
Abstract
Background
Diabetic nephropathy (DN) is a major contributor to chronic kidney disease. This study aims to identify immune biomarkers and potential therapeutic drugs in DN.
Methods
We analyzed two DN microarray datasets (GSE96804 and GSE30528) for differentially expressed genes (DEGs) using the Limma package, overlapping them with immune-related genes from ImmPort and InnateDB. LASSO regression, SVM-RFE, and random forest analysis identified four hub genes (EGF, PLTP, RGS2, PTGDS) as proficient predictors of DN. The model achieved an AUC of 0.995 and was validated on GSE142025. Single-cell RNA data (GSE183276) revealed increased hub gene expression in epithelial cells. CIBERSORT analysis showed differences in immune cell proportions between DN patients and controls, with the hub genes correlating positively with neutrophil infiltration. Molecular docking identified potential drugs: cysteamine, eltrombopag, and DMSO. And qPCR and western blot assays were used to confirm the expressions of the four hub genes.
Results
Analysis found 95 and 88 distinctively expressed immune genes in the two DN datasets, with 14 consistently differentially expressed immune-related genes. After machine learning algorithms, EGF, PLTP, RGS2, PTGDS were identified as the immune-related hub genes associated with DN. In addition, the mRNA and protein levels of them were obviously elevated in HK-2 cells treated with glucose for 24 h, as well as their mRNA expressions in kidney tissues of mice with DN.
Conclusion
This study identified 4 hub immune-related genes (EGF, PLTP, RGS2, PTGDS), as well as their expression profiles and the correlation with immune cell infiltration in DN.
Similar content being viewed by others


Background
As one of the most frequent and severe complications of diabetes, diabetic nephropathy (DN) is the leading cause of chronic kidney disease, accounting for nearly 50% of all end-stage renal diseases globally [1]. The progression of DN causes renal failure, whose management most often involves dialysis and kidney transplantation. Dialysis and kidney transplantation for many diabetic patients are associated with tremendous medical and economic burdens [2]. To prevent the occurrence and aggravation of DN, accumulating evidence has indicated that comprehensive prevention and novel treatment are of utmost importance [3]. The progression and therapeutic regimen for DN have been associated with numerous risk factors, including tight glycemic control, blood pressure control using RAAS inhibitors, and lipid-lowering agents [4]. Multidisciplinary studies on DN have substantially contributed to reducing the progression of risk and improving the clinical strategy for the prevention of DN [5]. Therefore, there is an urgent need to identify reliable markers for monitoring DN progression, which could then be used to develop novel therapeutic approaches to delay the development of DN.
Recent studies have confirmed that the immune system, including macrophages, T cells, cytokines, chemokines, extracellular matrix, growth factors, fibroblasts, and diseased cells, has an important role during DN [6, 7]. Existing research indicates substantial immune cell infiltration in renal tissues of DN patients, including macrophages, T cells, and B cells [8]. Such infiltration aligns closely with inflammatory responses and immune activation, exacerbating renal damage [9]. In murine models, the accumulation of macrophages has been correlated with DN progression [10]. Interestingly, a negative correlation was observed between the percentage of M1 macrophages and renal function, while M2 macrophages showed potential for mitigating kidney injury in DN mice [11]. Also, the cytokine landscape further accentuates its determinative role in DN advancement. Interleukin (IL)-6 levels are positively correlated with DN progression [12]. Moreover, the involvement of IL-1β, IL-18, and IL-17 A in the genesis and progression of DN has been well-documented [8]. Diminished TNF-α expression correlates with reduced urinary albumin excretion, indicating a significant relationship between TNF-α and DN pathogenesis [13]. Delving deeper into immune-related genes that regulate the functions of these immune cells and cytokines suggests that they might be of paramount importance as potential biomarkers. For instance, IDO1, a metabolic gene, has emerged as a novel immune biomarker for DN that is correlated with immune cell infiltration [14]. Besides, the activation of NF-κB, a master regulator of inflammation and pro-inflammatory chemokine transcription in tubular epithelial cells, is a marker for progressive DN [7, 15]. Interestingly, genome-wide transcriptome analyses of DN patients have unveiled the up-regulation of JAK1/2 and STAT1/3, underscoring their relevance [16]. Consequently, the endeavor to comprehend the pivotal immunomodulatory mechanisms underpinning the development and progression of DN elucidates the intricate nature of the disease, having great potential for identifying novel biomarkers that could potentially serve as diagnostic indicators and facilitate the design of innovative anti-inflammatory therapeutic strategies.
