, topography and stability provide different habitats for microorganisms—are colonized preferentially by different bacteria via surface-attachments, movements and complex interactions, resulting in spatial compositional variability [72]. Furthermore, oral bacterial communities exist in multiplex dynamic equilibrium states, with large and rapid changes in composition and activity in a temporal dimension in response to environmental conditions [44]. The microbial communities are in symbiosis with the host shaped by co-evolution, contributing to digestion and homeostasis, neurological signaling, regulating immune and endocrine functions, modifying metabolism and eliminating toxins [37, 41]. Nevertheless, under certain conditions, several pathogenic strains that are usually dominated by commensal bacteria can proliferate [84], and even some commensal bacteria can transit to a pathogenic lifestyle via complex changes involving gene expression patterns, the core genome and the pan-genome [70, 90, 115].
Several studies aiming to compare oral bacterial communities under health and disease conditions have been conducted [14, 38, 56], some of them focused on characterizing peri-implant and periodontal microbiota and its pathological changes [36]. The bacterial profile associated with peri-implant disease was reviewed by Pérez-Chaparro et al. [85], Rakic et al. [94], Sahrmann et al. [98], Butera et al. [15], Gazil et al. [43] and Rodríguez-Archilla and Palma-Casiano [96]. These authors revealed that the core peri-implantitis microbiome is enriched in periodontal-inflammation related taxa, including Fusobacterium nucleatum, Parvimonas micra and Aggregatibacter actinomycetemcomitans, Prevotella intermedia, Prevotella nigrescens, Treponema denticola, Tannerella forsythia, Campylobacter rectus and Porphyromonas gingivalis. According to most published research findings, oral bacterial communities from healthy implants, peri-implantitis and periodontitis sites show contrasting diversity and composition [28, 43, 105, 119]. However, conclusions regarding the role of individual taxa in oral pathogenesis are difficult to draw due to the presence of confounding factors, variability in experimental designs, interindividual variability and complex ecological communities. Furthermore, bacterial actions are often secondary to immunologic imbalance [4, 75].
Due to the irreversible nature of peri-implantitis, prognosis-based early therapeutic intervention is the best strategy to arrest the progression of the disease and prevent implant failure [114]. Thus, a wide range of early diagnosis methods have been suggested [19, 22, 95]. In this sense, molecular techniques—particularly metagenomic Next Generation Sequencing tests (mNGS)—are powerful approaches for the characterization of microbial communities in the oral cavity, and also for the detection and surveillance of obligate or facultative oral pathogens associated with peri-implantitis [50, 103]. Considering the high variability in microbiota composition between different oral microbial habitats at multiple scales, the selection of appropriate sampling methods is a key aspect of implant disease risk assessment [6]. Sample collection via direct removal of transepithelial abutment and bacterial DNA isolation from transepithelial abutment surface could be considered the most representative sampling strategy, but this is a destructive method and the replacement of the transepithelial abutment with another is required (thus making sampling harder, increasing the economic burden and preventing the possibility of long-term monitoring of the bacterial community in that particular transepithelial abutment).
Crevicular fluid collection via sterile paper points (either from an adjacent tooth or from the studied transepithelial abutment) was initially regarded as a promising approach, since it is nondestructive, fast and easy to carry out, and allows long-term sampling at different time points. The question is, could these alternative sampling strategies be considered representative of bacterial communities formed on transepithelial abutment surfaces? Are microbial communities of samples collected using these methods significantly different than those obtained by sampling directly transepithelial abutments? In what respect and to what level? Thus, the aim of this preliminary research was to select the most appropriate sampling approach to characterize bacterial biofilms from transepithelial abutment surfaces. For that, three sampling strategies were compared: crevicular fluid from an adjacent tooth (ToCF), crevicular fluid from transepithelial abutment (TACF) and transepithelial abutment (TA), with a particular focus on peri-implant or periodontal health and disease related taxa.
Methods
This research is part of a randomized clinical trial that was designed to assess the effect of transepithelial abutment surface on the biofilm formation. The trial was registered at Trial Registration ClinicalTrials.gov under the number NCT03554876. The study protocol and informed consent, in full accordance with the ethical principles of the Declaration of Helsinki of 1975, as revisited in 2000, were approved by the ethical committee of investigation with medicines of the Basque Country (FIBEA-06-EC/17/Multi-Im). This study constitutes a partial analysis of the results of this clinical trial aiming to assess the effect of sampling strategies on metagenomic outcomes. Additional information regarding the experimental design is provided as supplementary data (Supplementary Material 1). For the analysis of the effect of sampling strategy, transepithelial abutments with a machined surface were included. Patient selection for this randomized controlled clinical trial was based on the following criteria:
-
Inclusion criteria
-
Patients with an age ≥ 18 years.
-
Need for the placement of at least 3 dental implants.
-
Complete mouth plaque index ≤ 20% and absence of active periodontal disease.
-
Bleeding index ≤ 30%.
-
Pocket probing depth at the adjacent teeth < 4 mm.
-
No use of antibiotics in the last 6 months.
-
Nonsmoker.
-
Possibility of attending all the planned visits.
-
Signing of informed consent.
-
-
Exclusion criteria
-
Has severe hematological disease.
-
Has received or receiving in the last 30 days at least one of the following treatments: radiotherapy, chemotherapy, Immunosuppressive therapies, systemic corticoids, and anticoagulants.
-
Presence of malignancy, hemangioma or angioma at the site where dental implants will be placed.
-
Patients receiving bisphosphonates (oral or systemic).
-
Presence of metabolic osseous disease.
-
Presence of diseases that affect the oral mucosa.
-
Presence of diabetes mellites.
-
Severe parafunctional habits and/or temporomandibular joint disorders.
-
Pregnancy or breast-feeding.
-
Physical or mental disability to maintain good oral hygiene.
-
Participating in other study.
-
Other disabilities to participate in the study.
-
Clinical procedure
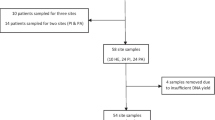
A total of 12 patients received professional oral hygiene and instructed how to maintain a good oral hygiene. After implant insertion, transepithelial abutments with machined surface were connected to the implants. Two months later, microbiome samples were acquired using different strategies. Sterile paper points size 30 (Maillefer, Ballaigues, Switzerland) were utilized to collect periodontal crevicular fluid from adjacent teeth (ToCF) and peri-implant crevicular fluid (TACF). These samples were collected from at least one adjacent healthy tooth and also from each previously connected transepithelial abutment with machined surface. After crevicular fluid collection, transepithelial abutments (TA) were removed for processing. The collected samples were stored at -80 °C until bacterial DNA isolation.
DNA extraction
Total microbial metagenomic DNA from each sample was extracted using the DNeasy PowerBiofilm DNA isolation kit (Qiagen, Germany) following the manufacturer’s instructions. The strips were homogenized (1 cycle at 6400 rpm for 30 s) with a Precellys 24 Tissue Homogenizer (Bertin Technologies, France) and the implants were homogenized with an IKA MS 3 digital vortex (IKA, Germany) for 10 min at 2250 rpm. DNA quantification and quality control was performed using a Nanodrop 8000 (Thermo Fisher Scientific, MA, USA) and a Qubit fluorometer (Thermo Fisher Scientific, MA, USA). Extracted DNA was kept frozen at − 30 °C until library preparation.
Library preparation and sequencing
16S rRNA library preparation workflow for MiSeq sequencing platform was performed as suggested by Illumina. The specific primers for the 16S rRNA gene (v3- v4 region) were selected from Klindworth et al. [57] and combined with Illumina adapter overhang nucleotide sequences to obtain a single amplicon of approximately ~ 460 bp. After PCR product purification, dual index barcodes and Illumina sequencing adapters were attached using the Nextera XT v2 index kit (Illumina, CA, USA) as a previous step to pooling, libraries were quantified by LabChip GX touch HT nucleic acid analyzer together with DNA 5 K/RNA/CZE chip (PerkinElmer, MA, USA) and diluted for an estimated sequencing depth of ~ 100.000 reads per sample. finally, pooled libraries were denatured with NaOH and diluted with hybridization buffer (library loading concentration = 6 pm) before MiSeq (llumina, CA, USA) sequencing. PhiX was included in each run to serve as an internal control for these low diversity libraries. Paired-end sequencing (2 × 300 bp) was performed using MiSeq v3 reagent kits (600 cycles) (Illumina, CA, USA).
Data processing
Secondary analysis was performed on BaseSpace using the 16S metagenomics application (Illumina, CA, USA). After assembling, full-length sequences from paired ends were referenced against the Illumina-curated version of Greengenes Consortium Database. The classification step is based on ClassifyReads, a high-performance implementation of the Ribosomal Database Project (RDP) Classifier [112].
Statistical analyses
Statistical analyses applied to explore the structure of bacterial communities with respect to their diversity, composition and bacterial association patterns across sampling methods and differential taxa abundance were performed in R [92]. Graphical data analysis was performed via ggplot2 [113] and fantaxtic R packages for data visualization [110].
Alpha diversity
In order to summarize the structure of the observed bacterial communities, a set of common alpha diversity metrics in mNGS data were computed for each sample at both the species and the genus level: observed richness, Chao index [20, 21], Abundance Coverage Estimator (ACE) [25, 77], Shannon index [101], Simpson indices [54] and Fisher’s alpha index [39] using vegan [78], phyloseq [73] andPMCMRplus [88] R packages. Generalized linear mixed-effects models were constructed and ANOVA tests were computed using lme4 to analyze the effect of sampling method on alpha diversity indices [7]. Assumptions underlying parametric in parametric statistics were checked in model residuals through visual inspection (QQ Plots and density distributions) and also by significance tests (Shapiro-Wilk and Levene’s test for assessing normality and homoscedasticity). Multiple comparisons were performed via Bonferroni corrected post hoc tests in the multcomp package [52].
Data normalization
Library size was standardized across samples using different normalization approaches available in metagenomeSeq [81] and NetCoMi [87] R packages: Total Sum Scaling – TSS [18], Cummulative Sum Scaling – CSS [82] and Centered Log-Ratio transformation – CLR [2].
Differential taxa abundance
Differential abundance testing was performed at different taxonomic ranks via differential expression analyses based on multivariate differential association computed by MaAsLin2 R package [71].
Beta diversity
The effect of sampling method on beta diversity was assessed via Permutational Analyses of Variance (PERMANOVA) tests (1000 permutations) based on Aitchison distance for the community composition at both the OTU and the genus level [1]. For visual inspection, dissimilarity networks (Aitchison distance) were also constructed using NetCoMi [87] R package.
Association networks
After constructing microbial association networks based on SparCC (Sparse Correlations for Compositional data) correlation measure [40], differential network analyses were conducted using the discordant method [104]. At both the OTU and the genus levels, differential plot networks were constructed and compared using the NetCoMi R package. A sparsification threshold of 0.5 was used for comparing global network properties, centrality measures and hub taxa in the NetCoMi package via permutation tests using 1000 permutations. Adjusted Rand Index (ARI) and Graphlet Correlation Distance (GCD) measures were calculated to assess whether the clustering solutions are more or less similar than expected at random or distance similarities [91, 116]. As a measure of conditional dependence, SPIEC-EASI (Sparse InversE Covariance estimation for Ecological Association and Statistical Inference) pipeline was also applied to non-transformed data. Association network properties were computed, and keystone taxa were selected from node degree (number of connections) and betweenness (node centrality) measures using SpiecEasi R package [65].
Results
The mean age of study participants was 56 years, ranging from 38 to 71 years, whereas the sex composition was 7 females (58%) and 5 males (42%). A total of 15 implants were placed in the following positions: 14 (1), 25 (1), 26 (1), 27 (1), 34 (1), 36 (2), 37 (2), 45 (1), 46 (2) and 47 (3). The number of dental implants in which transepithelial abutments with machined surface were placed ranged from one to two (only one transepithelial abutment with machined surface was placed in a total of nine patients, while a total of two transepithelial abutments with machined surface were placed in the remaining three patients).
As shown in Fig. 1, a significant effect of sampling method on most bacterial alpha diversity indices was detected, at both the OTU level (observed [p = 0.00090], Chao index [p = 0.0026] and Fisher’s alpha index [p = 2.4E−05]) and at the genus level (observed [p = 0.019], Chao index [p = 0.047], Fisher’s alpha index [p = 0.00038] and Simpson’s indices [p = 0.014]). After performing pairwise comparisons, significant differences in observed richness, Chao and Fisher’s alpha indices were identified between ToCF/TACF and TA at both the OTU and the genus level, and in Simpson’s indices at the genus level. No significant differences were detected in Shannon or Simpson’s diversity indices at the OTU (p = 0.81 and p = 0.20, in each case) or in the Shannon index at the genus level (p = 0.30).

Boxplot of alpha diversity indices reflecting taxa abundance and consistency in samples grouped by sampling method at: A) the OTU level, and B) the genus level
According to Fig. 2, Firmicutes (39–53%), Bacteroidota (9.4–21%), Actinobacteria (6.7–8.9%), Fusobacteria (5.9–9%) and Proteobacteria (5.5–9.5%) are the most abundant phyla in the studied samples, with a higher proportion of Firmicutes in ToCF (53%) and TACF (52%), and more Bacteroidota (21%), Fusobacteria (9.0%) and Synergistetes (1.6%) in TA samples. Considering the class level, Bacteroidia (7.4–19%) and Negativicutes (7.3-13%) were two main taxa in all cases. Nevertheless, while the proportion Actinomycetia (8.3%) was higher in ToCF and that of Bacilli was higher in both ToCF and TACF (37%), the abundance of Clostridia was higher in TACF (7.6%) and TA samples (10%). The orders Lactobacillales (12-33%), Bacteroidales (7.5-19%), Eubacteriales (7.5-10%) and Veillonellales (5.4-11%) reached the highest abundances in all sample types, together with Pasteurellales (3.5%) in TA and Actinomycetales in ToCF (5.4%) and Bacillales in TACF (4.1%) and ToCF (5.3%). On the other hand, Streptococcaceae (8.4-22%), Prevotellaceae (4.5-12%), Fusobacteriaceae (3.9-8.0%) and Veillonellaceae (5.4-11%) were the most represented families in all sample types, along with Selenomonadaceae (2.5%) in ToCF, Bifidobacteriaceae (2.3%) and Clostridiaceae (3.0%) in TACF, Enterococcaceae and Bacillaceae in TACF (9.4% and 2.5% in each case) and ToCF (11% and 2.6% in each case), and Peptostreptococcaceae (6.9%), Porphyromonadaceae (6.1%), Pasteurellaceae (3.5%) and Treponemaceae (1.2%) in TA samples. At the genus level, Streptococcus (8.3-11%) and Veillonella (4.5-6.9%) were abundant genera in all sample types. Nevertheless, ToCF and TACF samples were richer in Lactococcus (9.0% and 14% in each case) and Enterococcus (11%), whereas Prevotella (11%), Fusobacterium (8.1%), Porphyromonas (5.8%) and Peptostreptococcus (4.9%) reached higher frequencies in TA samples.