Molecular classifications based on RNA expression profiles related to patient prognosis enhance precision medicine during immune responses [17]. Due to the availability of various public cohorts providing bulk mRNA and single-cell RNA expression data, we can investigate the prognostic roles of immune-related genes in DN and their potential roles as drug targets [18, 19]. In the pursuit of identifying diagnostic biomarkers within the immune system for DN, the strategic application of advanced algorithms holds substantial promise. Support Vector Machine Recursive Feature Elimination (SVM-RFE), Random Forest, and Least Absolute Shrinkage and Selection Operator (LASSO) algorithms are pivotal tools for isolating key genes that can contribute significantly to disease diagnosis [8, 20]. SVM-RFE aids in selecting relevant features or genes from high-dimensional data [21]. By iteratively removing less informative features, SVM-RFE enhances the model’s discriminatory power, potentially unveiling key immune factors crucial for DN diagnosis [22]. The Random Forest algorithm can effectively handle complex and noisy datasets by aggregating multiple decision trees [23]. It identifies interactions and nonlinear relationships among immune factors, enhancing the potential to uncover intricate immune system dysregulation patterns associated with DN [24]. By imposing a penalty on regression coefficients and effectively shrinking less informative features to zero, LASSO is instrumental in feature selection as it mitigates the risk of overfitting, ensuring that only the most informative genes are considered potential biomarkers [25]. The integration of these algorithms empowers researchers to navigate the complexities of immune system interactions in DN [8]. The integrative approach enriches our understanding of the immunopathogenesis of DN, offering valuable insights for enhanced diagnostic strategies in the context of this debilitating condition.
In the present study, we acquired three distinct datasets from the Gene Expression Omnibus (GEO), including microarray data for DN and a single-cell RNA dataset. These datasets were used to identify DEGs between DN cases and healthy controls. Besides, the immune-related genes were found to be intersected with the immune gene database. We systematically sieved through potential diagnostic biomarkers intrinsic to DN by applying three machine-learning methodologies. Furthermore, the third DN dataset was employed as a validation cohort to affirm the viability of candidate genes, with the construction of receiver operating characteristic (ROC) curves. Moreover, the differential immune infiltration within 22 immune cell subsets from DN patients and normal controls was dissected employing CIBERSORT, unraveling the intricate relationship between the identified biomarkers and immune cells. Also, molecular docking was used to unearth potential therapeutic agents. By employing these comprehensive analyses, we aimed to identified the hub immune-related genes, as well as their expression profiles and the correlation with immune cell infiltration in DN, furthering the current understanding of the molecular intricacies of DN, expediting diagnostic advancements, and potentially facilitating novel therapeutic avenues.
Methods
Data gathering
mRNA expression data and clinical parameters were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo). Microarray datasets GSE96804, GSE30528, and GSE142025, as well as scRNA-seq dataset GSE183276, were included in this study. According to microarray data processing standards: (1) samples without gene expression were excluded; (2) the probe was converted into symbol according to the probe correspondence of the platform; (3) if one probe corresponded to multiple symbols, it was deleted; (4) if multiple probes corresponded to the same symbol, the median count was taken (Table 1).
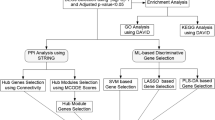
The immune-related genes were downloaded from the IMMPORT database (https://www.immport.org/home) and InnateDB database (https://www.innatedb.com/). The microRNA-lncRNA interaction relations were downloaded from ENCORI database (https://rnasysu.com/encori/). The gene and transcriptional regulation factor interaction relations were downloaded from TRRUST database (https://www.grnpedia.org/trrust/). Ethical approval was not necessary for this study because all data were retrieved from a public database. And the data processing process was showed in Fig. 1. Briefly, the same DEGs were obtained from the datasets of GSE96804 and GSE30528. After intersecting with the immune-related genes, the immune-related DEGs were used for SVM-REF, randomForest, and LASSO analyses to get the hub genes. Then the dataset of GSE142025 was used for model validation. And the dataset of GSE183276 was used to identify the expressions of hub genes in different cell types. Finally, the correlation of hub genes and immune cell infiltration and the drugs targeted to the hub genes were analyzed.

The flow chart of data processing process
Analysis of immune-related differentially expressed genes (irDEGs)
Hierarchical clustering on the samples in the datasets GSE96804 and GSE30528 was conducted to detected whether there are outlier samples. Then, by using the limma package (v3.50.3) of R (v4.1.3) (http://www.bioconductor.org/), we screened the DEGs in the dataset of GSE96804 and GSE30528 with the criteria of p-value < 0.05 and |log fold change|>1. To obtain immune-related differentially expressed gene (irDEGs), DEGs were overlapped with the immune-related genes obtained from the ImmPort database and the InnateDB database. The synergistically up-regulated and down-regulated genes in the two datasets were selected as the common irDEGs for subsequent analysis. Functional enrichment analysis was performed using the “clusterProfiler” R package (v4.7.1). Different immune-related genes enriched in Gene Ontology (GO) and KEGG pathway categories were identified. The GO terms and KEGG pathways were plotted using the R package ggplot2.