Stacked bar graphs representing cumulative abundances of taxa that represent up to 10 most abundant taxa nested by Phylum (only top 10 phyla were plotted) and grouped by sampling method (TSS normalized data). Taxonomic ranks nested by Phylum were: A) the Class level, B) the Order level, C) the Family level, and D) the Genus level
As shown in Fig. 3, significant differences in taxa abundances between sampling methods were identified in 0.58% (10/1732) of the OTUs shared by ToCF and TA, and in 0.40% of the OTUs shared by TCFA and TA (7/1732). Significant changes were also detected after comparing TA and ToCF in 3.1% of the families (10/322), 4.1% of the orders (6/145), 8.7% of the classes (6/69) and 6.3% of the phyla (2/32); and TA and TACF in 0.60% (5/830) of the genera, 2.0% (6/306) of the families and in 1.4% (2/141) of the orders. Pairwise comparisons revealed significant changes in the frequency of some peri-implant or periodontal disease related taxa between different sampling methods. In TA samples, Synergistetes, Slackia, Peptostreptococcus, Atopobium, Mogibacterium, Slackia exigua or Peptostreptococcus stomatis showed increased abundance when compared to ToCF, whereas those of Lactococcus, Enterococcus, Cellulomonas, Corynebacterium, Exiguobacterium, Bacillus, Microbacterium and Actinomyces naturae were reduced. In contrast to TACF, TA samples also showed lower abundances of several taxa, including Bacilli, Exiguobacterium, Enterococcus, Cellulomonas, Acinetobacter, Microbacterium, Paenibacillus, Lactococcus and Actinomyces naturae.

Differentially abundant taxa between pairwise sampling method (SM) comparison identified through MaAsLin2 multivariate association testing: A) Phylum level, B) Class level, C) Order level, D) Family level, E) Genus level and F) OTU level
Figure 4 shows compositional beta diversity biplot generated through Aitchison distance matrix considering taxa with at least 0.1% of sequencing reads. PERMANOVA analyses based on Aitchison distance showed that the effect of sampling method on bacterial community composition was statistically significant at both the OTU (p = 0.0010) and the genus level (p = 0.0010). Multilevel pairwise comparisons revealed significant differences between TA and ToCF (OTU level: p = 0.0010; genus level: p = 0.0010) and between TACF and TA (OTU level: p = 0.0020; genus level: p = 0.0030), but not between TACF and ToCF (OTU level: p = 0.837; genus level: p = 0.797).

Dissimilarity network based on Aitchison distance matrix representing the beta-diversity at: A) the OTU level, and B) the genus level retaining taxa with at least 0.1% of sequencing reads (zeros were replaced via multiplicative simple replacement and k-nearest neighbor was used as sparsification method)
Bacterial association networks of ToCF, TACF and TA communities constructed from SparCC correlations retaining taxa with at least 0.1% of sequencing reads are represented in Fig. 5, where nodes represent bacterial taxa and edges represent either co-presence (positive association) or mutual exclusion (negative association) relationships. At both the OTU and the genus levels, pairwise network comparisons revealed high clustering similarities in terms of ARI and GCD, and no significant changes in global network properties when comparing different sampling methods (Table 1). However, significant differences were detected in terms of hub taxa between ToCF and TA samples at the OTU level (Jaccard Index = 0.00, p = 0.017), with a higher influence of Streptococcus spp., Veillonella atypica and Neisseria mucosa in ToCF, and a particular importance of Porphyromonas pasteri, Centipeda periodontii, Selenomonas sputigena, Veillonella tobetsuensis or Parvimonas micra in TA samples (Table 2). Significant differences were also identified in betweenness centrality after comparing ToCF and TA sampling methods at the OTU level (Jaccard Index = 0.13, p = 0.0033). Nevertheless, no significant differences were observed after adjusting permutation p-values of the tests for differential centrality values for multiple testing. Further information on these measures is available in Supplementary Material 2.

Bacterial association networks from ToCF, TACF and TA at the OTU and genus levels constructed using SparCC correlation coefficients for compositional data retaining taxa with at least 0.1% of sequencing reads. For network sparsification, only edges corresponding to an absolute association greater or equal than 0.7 (OTU level) and 0.6 (genus level) were represented in order to improve network readability
After constructing SPIEC-EASI based ecological networks, the highest rated keystone OTUs attending to their node degree and betweenness were Prevotella multisaccharivorax, Streptococcus dentapri, Peptococcus niger, Porphyromonas circumdentaria, Mycoplasma salivarium and Streptococcus gallinaceus in ToCF, Streptococcus oligofermentans, Actinomyces odontolyticus, Bacteroides heparinolyticus, Acidaminococcus intestini, Moryella indoligenes and Leptotrichia wadei in TACF, and Rothia aeria, Streptococcus sobrinus, Moryella indoligenes, Actinobaculum massiliense, Prevotella paludivivens and Megasphaera sueciensis in TA bacterial communities. At the genus level, keystone genera were Butyrivibrio, Flavobacterium, Leptotrichia, Paracoccus, Pasteurella and Sphingomonas in ToCF, Haloactinobacterium, Porphyromonas, Sphingobacterium, Shuttleworthia, Moraxella and Propionibacterium in TACF, and Bacillus, Alloprevotella, Campylobacter, Pontibacillus, Eikenella and Parascardovia in TA.
Differential network analyses (Fig. 6) revealed correlation changes between certain genera following pairwise comparisons. When comparing association patterns from TACF and TA samples, the differential network analysis revealed significant correlation changes between a large number of genera, including Clostridium and Bifidobacterium, Bifidobacterium and Dialister, Fusobacterium and Haemophilus or Actinomyces odontolyticus and Veillonella atypica (inversely correlated in TACF and no correlated in TA), and Peptostreptococcus and Haemophilus, Campylobacter and Oribacterium or Bifidobacterium and Porphyromonas (no correlated in TACF and negatively correlated in TA). Pairwise comparisons between TA and ToCF sampling methods also showed strong positive correlations in ToCF samples that remained no significant in TA samples (Fusobacterium and Selenomonas, Alkaliphilus and Dysgonomonas, Dysgonomonas and Actinomyces, Gemella and Haemophilus, Rothia and Streptococcus, Rothia and Parascardovia, or Haemophilus and Lautropia) and viceversa (Megasphaera and Prevotella). Moreover, some negative correlations between taxa detected in ToCF samples were not identified in TA samples (Treponema and Rothia, and Selenomonas and Rothia) and viceversa (Lautropia mirabilis and Haemophilus parainfluenciae, Actinomyces odontolyticus and Prevotella maculosa, or Atopobium parvulum and Streptococcus cristatus). No significant differential correlations were observed between ToCF and TACF samples.

Differential association networks based on Fig. 5: A) OTU level and B) Genus level
The effect of sampling strategy on the metagenomic profile of transepithelial abutments at different levels of analysis, including alpha diversity, beta diversity, differential taxa abundance and association network properties, was summarized in Table 3.
Discussion
When considering healthy peri-implant environments, bacterial colonization of the implant surface tends to be like healthy surrounding periodontal sites, with lower diversity [49]. However, it evolves toward the establishment of organized biofilms within the next two weeks [9]. Despite the fact that salivary pellicles adsorbed to implant surfaces promote the adhesion of microbes, its molecular features (chemical composition) and the immunological microenvironment determine the microbial colonization process [33]. In the present study, certain alpha diversity metrics (Observed taxa, Chao index, Fisher’s alpha index and Simpson’s indices) have revealed significant differences between crevicular fluid samples (ToCF and/or TACF) and transepithelial abutments (TA). Differential statistical significance between diversity metrics could be explained by the varying influence of richness, evenness and dominance in each index. Considering that TA samples, as opposed to crevicular fluid samples, account not only free-living bacteria around transepithelial abutments, but also most biofilm forming surface adhered bacteria, these results suggest that tooth surface could harbor a richer microbiota than TA surface. These results – indicating a lower diversity in peri-implant microbiota—are in agreement with those of Dabdoub et al. [28] and Payne et al. [83]. Nevertheless, bacterial diversity in peri-implantitis sites is usually higher than that of healthy periodontal sites [106, 120], but lower than that of periodontitis environments [8].
According to the consulted bibliography, differentially abundant taxa related to healthy sites include mainly gram-positive cocci and non-motile bacilli, with higher frequencies of lactic acid bacteria (Lactobacillales and Bifidobacterium) [47], Lactococcus [15], Haemophilus [97], Veillonella [97, 102, 109], Streptococcus [97, 109], Neisseria [97], Rothia [97], Prevotella [42, 109], Actinomyces [64, 109]), Leptotrichia [15, 64], Gemella spp. [102], Vibrio [42], Brevundimonas [15, 118], Pseudomonas [118], Oribacterium spp., Selenomonas spp. and Cardiobacterium spp. [28], Staphylococcus [105], Granulicatella adjacens, Veillonella dispar, Actinomyces meyeri and Streptococcus mitis [27], Propionibacterium acnes [120], Acinetobacter, Paracoccus and Moraxella [45], Dialister [61], Abiotrophia defectiva [111], Microbacterium [108], Corynebacterium ([105]), Novosphingobium capsulatum [36], Propionibacter, Lautropia, Chitinophagaceae, Brevundimonas nasdae, Delfitia acidivorans, Rothia aeria, Anaerofilum pentosovorans, Anaerofilum agile, Pseudoramibacter alactolyticus and Porphyromonas HMT-277/278 [15]. Moreover, Kroeger et al. [63] found higher abundances of Lautropia mirabilis, Rhodobacteriaceae or Bergeyella in shallow peri-implant pockets.
On the other hand, most published research findings confirm that peri-implantitis associated bacteria consist mainly of gram-negative motile anaerobic periopathogens and opportunistic bacteria, including higher abundancies of Porphyromonas gingivalis ([3, 10, 55, 68, 86, 97, 103, 107, 109, 117]), Aggregatibacter actinomycetemcomitans ([53, 68, 117]), Prevotella intermedia ([55, 68, 74, 109]), Prevotella nigrescens [68, 74], Treponema denticola ([17, 85, 103]), Tannerella forsythia ([3, 10, 74, 86, 103, 117]), Fusobacterium nucleatum ([3, 10, 55, 109]), Parvimonas micra [17, 62, 109], Eubacterium [64, 109], Butyrivibrio [28, 64], Filifactor alocis [97, 109], Pseudoramibacter [60], Desulfobulbus [16, 34], Streptococcus ([27, 55, 109]), Exiguobacterium [24], Streptococcus mutans and Peptococcus [28, 64]. Koyanagi et al. [62] reported that Chloroflexi, Tenericutes, Synergistetes, Peptostreptococcus stomatis and Solobacterium moorei were detected only in peri-implantitis sites.
Peri-implantitis has been also associated with higher relative frequencies of Eikenella corrodens [17], Streptococcus intermedius, Streptococcus mitis, Haemophilus influenzae and Treponema socranskii [86], Campyblobacter gracilis, Dialister invisus, Eubacterium infirmum and Mitsuokella (da Silva et al. 2013), Campylobacter rectus [17, 26], Fusobacterium [64, 74], Slackia exigua, Parascardovia denticolens and Centipeda periodontii [109], Mycoplasma and Treponema [64], Streptococcus non-mutans [28], Neisseria, Kingella, Enterococcus, Fretibacterium and Bacillus [55], Propionibacterium, Paludibacter, Staphylococcus, Filifactor and Mogibacterium [105], Treponema maltophilum [97], Olsenella and Sphingomonas [76], Veillonella [30, 64], Treponema [63, 117], Prevotella tannerae [35], Actinomyces [28], Fretibacterium fastidiosum [97], Pseudoramibacter alactolyticus [28, 62], Campylobacter [64] and Staphylococcus aureus [86], Stenotrophomonas, Leuconostoc, Faecalibacterium prausnitzii, Haemophilus parainfluenzae, Prevotella copri, Bacteroides vulgatus and Bacteroides stercoris [80]. Furthermore, the following taxa are also strongly associated with subgingival plaque of periodontitis: Alloprevotella, Phocaeicola, Johnsonella and Mycoplasma [24].
Oral micro-habitats, including teeth, transepithelial abutments and crevicular fluid, provide unique biological niches that harbors specific bacterial communities. Compositional variation in oral microbiota related to sampling origin has been highlighted by multiple studies, including crevicular fluid around dental prostheses that were fabricated by various biomaterials and fabrication techniques [13, 48], supra and subgingival biofilm [103, 117] and different peri-implantitis lesions [63, 89]. Moreover, dental intervention-related perturbations lead to significant environmental changes at the microscale level, including surface topology, chemistry and immunological response, resulting in dysbiosis and structural disruption of the oral microbiota. Compositional variation was higher between bacterial communities from transepithelial abutments (TA) and periodontal crevicular fluid (ToCF). In this sense, a significant reduction in certain peri-implant or periodontal health related taxa was observed in TA when compared to ToCF (Bacilli, Actinomycetia and Lactococcus), while several of those related with oral dysbiosis showed increased relative frequencies (Synergistetes, Peptostreptococcus, Slackia, Atopobium and Mogibacterium).
Considering higher taxonomic ranks, higher abundances of certain taxa after implant insertion (Bacteroidota, Synergistetes and Coriobacteriaceae) and lower frequencies of Actinomycetia, Bacillaceae, Cellulomonadaceae and Enterococcaceae were identified in TA samples. In this context, Heyman et al. [51] reported analogous population trends for Bacteroidota and Coriobacteriaceae after implant placement using a murine model. On the other hand, certain peri-implant and periodontal disease related taxa, including Mogibacterium, Slackia, Peptostreptococcus, Atopobium, Slackia exigua and Peptostreptococcus stomatis showed increased abundances in transepithelial abutments. Similar results were obtained after comparing crevicular fluid from transepithelial abutments (TACF) with transepithelial abutments (TA), with lower frequencies of several opportunistic commensal enterococci, Cellulomonas, Lactococcus and Actinomyces naturae in TA samples. When compared with crevicular fluid samples extracted transepithelial abutments, TA were also depleted in other taxa, including Bacilli, Paenibacillus, Exiguobacterium, Microbacterium and Corynebacterium. These results do not agree with those of Schaumann et al. [100], who concluded that peri-implant and periodontal tissues share a similar biofilm composition at the genus level.