Multiple machine learning algorithms to identify the hub genes
To select reliable hub genes for constructing the DN prediction model, three machine learning algorithms, including Least Absolute Shrinkage and Selection Operator (LASSO), randomForest (RF), and support vector machine-recursive feature elimination (SVM-REF), were applied to analyze the GSE96084 dataset by “glmnet (v4.1)”, “randomForest (v4.7)”, and caret (v6.0) packages of R software, respectively. The overlapping hub genes from the three algorithms were obtained as the hub genes for constructing the comprehensive prediction model by logistic regression. The model score was calculated according to the following formula. Score = (4.825259 * PTGDS) + (1.384 * EGF) + (1.456 * RGS2) + (1.475 * PLTP). The prediction performance of the constructed model indicated by ROC was analyzed on the training dataset GSE96084 and GSE30528 and the independent validation dataset GSE142025, respectively by using the “pROC (v1.18)” package of R language.
Hub gene correlation analysis
The interaction network of competing endogenous RNAs (ceRNAs) was constructed using data obtained from the ENCORI database for lncRNA-miRNA-mRNA interactions. Additionally, gene-to-transcription factor interactions were sourced from the TRRUST database. Cytoscape software was used for the visualization of this network. Spearman analysis was performed on the GSE96084 dataset to evaluate the correlations among the identified hub genes, namely EGF, PLTP, RGS2, and PTGDS. The strength and significance of these correlations were depicted graphically using the R package corrplot (v0.92).
Single-cell RNA analysis of the hub genes
The single-cell dataset GSE183276 was preprocessed using the R-package Seurat (v4.2). A total of 12 DN and 18 normal samples were included in subsequent analysis. Each gene was found to be expressed in at least 3 cells. The cells expressing > 500 but ≤ 5,000 genes and the counts ranging from 600 to 40,000 were selected. Meanwhile, 50% of mitochondrial genes were set as cut-off values for further filtration. After identifying 2,000 hypervariable genes by the FindVariableFeatures function for analysis, the number of principal components (PCs) was adjusted to 50 to generate cell clusters annotated by original publications and exhibited using the “uMAP” function. The hub genes were visualized in each cell cluster.
Correlation analysis between hub genes and immune cell infiltration
CIBERSORT was used to estimate the abundances of member cell types in a mixed cell population using gene expression data on the GSE96084 dataset. With CIBERSORT in R software, the proportion of infiltrating immune cells was evaluated in DN and normal groups and visualized by the “ggpubr v0.4” package of R language. Principal component analysis (PCA) was used to identify the difference in the immune cell infiltration between the DN and normal groups. The Spearman correlation analysis of immune cells and gene expression was performed and visualized by the R package “corrplot (v0.92)”. The Wilcoxon test was used to compare the levels of immune infiltration between the disease and the control, and a p-value < 0.05 indicated a significant difference. Violin plots were generated using the “vioplot” package in R to visualize the differences in immune cell infiltration expression levels between the DN and control groups.
Molecular docking screening of potential therapeutic drugs
The corresponding PDB files of the hub genes were downloaded from the RCSB PDB database (https://www.rcsb.org/). The structural information of corresponding small molecule compounds was downloaded from any ZINC database file (https://zinc15.docking.org/substances/subsets/fda/). After setting other relevant parameters, autodock-vina was used to separately dock with the small molecule compounds and search for the minimum-energy docking conformations. PyMol demonstrated docking conformation for proteins and compounds.
Cell culture
The HK-2 cells were cultured in DMEM/F12(1:1) medium (SH30243.FS, c HyClone, UT, USA) containing 10% FBS (16000-044, GIBCO, MA, USA) in a humidified atmosphere with 5% CO2 at 37 ℃. Glucose (G8150, Solarbio, Beijing, China) with different concentration (5 mM, 20 mM, 30 mM) was used to stimulate HK-2 cells for 24 h.
Animal models
Twelve 8-week-old C57/J male mice were used to establish DN model induced by streptozotocin (STZ) as previously described [26]. The mice were divited into DN group (n = 6) and NC group (n = 6) and kept in the environment with constant temperature and humidity. After fasting for 12 h, the mice in the DN group were intraperitoneally injected with STZ (150 mg/kg), then the mice were fed with water with 10% sucrose for 3 days. The fasting blood-glucose (FBG) level was detected every 3 days after surgery and the FBG level >8.3 mM for 3 consecutive times meant the diabetes model was successfully constructed. Then the mice were kept for another 8 weeks after successful establishment of the diabetes model. After anesthesia, the mice were perfused via heart, and the kidney tissues were obtained for further studies.