Dental intervention-related perturbations lead to significant environmental changes at the microscale level, including surface topology and chemistry, resulting in dysbiosis and structural disruption of the oral microbiota. Abundance-based associations also revealed differential correlation across sampling methods, particularly when TA was compared to ToCF. More specifically, differential associations affecting mainly peri-implant health and peri-implantitis related taxa (Bifidobacterium, Haemophilus, Campylobacter, Peptostreptococcus, Porphyromonas, Treponema, Selenomonas, Fusobacterium, Rothia and Prevotella) were detected after performing pairwise comparisons. Moreover, significant variations regarding hub OTUs were identified between ToCF and TA. These findings suggest that both peri-implant and crevicular fluid bacterial communities are characterized by a particular distinctive associational footprint as opposed to that of the adjacent teeth or transepithelial abutments and also that environmental gradients and complex ecological interactions contribute to variations in ecosystem structure and function.
According to the results found in the present study, the abundances of most peri-implantitis or periodontal disease related taxa were higher in TA samples, whereas those of peri-implant health associated bacteria were higher in ToCF or TACF. Thus, current data suggest that implant micro-habitats are usually characterized by a dysbisotic shift, enriched in Synergistetes, Bacteroidota, Atopobium, Slackia, Peptostreptococcus, Mogibacterium and Slackia exigua, and depleted in Bacillus, Paenibacillus, Enterococcus, Lactococcus, Microbacterium, Corynebacterium and Actinomyces naturae. Differential abundance patterns between crevicular fluid from transepithelial abutments and transepithelial abutments, not detected when comparing crevicular fluid samples from different origins, reflect the particularities of specific oral niches [69]. Furthermore, sampling method was a significant source of bacterial community dissimilarity. In this sense, significant differences were identified at the community level between TA and ToCF and also between TACF and TA, but not between ToCF and TACF. With respect to network properties, sampling procedures did not significantly differ in terms of global network properties or centrality measures. However, variations in hub OTUs were identified between ToCF and TA. By contrast, TACF and TA shared the most relevant hub taxa at both the OTU and the genus level.
As stated by Berry and Widder [12], hub taxa are not necessarily keystones in the microbial community, but ecologically relevant hub taxa are likely to be. Thus, different keystone taxa were also identified in bacterial communities from different sampling origins. Considering node degree and betweenness, Streptococcus oligofermentans, Actinomyces odontolyticus, Bacteroides heparinolyticus, Acidaminococcus intestini, Moryella indoligenes, Leptotrichia wadei, Haloactinobacterium, Porphyromonas, Sphingobacterium, Shuttleworthia, Moraxella and Propionibacterium have a pivotal role in TACF samples, whereas Rothia aeria, Streptococcus sobrinus, Moryella indoligenes, Actinobaculum massiliense, Prevotella paludivivens, Megasphaera sueciensis, Bacillus, Alloprevotella, Campylobacter, Pontibacillus, Eikenella and Parascardovia showed a higher influence in TA samples. On the other hand, Prevotella multisaccharivorax, Streptococcus dentapri, Peptococcus niger, Porphyromonas circumdentaria, Mycoplasma salivarium, Streptococcus gallinaceus, Butyrivibrio, Flavobacterium, Leptotrichia, Paracoccus, Pasteurella and Sphingomonas were identified as drivers of microbiome structure and functioning in ToCF samples. With the exception of Flavobacterium (ToCF), Sphingobacterium (TACF), Rothia aeria (TA), Moraxella (TACF), Sphingomonas (ToCF) and Rothia mucilaginosa (TA), most keystone taxa were obligate (eg. Bacteroides heparinolyticus, Moryella indoligenes, Acidaminococcus intestine, Peptococcus niger, Megasphaera sueciensis, Shuttleworthia, Campylobacter, Butyrivibrio, Actinomyces, Propionibacterium) or facultative anaerobes (eg. Streptococcus spp., Parascardovia, Alloprevotella, Bacillus, Paracoccus, Leptotrichia, Eikenella, Porphyromonas, Pasteurella, Mycoplasma salivarium or Actinobaculum massiliense).
Conclusions
According to these findings, it should be highlighted that bacterial community assessment via mNGS could be considered a promising strategy for peri-implant and periodontal health surveillance and early peri-implant disease diagnosis. Despite the fact that, regardless of sampling approach, association network properties and centrality measures from transepithelial bacterial communities were similar, significant differences were detected in terms of hub taxa, alpha and beta diversity and individual taxa abundance, including some peri-implant and periodontal health related taxa. In most aspects, crevicular fluid samples (ToCF and TACF) are not representative of bacterial communities developed on transepithelial surfaces. As a result, it can be concluded that the choice of sampling strategy can deeply affect the results of oral microbiota profiling, with a particular emphasis on peri-implant health related bacterial taxa.
Availability of data and materials
Sequence Read Archive (SRA) data corresponding to the National Center for Biotechnology Information (NCBI) BioProject PRJNA1138960 are available at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1138960.
Abbreviations
- CLR:
-
Centered Log-Ratio transformation
- CSS:
-
Cummulative Sum Scaling
- mNGS:
-
Metagenomics Next Generation Sequencing
- OTU:
-
Operational Taxonomic Unit
- SM:
-
Sampling method
- TA:
-
Transepithelial abutment
- TACF:
-
Crevicular fluid from transepithelial abutment
References
Anderson JA, et al. Permutational Multivariate Analysis of Variance (PERMANOVA). Wiley StatsRef: Statistics Reference Online. 2017. p. 1–15. https://doi.org/10.1002/9781118445112.stat07841.
Aitchison J. The Statistical Analysis of Compositional Data, Chapman and Hall, reprinted in 2003 with additional material by The Blackburn Press. 1986.
Al-Ahmad A, et al. Shift of microbial composition of peri-implantitis-associated oral biofilm as revealed by 16S rRNA gene cloning. J Med Microbiol. 2018;67:332–40. https://doi.org/10.1099/jmm.0.000682.
Albrektsson T, et al. An imbalance of the immune system instead of a disease behind marginal bone loss around oral implants: position paper. Int J Oral Maxillofac Implants. 2020;35(3):495–502. https://doi.org/10.11607/jomi.8218.
Atieh MA, et al. The frequency of peri-implant diseases: a systematic review and meta-analysis. J Periodontol. 2013;84(11):1586–98. https://doi.org/10.1902/jop.2012.120592.
Bang E, et al. Factors influencing oral microbiome analysis: from saliva sampling methods to next-generation sequencing platforms. Sci Rep. 2023;13(1):10086. https://doi.org/10.1038/s41598-023-37246-2.
Bates D, et al. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1–48. https://doi.org/10.18637/jss.v067.i01.
Belibasakis GN. Microbiological and immuno-pathological aspects of peri-implant diseases. Arch Oral Biol. 2014;59:66–72. https://doi.org/10.1016/j.archoralbio.2013.09.013.
Belibasakis GN, et al. Peri-implant infections of oral biofilm etiology. Adv Exp Med Biol. 2015;830:69–84. https://doi.org/10.1007/978-3-319-11038-7_4.
Belkacemi SG, et al. Periimplantitis-associated methanogens: a preliminary report. Sci Rep. 2018;8:9447. https://doi.org/10.1038/s41598-018-27862-8.
Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat. 2000;25(1):60–83. https://doi.org/10.2307/1165312.
Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:219. https://doi.org/10.3389/fmicb.2014.00219.
Blank E, et al. Evaluation of biofilm colonization on multi-part dental implants in a rat model. BMC Oral Health. 2021;21:313. https://doi.org/10.1186/s12903-021-01665-2.
Burcham ZM, et al. Patterns of oral microbiota diversity in adults and children: a crowdsourced population study. Sci Rep. 2020;10:2133. https://doi.org/10.1038/s41598-020-59016-0.
Butera A, et al. Oral microbiota in patients with peri-implant disease: a narrative review. Appl Sci. 2022;12:3250. https://doi.org/10.3390/app12073250.
Camelo-Castillo A, et al. Relationship between periodontitis-associated subgingival microbiota and clinical inflammation by 16S pyrosequencing. J Clin Periodontol. 2015;42(12):1074–82. https://doi.org/10.1111/jcpe.12470.
Canullo L, et al. Microbiologic and clinical findings of implants in healthy condition and with peri-implantitis. Int J Oral Maxillofac Implants. 2015;30:834–42. https://doi.org/10.11607/jomi.3947.
Cao Y. microbiomeMarker: microbiome biomarker analysis toolkit_. R package version 1.2.2. 2022. https://github.com/yiluheihei/microbiomeMarker.
Chang HY, et al. Early radiographic diagnosis of peri-implantitis enhances the outcome of peri-implantitis treatment: a 5-year retrospective study after non-surgical treatment. J Periodontal Implant Sci. 2015;45(3):82–93. https://doi.org/10.5051/jpis.2015.45.3.82.
Chao A. Non-parametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–70. https://doi.org/10.2307/4615964.
Chao A, Chun-Huo C, Jost L. Phylogenetic diversity measures and their decomposition: a framework based on hill numbers. biodiversity conservation and phylogenetic systematics. Springer International Publishing. 2016. pp. 141–172.
Chaparro A, et al. Molecular biomarkers in peri-implant health and disease: a cross-sectional pilot study. Int J Mol Sci. 2022;23:9802. https://doi.org/10.3390/ijms23179802.
Charalampakis G, Belibasakis GN. Microbiome of peri-implant infections: lessons from conventional, molecular and metagenomic analyses. Virulence. 2015;6(3):183–7. https://doi.org/10.4161/21505594.2014.980661.
Chen C, et al. Oral microbiota of periodontal health and disease and their changes after nonsurgical periodontal therapy. ISME J. 2018;12:1210–24. https://doi.org/10.1038/s41396-017-0037-1.
Chiu CH, et al. Improved nonparametric lower bound of species richness via a modified Good-Turing frequency formula. Biometrics. 2014;70:671–82. https://doi.org/10.1111/biom.12200.
Cortelli SC, et al. Frequency of periodontal pathogens in equivalent peri-implant and periodontal clinical statuses. Arch Oral Biol. 2013;58(1):67–74. https://doi.org/10.1016/j.archoralbio.2012.09.004.
da Silva ES, et al. Microbiological diversity of peri-implantitis biofilm by Sanger sequencing. Clin Oral Implant Res. 2014;25:1–8. https://doi.org/10.1111/clr.12231.
Dabdoub SM, Tsigarida AA, Kumar PS. Patient-specific analysis of periodontal and peri-implant microbiomes. J Dent Res. 2013;92:168S–175S. https://doi.org/10.1177/0022034513504950.
Daubert DM, et al. Prevalence and predictive factors for peri-implant disease and implant failure: a cross-sectional analysis. J Periodontol. 2015;86:337–47. https://doi.org/10.1902/jop.2014.140438.
Daubert D, et al. Titanium as a modifier of the peri-implant microbiome structure. Clin Implant Dent Relat Res. 2018;20(6):945–53. https://doi.org/10.1111/cid.12676.
Deo PN, Deshmukh R. Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol. 2019;23(1):122–8. https://doi.org/10.4103/jomfp.JOMFP_304_18.
Derks J, Tomasi C. Peri-implant health and disease. A systematic review of current epidemiology. J Clin Periodontol. 2015;42(Suppl 16):S158–71. https://doi.org/10.1111/jcpe.12334.
Dhir S. Biofilm and dental implant: the microbial link. J Indian Soc Periodontol. 2013;17(1):5–11. https://doi.org/10.4103/0972-124X.107466.
Dingsdag S, Nelson S, Coleman NV. Bacterial communities associated with apical periodontitis and dental implant failure. Microb Ecol Health Dis. 2016;27:31307. https://doi.org/10.3402/mehd.v27.31307.
Ebadian AR, et al. Bacterial analysis of peri-implantitis and chronic periodontitis in Iranian subjects. Acta Med Iran. 2012;50(7):486–92.
Esparbès P, et al. Subgingival microbiota and cytokines profile changes in patients with periodontitis: a pilot study comparing healthy and diseased sites in the same oral cavities. Microorganisms. 2021;9:2364. https://doi.org/10.3390/microorganisms9112364.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19:55–71. https://doi.org/10.1038/s41579-020-0433-9.
Faveri M, et al. Microbiological diversity of peri-implantitis biofilms. Adv Exp Med Biol. 2015;830:85–96. https://doi.org/10.1007/978-3-319-11038-7_5.
Fisher RA, Corbet AS, Williams CB. The relation between the number of species and the number of individuals in a random sample of animal population. J Anim Ecol. 1943;12:42–58. https://doi.org/10.2307/1411.
Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol. 2012;8(9):e1002687. https://doi.org/10.1371/journal.pcbi.1002687.
Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20(2):145–55. https://doi.org/10.1038/nn.4476.
Gao X, et al. Diversity analysis of subgingival microbial bacteria in peri-implantitis in Uygur population. Medicine (Baltimore). 2018;97(5):e9774. https://doi.org/10.1097/MD.0000000000009774.
Gazil V, et al. Current data on oral peri-implant and periodontal microbiota and its pathological changes: a systematic review. Microorganisms. 2022;10:2466. https://doi.org/10.3390/microorganisms10122466.
Giordan-Kelhoffer B, et al. Oral microbiota, its equilibrium and implications in the pathophysiology of human diseases: a systematic review. Biomedicines. 2022;10:8. https://doi.org/10.3390/biomedicines10081803.
Griffen AL, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6(6):1176–85. https://doi.org/10.1038/ismej.2011.191.
Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95–108. https://doi.org/10.1038/nrmicro821.
Hashimoto Y, et al. Microbial differences between active and remission peri-implantitis. Sci Rep. 2022;12:5284. https://doi.org/10.1038/s41598-022-09192-y.
Heboyan A, et al. Bacteriological evaluation of gingival crevicular fluid in teeth restored using fixed dental prostheses: an in vivo study. Int J Mol Sci. 2021;22(11):5463. https://doi.org/10.3390/ijms22115463.
Heitz-Mayfield LJA, Salvi GE. Peri-implant mucositis. J Clin Periodontol. 2018;45(20):237–45. https://doi.org/10.1111/jcpe.12953.
Heydenrijk K, et al. Microbiota around root-form endosseous implants: a review of the literature. Int J Oral Maxillofac Implants. 2002;17(6):829–38.
Heyman O, et al. Niche specific microbiota-dependent and independent bone loss around dental implants and teeth. J Dent Res. 2020;99(9):1092–101. https://doi.org/10.1177/0022034520920577.
Hothorn T, Bretz F, Westfall P. Simultaneous inference in general parametric models. Biom J. 2008;50(3):346–63. https://doi.org/10.1002/bimj.200810425.
Hultin M, et al. Microbiological findings and host response in patients with peri-implantitis. Clin Oral Implant Res. 2002;13:349–58. https://doi.org/10.1034/j.1600-0501.2002.130402.x.
Hurlbert SH. The nonconcept of species diversity: a critique and alternative parameters. Ecology. 1971;52:577–86. https://doi.org/10.2307/1934145.
Jakobi M, et al. The peri-implant and periodontal microbiota in patients with and without clinical signs of inflammation. Dentistry Journal. 2015;3:24–42. https://doi.org/10.3390/dj3020024.