qPCR analysis
Total RNA was isolated from HK-2 cells and kidney tissues by using Total RNA extraction solution (10606ES60, Yeasen Biotechnology Co., Ltd, Shanghai, China). Then equal amount of RNA was reverse transcribed into cDNA by employing Hifair® II 1st Strand cDNA Synthesis Kit (11119ES60, Yeasen Biotechnology Co., Ltd, Shanghai, China). qPCR experiments were performed using the Fluorescent quantitative PCR instrument (CG-05, HANGZHOUJINGGE scientific instrument Co., Ltd, Hangzhou, China) with Hieff® qPCR SYBR Green Master Mix (11203ES03, Yeasen Biotechnology Co., Ltd, Shanghai, China). β-actin was used as the internal control. And the primers were showed in Table S4.
Western blot analysis
The protein was collected from HK-2 cells by using RIPA buffer. After detecting the protein concentration by using BCA kit, an equal amount of protein was separated by SDS-PAGE. Then the proteins on the gels were transferred to NC membranes. The membranes were blocked by 5% skim milk powder and incubated with primary antibodies (Table S5), following by being incubated with the secondary antibodies (Table S5). The signals on membranes were visualized by ECL reagent.
Statistical analysis
All statistical analyses were based on R 4.1.2. Wilcoxon rank-sum test was used to compare the differences in various values (expression amount, infiltration proportion, and various eigenvalues, etc.) between the DN and normal group. NS indicates p > 0.05, * indicates p < 0.05, ** indicates p < 0.01, *** indicates p < 0.001, **** indicates p < 0.0001, otherwise specified.
Results
Identification of the irDEGs and exploring their functions in DN disease
To identify and characterize the irDEGs between a disease group (DN group) and a normal group, we conducted a comprehensive bioinformatics analysis focusing on immune-associated genes (Table S1). Using stringent criteria (|log2FC| > 1, corrected p-value < 0.05), we screened the GSE96804 and GSE30528 datasets after making sure there are no outlier samples in these two datasets (Fig. 2A). Our analysis showed 644 DEGs (95 immune-associated) in GSE96804 (Table S2) and 427 DEGs (88 immune-associated) in GSE30528 (Table S3), depicted in Fig. 2B-E. The intersection of immune-associated DEGs between these two datasets yielded 14 co-expressed immune-related differentially expressed genes (co-irDEGs), with 4 up-regulated and 10 down-regulated co-irDEGs (Fig. 2F). Enrichment analysis uncovered functional pathways such as positive regulation of the MAPK cascade, EGFR tyrosine kinase inhibitor resistance, and Ras signaling (Fig. S1).

Differential expression analysis of genes in diabetic nephropathy (DN) and normal controls. (A) Hierarchical clustering on the samples of the datasets GSE96804 and GSE30528. (B) Volcano plot depicting the distribution of differentially expressed genes in the GSE30528 dataset. (C) Heatmap illustrating the expression patterns of the top 50 immune-related differentially expressed genes in GSE30528. (D) Volcano plot displaying the distribution of differentially expressed genes in the GSE96804 dataset. (E) Heatmap showing the expression profiles of the top 50 immune-related DEGs in GSE96804. (F) Venn diagram illustrating the intersection of up-regulated and down-regulated genes differentially expressed in both GSE30528 and GSE96804 datasets
Identification of immune-related hub genes in DN disease by using machine learning methods
To identify the specific genes among the 14 co-irDEGs associated with DN, we employed a combination of LASSO regression, SVM-RFE algorithm, and Random Forest analysis. Initially, we fine-tuned the ntree and mtry parameters of the Random Forest algorithm, identifying the optimal values as mtry = 13 and ntree = 150, which led to the lowest and most stable model error rate (Fig. 3A, B). Utilizing these parameters, the Random Forest model was trained, and gene weight coefficients were derived (Fig. 3C). Subsequently, based on the MeanDecreaseGini ranking, the top 5 genes were selected as candidate genes. Furthermore, employing the SVM-RFE algorithm, we determined that the model achieved its highest accuracy with 6 candidate genes (Fig. 3D). The LASSO algorithm analysis yielded eight non-zero features, highlighting the key genes associated with DN (Fig. 3E-F).

Screening of hub genes for diabetic nephropathy diagnosis. (A) Parameter optimization (mtry) training of the random forest algorithm. (B) Parameter optimization (ntree) training of the random forest algorithm. (C) Weight coefficient distribution analysis using random forest, aiding the selection of potential diagnostic genes. (D) Support Vector Machine Recursive Feature Elimination (SVM-RFE) algorithm employed for potential diagnostic gene selection. (E) Analysis of coefficient distribution through LASSO regression. (F) Application of LASSO regression for the selection of potential diagnostic genes. (G) Venn diagram showcasing the four specific genes identified through the convergence of the three algorithms. H. ROC curve of the hub gene and integrated model in GSE96804. (I) ROC curve of the hub gene and integrated model in the GSE30528 dataset. (J) ROC curve of the hub gene and integrated model in the GSE142025 dataset
Through the intersection of candidate genes, four hub genes, i.e., epidermal growth factor (EGF), phospholipid transfer protein (PLTP), regulator of G protein signaling 2 (RGS2), and glycoprotein prostaglandin D2 synthase (PTGDS), were selected by Random Forest, SVM-RFE, and LASSO algorithms (Fig. 3G). To establish the diagnostic utility of these hub genes, we constructed a logistic regression model on GSE96804 and GSE30528 datasets. Next, ROC curve analyses were conducted to evaluate the sensitivity and specificity of the four hub genes and the integrated model in DN diagnosis. EGF, PLTP, RGS2, PTGDS, and the comprehensive model exhibited substantial diagnostic potential in distinguishing DN patients from healthy individuals. The area under the curve (AUC) for the comprehensive model reached an impressive 0.995 on GSE96804 and 1.00 on GSE30528 (Fig. 3H, I). In order to enhance the reliability of our findings, we validated the model using the GSE142025 dataset, revealing consistent AUC values > 0.9, which was indicative of significant and meaningful diagnostic capacity in DN (Fig. 3J). Furthermore, we measured the relevant metrics including Precision, Recall, and F1-score of the model, the results showed the Precision, Recall, and F1-score of the model were 1, 0.93, and 0.96, respectively for GSE96804, and 1, 1, and 1, respectively for GSE30528 and GSE142025. These findings underscore the robustness of our integrated approach in identifying and validating key hub genes for potential use as diagnostic markers in DN.
We constructed a multifactor network around the identified hub genes using data sourced from the ENCORI database for lncRNA-miRNA-mRNA interactions and the TRRUST database for gene-transcriptional regulator interactions. The final network encompassed 4 hub genes, 17 miRNAs, 5 lncRNAs, and 6 transcription factors (TFs) (Fig. S2A). Notably, interactions with miRNAs were absent in hub genes EGF, PLTP, and PTGDS; however, each demonstrated at least one associated TF in transcriptional regulation. On the other hand, while RGS2 lacked an associated TF, RGS2 exhibited multiple interactions as competing endogenous RNA. Additionally, Spearman correlation analysis among the hub genes revealed significant correlations, particularly for PTGDS, displaying significant associations with the remaining three hub genes (Fig. S2B). This intricately woven multifactor network offers insights into the regulatory complexities underlying the identified hub genes, highlighting potential cross-talk among various regulatory layers.
Analysis of the expression profiles of hub genes among different cell subsets
In order to gain deeper insights into the expression profiles of the identified hub genes, an examination of their expression across various cell types was conducted using single-cell RNA sequencing data (GSE183267), which included 12 DN and 18 normal samples. After quality control and filtering, the remaining 64,726 cells in 30 samples were used for further analysis. By using sub-clustering and annotation, these cells were categorized into distinct cell types, namely endothelial cells, epithelial cells, immune cells, and stromal cells (Fig. 4A). Notably, Fig. 4B highlights a relatively lower proportion of epithelial cells than other cell types. Remarkably, Fig. 4C underscores the enriched expression of the four hub genes within epithelial cells. Further details regarding single-cell data preprocessing are provided in Fig. S1.

Single-cell analysis of hub genes. (A) Two-dimensional UMAP projection depicting the cell subsets derived from scRNA-seq data. (B) Distribution representation of individual cell clusters as defined through scRNA-seq data analysis. (C) Expression patterns of hub genes across different cell clusters showcased in the UMAP projection
The correlation of hub gene and immune infiltration
The immune microenvironment has a profound impact on the diagnosis of DN. Using CIBERSORT to calculate the proportions of 22 immune cell types, these values were projected onto PCA plots, demonstrating a distinct separation between DN and normal groups (Fig. 5A). Moreover, intricate interactions among immune cells were explored through Spearman correlation analysis, indicating positive associations among T cells, CD4 T memory-activated cells, and activated dendritic cells (Fig. 5B). The present study also identified significant differences in the proportions of macrophage M1, macrophage M2, CD8+ T cells, and neutrophils between DN and normal groups using the Wilcoxon test (Fig. 5C), indicating the relevance of specific immune cell subsets to DN pathogenesis. Furthermore, the correlation analysis unveiled a positive association between the expression levels of all four identified hub genes and neutrophil infiltration (Fig. 5D). Collectively, these findings illuminate the intricate immune cell dynamics in DN.