Kilian M, et al. The oral microbiome – an update for oral healthcare professionals. Br Dent J. 2016;221:657–66. https://doi.org/10.1038/sj.bdj.2016.865.
Klindworth A, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41(1):e1. https://doi.org/10.1093/nar/gks808.
Klinge B, et al. Peri-implant diseases. Eur J Oral Sci. 2018;126(21):88–94. https://doi.org/10.1111/eos.12529.
Konstantinidis IK, et al. Cross-sectional study on the prevalence and risk indicators of peri-implant diseases. Eur J Oral Implantol. 2015;8:75–88.
Korsch M, et al. Microbiological findings in early and late implant loss: an observational clinical case-controlled study. BMC Oral Health. 2021;21(1):112. https://doi.org/10.1186/s12903-021-01439-w.
Kotsakis GA, Olmedo DG. Peri-implantitis is not periodontitis: Scientific discoveries shed light on microbiome-biomaterial interactions that may determine disease phenotype. Periodontol. 2021;2000(86):231–40. https://doi.org/10.1111/prd.12372.
Koyanagi T, et al. Analysis of microbiota associated with peri-implantitis using 16S rRNA gene clone library. J Oral Microbiol. 2010;2:5104–10. https://doi.org/10.3402/jom.v2i0.5104.
Kroeger A, et al. The severity of human peri-implantitis lesions correlates with the level of submucosal microbial dysbiosis. J Clin Periodontol. 2018;45(12):1498–509. https://doi.org/10.1111/jcpe.13023.
Kumar PS, et al. Pyrosequencing reveals unique microbial signatures associated with healthy and failing dental implants. J Clin Periodontol. 2012;39:425–33. https://doi.org/10.1111/j.1600-051X.2012.01856.x.
Kurtz ZD, et al. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015;11(5). https://doi.org/10.1371/journal.pcbi.1004226.
Langaas M, Lindqvist BH, Ferkingstad E. Estimating the proportion of true null hypotheses, with application to DNA microarray data. J Royal Stat Soc Ser B Stat Methodol. 2005;67(4):555–72.
Lenartova M, et al. The oral microbiome in periodontal health. Front Cell Infect Microbiol. 2021;11:629723. https://doi.org/10.3389/fcimb.2021.629723.
Leonhardt A, Dahlén G, Renvert S. Five-year clinical, microbiological, and radiological outcome following treatment of peri-implantitis in man. J Periodontol. 2003;74:1415–22. https://doi.org/10.1902/jop.2003.74.10.1415.
Li X, et al. The oral microbiota: community composition, influencing factors, pathogenesis, and interventions. Front Microbiol. 2022;13:895537. https://doi.org/10.3389/fmicb.2022.895537.
Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10(4):311–23. https://doi.org/10.1016/j.chom.2011.10.004.
Mallick H, et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol. 2021;17(11):e1009442. https://doi.org/10.1371/journal.pcbi.1009442.
Mark-Welch JLM, Ramírez-Puebla ST, Borisy GG. Oral microbiome geography: micron-scale habitat and niche. Cell Host Microbe. 2020;28(2):160–8. https://doi.org/10.1371/journal.pcbi.1009442.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. https://doi.org/10.1371/journal.pone.0061217.
Mombelli A, et al. Treatment of peri-implantitis by local delivery of tetracycline. Clinical, microbiological and radiological results. Clin Oral Implant Res. 2001;12:287–94. https://doi.org/10.1034/j.1600-0501.2001.012004287.x.
Mombelli A, Décaillet F. The characteristics of biofilms in peri-implant disease. J Clin Periodontol. 2011;38(Suppl. 11):203–13. https://doi.org/10.1111/j.1600-051X.2010.01666.x.
Maruyama N, et al. Intraindividual variation in core microbiota in peri-implantitis and periodontitis. Sci Rep. 2014;13(4):6602. https://doi.org/10.1038/srep06602.
O’Hara RB. Species richness estimators: how many species can dance on the head of a pin? J Anim Ecol. 2005;74:375–86. https://doi.org/10.1111/j.1365-2656.2005.00940.x.
Oksanen J, et al. _vegan: Community Ecology Package_. R package version 2.6-4. 2022. https://CRAN.R-project.org/package=vegan.
Padial-Molina M, et al. Microbial profiles and detection techniques in peri-implant diseases: a systematic review. J Oral Maxillofac Res. 2016;7(3):e10. https://doi.org/10.5037/jomr.2016.7310.
Pallos D, et al. Salivary microbial dysbiosis is associated with peri-implantitis: a case-control study in a brazilian population. Front Cell Infect Microbiol. 2022;5(11):696432. https://doi.org/10.3389/fcimb.2021.696432.
Paulson JN, et al. metagenomeSeq: Statistical analysis for sparse high-throughput sequencing. Bioconductor package. 2013a. http://www.cbcb.umd.edu/software/metagenomeSeq.
Paulson JN, et al. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10(12):1200–2. https://doi.org/10.1038/nmeth.2658.
Payne JB, et al. Subgingival microbiome colonization and cytokine production during early dental implant healing. mSphere. 2017;2(6):e00527–17. https://doi.org/10.1128/mSphereDirect.00527-17.
Pennisi E. A mouthful of microbes. Science. 2005;307(5717):1899–901. https://doi.org/10.1126/science.307.5717.1899.
Pérez-Chaparro PJ, et al. The current weight of evidence of the microbiologic profile associated with peri-implantitis: a systematic review. J Periodontol. 2016;87:1295–304. https://doi.org/10.1902/jop.2016.160184.
Persson GR, Renvert S. Cluster of bacteria associated with peri-implantitis. Clin Implant Dent Relat Res. 2014;16(6):783–93. https://doi.org/10.1111/cid.12052.
Peschel S. NetCoMi: network construction and comparison for microbiome data. R package version 1.0.3. 2022.
Pohlert T. PMCMRplus: calculate pairwise multiple comparisons of mean rank sums extended. R package version 1.9.6. 2022. https://CRAN.R-project.org/package=PMCMRplus.
Polymeri A, et al. Non-surgical peri-implantitis treatment with or without systemic antibiotics: a randomized controlled clinical trial. Clin Oral Implant Res. 2022;33(5):548–57. https://doi.org/10.1111/clr.13914.
Proença JT, Barral DC, Gordo I. Commensal-to-pathogen transition: one-single transposon insertion results in two pathoadaptive traits in Escherichia coli -macrophage interaction. Sci Rep. 2017;7:4504. https://doi.org/10.1038/s41598-017-04081-1.
Qannari EM, Courcoux P, Faye P. Significance test of the adjusted Rand index. Application to the free sorting task. Food Qual Prefer. 2014;32:93–7. https://doi.org/10.1016/j.foodqual.2013.05.005.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2022. https://www.R-project.org/.
Radaic A, Kapila YL. The oralome and its dysbiosis: new insights into oral microbiome-host interactions. Comput Struct Biotechnol J. 2021;19:1335–60. https://doi.org/10.1016/j.csbj.2021.02.010.
Rakic M, Grusovin MG, Canullo L. The microbiologic profile associated with peri-implantitis in humans: a systematic review. Int J Oral Maxillofac Implants. 2016;31(2):359–68. https://doi.org/10.11607/jomi.4150.
Ritzer J, et al. Diagnosing peri-implant disease using the tongue as a 24/7 detector. Nat Commun. 2017;8:264. https://doi.org/10.1038/s41467-017-00340-x.