Immune cell interactions and correlations with hub genes in diabetic nephropathy (DN). (A) Principal Component Analysis (PCA) plot illustrating distinct immune cell infiltration distributions between the DN and normal groups. (B) Heat map displaying the correlation analysis results among various immune cell types. (C) Box diagrams depicting the differential immune cell infiltration between the DN and normal groups, with statistical significance denoted (p < 0.05). (D) Correlation analysis revealing the associations between the four identified hub genes and infiltrating immune cells
Identification of potential therapeutic drugs targeted the hub genes by using molecular docking
We employed Molecular Docking to identify potential therapeutic options for DN, i.e., to assess the interactions between three of the identified hub genes and various compounds. PLTP was excluded from the molecular docking analysis due to the absence of protein structure information. Consequently, molecular docking was exclusively conducted for the remaining three hub genes. This analysis identified compounds displaying the lowest binding energies, indicative of favorable interactions, as promising candidates. Notably, cysteamine exhibited the lowest binding energy (-2.3) to the EGF protein (Fig. 6A), eltrombopag demonstrated the most favorable interaction with the PTGDS protein, with a binding energy of -10.1 (Fig. 6B), and DMSO exhibited the smallest binding energy of -2.1 with the RGS2 protein (Fig. 6C). These findings collectively suggest that cysteamine, eltrombopag, and DMSO could be used as therapeutic agents for addressing the complexities of DN.

The docking conformation analysis of hub genes with their docked compounds. (A) Analysis of the docking conformation and EGF and Cysteamine. (B) Analysis of the docking conformation and PTGDS and eltrombopag. (C) Analysis of the docking conformation and RGS2 and DMSO. PyMol shows docking conformation and hydrogen bonding with the color symbols of cyan for small molecules and blue for amino acid residues forming hydrogen bonds with small molecules
Validation of the expressions of EGF, PLTP, RGS2, and PTGDS in vivo and in vitro
To further validate the expressions of EGF, PLTP, RGS2, and PTGDS in DN, we stimulated HK-2 cells with different concentration of glucose for 24 h. As shown in Fig. 7, the mRNA levels of EGF, PLTP, RGS2, and PTGDS were significantly increased in HK-2 cells treated with glucose compared with those in the vehicle group (Fig. 7A). In addtion, the protein expressions of EGF, PLTP, RGS2, and PTGDS were elevated in HK-2 cells treated with glucose in a dose dependent manner (Fig. 7B, C). Furhtermore, we established a mice DN model with STZ and the kidney tissues were collected. The results revealed the mRNA levels of EGF, PLTP, RGS2, and PTGDS were observably increased in the kidney tissues of mice with DN compared with those in the control group. These results indicated that the expressions of EGF, PLTP, RGS2, and PTGDS were up-regulated both in HK-2 cells treated with glucose and kidney tissues of mice with DN.

The expressions of EGF, PLTP, RGS2, PTGDS in vivo and in vitro. HK-2 cells were treated with different concentration of glucose (5 mM, 20 mM, 30 mM) for 24 h, (A) mRNA expressions of EGF, PLTP, RGS2, PTGDS were detected. N = 3, **p < 0.01 vs. Vehicle, ***p < 0.001 vs. Vehicle. (B-C) Representative western blotting images (B) and quantifications (C) of EGF, PLTP, RGS2, PTGDS were showed. N = 3, *p < 0.05 vs. Vehicle, **p < 0.01 vs. Vehicle, ***p < 0.001 vs. Vehicle. (D) mRNA expressions of EGF, PLTP, RGS2, PTGDS were detected in the kidney tissues of mice with DN or the control mice. N = 6 per group, ***p < 0.001 vs. Ctrl
Discussion
DN is among the most common chronic complications of diabetes mellitus, posing a serious threat to human health. Due to the lack of specific indicators for the early diagnosis of DN, some DN patients miss the opportunity for early diagnosis and treatment. In this study, 14 co-irDEGs were obtained. Among these co-irDEGs, four hub genes, including EGF, PLTP, RGS2, and PTGDS, were pinpointed as promising candidates capable of accurately predicting DN, which expressions were validated to be significantly increased in HK-2 cells treated with glucose and the kidney tissues of mice with DN. Notably, these four hub genes exhibited robust expression profiles within epithelial cells, suggesting their probable significance in renal physiology. Intriguingly, the expression levels of these hub genes were correlated with the infiltration of neutrophils, thus highlighting a potential interplay between gene expression patterns and immune cell dynamics. Beyond their diagnostic potential, this study identified cysteamine, eltrombopag, and dimethyl sulfoxide (DMSO) as conceivable therapeutic agents for DN.
The 14 co-irDEGs, comprising 4 up-regulated and 10 down-regulated genes identified in the present study, constitute a pivotal step in unraveling the genetic underpinnings of DN. The subsequent enrichment analyses provided invaluable insights into the functional context of these differentially expressed genes. Notably, the enrichment results showcased pronounced associations with processes such as positive regulation of the MAPK cascade, EGFR tyrosine kinase inhibitor resistance, and activation of dendritic cells. These enriched pathways, resonating with immune and inflammatory responses, underline the intricate interplay between immune dynamics and DN progression [27]. This aligns seamlessly with recent research affirming the role of dendritic cells in DN. Demonstrably, hyperglycemia-induced up-regulation of CD83 and CD86 in dendritic cells triggers cascades of cytokines, including IL-6 and IL-12, through signaling pathways that discernibly impact kidney function [28]. Not only MAPK-pathway, our results also revealed EGFR tyrosine kinase inhibitor resistance, and Ras signaling were enriched in the DEGs between DN and normal controls. Previous studies showed several EGFR ligands were elevated in DN. Inhibiting the tyrosine kinase of EGFR could alleviate the development of DN [29]. However, our study found EGF, the ligand of EGFR, was down-regulated in DN. Others showed EGF increased the degradation of EGFR to inhibit its activation [30]. Furthermore, EGF was reported to protect podocytes from high glucose-induced injury [31]. Our study provided a potential role of EGF-EGFR signaling in DN formation. In addition, RGS2 was revealed to inhibit Ras signaling [32], and RAS inhibitors were the first-line drugs for DN. However, the role of RGS2 in DN development is unknown. We found that the expression of RGS2 was decreased in DN, further functional assays are necessary to confirm its effects on DN. Collectively, these findings fortify the understanding of the relationships of hub genes and regulated sinalings in DN development and progression, elucidating a crucial facet of the complex pathogenesis of the disease.
Based on the LASSO, SVM-RFE algorithms, and RF analysis, four hub genes, including EGF, PLTP, RGS2, and PTGDS, were identified. This integrative approach harnesses the power of machine learning to refine our understanding of DN-related gene selection. EGF is a polypeptide with 53 amino acids and multiple roles, such as regulating cell growth, proliferation, and tissue repair [33]. It has been linked to kidney damage biomarkers [34], correlating with estimated glomerular filtration rate (eGFR) loss and predicting renal damage [35]. In addition, EGF protects podocytes from high glucose-induced apoptosis [36] by modulating autophagy and the PI3K/AKT/mTOR signaling pathway [31]. EGFR deletion delays the recovery of renal injury [37], although the EGFR pathway is activated in diabetic kidney diseases [38]. PLTP, a widely expressed key lipid transfer protein [39], can alter responses to pro-inflammatory stimuli [40]. Increased plasma PLTP activity in diabetics correlates with CRP levels [41], suggesting an influence on DN through inflammation modulation rather than direct progression control. RGS2 inhibits Gαq and Gαi signaling [42], particularly in kidney tubules [43]. RGS2 deletion exacerbates kidney fibrosis via AngI/AT1R signaling [44]. RGS2 regulates chemokine receptors like CXCR4 [45], with higher CXCR4 expression in RGS2 knockout mice. RGS2 lacks associated TFs but has competing endogenous RNA interactions. PTGDS, a lipocalin superfamily member [46], influences kidney failure progression [47]. Its overexpression impacts metabolite levels in serum and urine, potentially causing renal lipid toxicity and damage [48]. While PTGDS is involved in DN, the exact mechanism remains unclear. These findings potentially unravel intricate molecular mechanisms underlying the condition and facilitating the discovery of novel therapeutic avenues.
Our single-cell analysis yielded insightful findings on the composition of different cell types within the studied context. Interestingly, the proportion of epithelial cells was relatively low, underscoring the cellular diversity and complexity inherent to the microenvironment under investigation [49]. Within this intricate landscape, the emergence of four hub genes that were abundantly expressed within epithelial cells is particularly noteworthy. It was found that the EGF can activate EGFR signaling in renal epithelial cells, thus exacerbating diabetic kidney injury [50]. This aligns intriguingly with their role as potential biomarkers for predicting DN, potentially suggesting a specialized function or regulatory role within the epithelial cell population. Observing enriched expression of these hub genes within a subset of cells highlights the nuanced interplay between gene expression patterns and cellular dynamics [51], potentially elucidating their functional significance in DN development and progression. Moreover, the lower proportion of epithelial cells prompts consideration of their interactions with other cell types, such as immune cells or stromal cells within the microenvironment [51]. Further exploration of these intercellular communication networks could offer insights into the broader regulatory mechanisms that underlie DN and its associated molecular signatures [52]. The convergence of our single-cell analysis data suggests an intricate relationship between epithelial cells and the identified hub genes in the context of DN.