Rodriguez-Archilla A, Palma-Casiano B. Changes in the oral microbiota induced by peri-implantitis: a metaanalysis. J Inflamm Dis. 2022;25(4):3–11. https://doi.org/10.32598/JID.25.4.1.
Sanz-Martin I, et al. Exploring the microbiome of healthy and diseased peri-implant sites using Illumina sequencing. J Clin Periodontol. 2017;44(12):1274–84. https://doi.org/10.1111/jcpe.12788.
Sahrmann P, et al. The microbiome of peri-implantitis: a systematic review and meta-analysis. Microorganisms. 2020;8(5):661. https://doi.org/10.3390/microorganisms8050661.
Scarano A, et al. Bacterial adhesion on commercially pure titanium and zirconium oxide disks: an in vivo human study. J Periodontol. 2004;75(2):292–6. https://doi.org/10.1902/jop.2004.75.2.292.
Schaumann S, et al. Pyrosequencing of supra- and subgingival biofilms from inflamed peri-implant and periodontal sites. BMC Oral Health. 2014;14:157. https://doi.org/10.1186/1472-6831-14-157.
Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379–423. https://doi.org/10.1002/j.1538-7305.1948.tb01338.x.
Shiba T, et al. Distinct interacting core taxa in co-occurrence networks enable discrimination of polymicrobial oral diseases with similar symptoms. Sci Rep. 2016;6:30997. https://doi.org/10.1038/srep30997.
Shibli JA, et al. Composition of supra- and subgingival biofilm of subjects with healthy and diseased implants. Clin Oral Implant Res. 2008;19(10):975–82. https://doi.org/10.1111/j.1600-0501.2008.01566.x.
Siska C, Bowler R, Kechris K. The discordant method: a novel approach for differential correlation. Bioinformatics. 2016;32(5):690–6. https://doi.org/10.1093/bioinformatics/btv633.
Sousa V, et al. Peri-implant and periodontal microbiome diversity in aggressive periodontitis patients: a pilot study. Clin Oral Implant Res. 2017;28:558–70. https://doi.org/10.2307/1268034.
Sousa V, et al. Oral microcosm biofilms grown under conditions progressing from peri-implant health, peri-implant mucositis, and peri-implantitis. Int J Environ Res Public Health. 2022;19(21):14088. https://doi.org/10.3390/ijerph192114088.
Stokman MA, et al. Bacterial colonization of the peri-implant sulcus in dentate patients: a prospective observational study. Clin Oral Invest. 2016;21:717–24. https://doi.org/10.1007/s00784-016-1941-x.
Sun F, et al. Shift in the submucosal microbiome of diseased peri-implant sites after non-surgical mechanical debridement treatment. Front Cell Infect Microbiol. 2023;12:1091938. https://doi.org/10.3389/fcimb.2022.1091938.
Tamura N, et al. Analysis of bacterial flora associated with peri-implantitis using obligate anaerobic culture technique and 16S rDNA gene sequence. Int J Oral Maxillofac Implants. 2013;28(6):1521–9. https://doi.org/10.11607/jomi.2570.
Teunisse GM. fantaxtic: Fantaxtic Plots for Phyloseq Data. R package version 0.2.0. 2022. https://github.com/gmteunisse/Fantaxtic.
Tsigarida AA, et al. The influence of smoking on the peri-implant microbiome. J Dent Res. 2015;94(9):1202–1127. https://doi.org/10.1177/0022034515590581.
Wang Q, et al. Naive bayesian classifier for rapid assignment of rRNA Sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. https://doi.org/10.1128/AEM.00062-07.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Wong RL, et al. Early intervention of peri-implantitis and periodontitis using a mouse model. J Periodontol. 2018;89(6):669–79. https://doi.org/10.1002/JPER.17-0541.
Wu T, et al. Zinc exposure promotes commensal-to-pathogen transition in pseudomonas aeruginosa leading to mucosal inflammation and illness in mice. Int J Mol Sci. 2021;22(24):13321. https://doi.org/10.3390/ijms222413321.
Yaveroǧlu ÖN, et al. Revealing the hidden language of complex networks. Sci Rep. 2014;4:4547. https://doi.org/10.1038/srep04547.
Yu X, et al. Intra-oral single-site comparisons of periodontal and peri-implant microbiota in health and disease. Clin Oral Implant Res. 2019;30:760–76. https://doi.org/10.1111/clr.13459.
Zhang Q, et al. Comparison of subgingival and peri-implant microbiome in chronic periodontitis. Chin J Dent Res. 2015;18(3):155–62.
Zhang Y, et al. Periodontal and peri-implant microbiome dysbiosis is associated with alterations in the microbial community structure and local stability. Front Microbiol. 2022;12: 785191. https://doi.org/10.3389/fmicb.2021.785191.
Zheng H, et al. Subgingival microbiome in patients with healthy and ailing dental implants. Sci Rep. 2015;5:10948. https://doi.org/10.1038/srep10948.
Acknowledgements
The authors thank for technical and human support provided by SGIker (UPV/EHU/ ERDF, EU).
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Eduardo Anitua and Mohammad Hamdan Alkhraisat designed the study; Eduardo Anitua, Alia Murias-Freijo and Ricardo Tejero performed the clinical procedures and collected/managed the samples; Roberto Tierno analyzed the data and prepared the manuscript, and Eduardo Anitua and Mohammad Hamdan Alkhraisat revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This research is part of a randomized, parallel-group, evaluators-blinded, and controlled randomized clinical trial that was designed to assess the effect of transepithelial abutment surface on the biofilm formation. The trial was registered at Trial Registration ClinicalTrials.gov under the number NCT03554876. All participants provided a written informed consent to participate. The study protocol and informed consent, in full accordance with the ethical principles of the Declaration of Helsinki of 1975, as revised in 2013, were approved by the ethical committee of investigation with medicines of the Basque Country (FIBEA-06-EC/17/Multi-Im).
Consent for publication
Not applicable.
Competing interests
Dr. Anitua reports other from BTI Biotechnology Institute, during the conduct of the study. Other from BTI Biotechnology Institute, outside the submitted work. In addition, Dr. Anitua has a patent US8123524B2 issued to BTI Biotechnology Institute. Dr. Murias-Friejo has nothing to disclose. Dr. Tierno reports personal fees from BTI Biotechnology Institute, during the conduct of the study. Personal fees from BTI Biotechnology Institute, outside the submitted work. Dr. Tejero reports personal fees from BTI Biotechnology Institute, during the conduct of the study. Personal fees from BTI Biotechnology Institute, outside the submitted work. Dr. Alkhraisat reports personal fees from BTI Biotechnology Institute, during the conduct of the study. Personal fees from BTI Biotechnology Institute, outside the submitted work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12903_2024_4675_MOESM2_ESM.docx
Supplementary Material 2. Results summary from testing centrality measures of the networks in Figure 5 for group differencesat: A) the OTU level, and B) the genus level. The absolute differences between sampling procedures and the p-values are shown. The p-values were adjusted for multiple testing using the adaptive Benjamini-Hochberg method [11], where the portion of true null hypothesis is determined according to Langaas et al. [66]. Only the 10 taxa with the highest absolute group difference are included in the summary table. All measures were normalized to [0,1].
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Anitua, E., Murias-Freijo, A., Tierno, R. et al. Assessing peri-implant bacterial community structure: the effect of microbiome sample collection method. BMC Oral Health 24, 1001 (2024). https://doi.org/10.1186/s12903-024-04675-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-024-04675-y