By scrutinizing the involvement of various immune cell types, we gained a deeper understanding of their intricate interplay in relation to disease advancement. The proportion of macrophage M1, macrophage M2, CD8 + T cells, and neutrophils was significantly different between DN and the normal group, thus suggesting that these immune cells may be involved in the occurrence and development of DN. Immune cell infiltration is a significant feature in diabetic samples that gradually became an identified biomarker in the early prediction of DN [53]. There was a phenotypic imbalance of M1/M2 macrophages in DN renal tissues, with a predominance of M1-type infiltration, which leads to inflammation and fibrosis [54]. Spearman correlation analysis detected a significant positive correlation with the infiltration level of neutrophils, thus suggesting that the expression level of these four hub genes may be among the factors affecting neutrophils infiltration levels in DN patients [53]. Neutrophil counts are closely associated with DKD in patients with autoimmune diabetes [55]. The ratio of neutrophils could be higher in normal controls, but with the disease progress, these inflammatory cells substantially increase in DN patients. Neutrophil-secreting enzymes and oxidation products can damage the local microenvironment, resulting in tissue injury [56]. Besides, our findings revealed that EGF, PLTP, RGS2, and PTGDS genes were associated with neutrophil infiltration. Consistently, the EGF-induced chemotactic activity involved RhoA signaling in neutrophils to produce pro-neutrophil activities in asthma patients [57]. PLTP can prevent neutrophil degranulation but serine proteases [58]. In a broader context, the immune system and its related genes significantly affect the pathogenesis and progression of DN. An in-depth exploration of immune cell infiltration and immune factor modulation offers insights into their precise roles in the etiology of DN, thereby furnishing valuable insights for developing novel therapeutic strategies.
Previous studies reported JAK/STAT pathways aggravated the formation of diabetic kidney disease [59]. Furthermore, NF-κB activation was involved in the development of diabetic nephropathy [60]. Interestingly, cysteamine, which was the predicted drug for EGF, a hub gene identified in our study, decreased the activation of NF-κB and the expression of iNOS [61]. In addition, eltrombopag, another candidate drug that targeted PTGDS protein, would induce phosphorylation of STAT5 [62]. These researches indicate cysteamine and cysteamine may be associated with the molecular mechanisms of DN.
Although this study offers valuable insights into potential biomarkers and mechanisms associated with DN, the limitations stemming from data variability and the reliance on bioinformatic analyses underscore the need for future research combining basic experiments with computational approaches. Furthermore, even though we identified some candidate drugs targeted the hub genes, the validation study was not performed to confirm the effects of them on DN therapy, further studies are necessary to unveil their functions both in animals and clinical trials. This multifaceted approach enhances the robustness of the reported findings and lays the foundation for informed and effective strategies for diagnosing and treating DN.
Conclusion
The present study represents a pivotal advancement in the realm of DN research, ranging from the early detection of the disease to identifying potential therapeutic strategies. Identifying the four hub genes as accurate predictors of DN enables unraveling previously unexplored immune-related mechanisms and can be potentially used to guide future therapeutic strategies.
Data availability
All data generated or analyzed during this study are included in this article and supplementary information files.
Access links to the public data sets analyzed in the article are as follows:
GSE96804(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96804).
GSE30528(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30528).
GSE142025(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE142025).
GSE183276(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183276).
Abbreviations
- DN:
-
Diabetic nephropathy
- DEGs:
-
Differentially expressed genes
- SVM-RFE:
-
Support Vector Machine Recursive Feature Elimination
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- GEO:
-
Gene Expression Omnibus
- ROC:
-
Receiver operating characteristic
- irDEGs:
-
Immune-related differentially expressed genes
- GO:
-
Gene Ontology
- RF:
-
RandomForest
- ceRNAs:
-
Competing endogenous RNAs
- PCA:
-
Principal component analysis
- EGF:
-
Epidermal growth factor
- PLTP:
-
Phospholipid transfer protein
- RGS2:
-
Regulator of G protein signaling 2
- PTGDS:
-
Prostaglandin D2 synthase
- AUC:
-
Area under the curve
- TFs:
-
Transcription factors
- eGFR:
-
Estimated glomerular filtration rate
References
Kos I, Prkačin I, [diabetic, nephropathy as a cause of chronic kidney disease]. Acta Med Croatica. 2014;68:375–81.
Vaidya SR, Aeddula NR. Chronic renal failure. StatPearls. Treasure Island (FL) with ineligible companies. Disclosure: Narothama Aeddula declares no relevant financial relationships with ineligible companies.: StatPearls Publishing Copyright © 2023. StatPearls Publishing LLC.; 2023.
Samsu N. Diabetic Nephropathy: challenges in Pathogenesis, diagnosis, and treatment. Biomed Res Int. 2021;2021:1497449.
Wang G, Ouyang J, Li S, Wang H, Lian B, Liu Z, et al. The analysis of risk factors for diabetic nephropathy progression and the construction of a prognostic database for chronic kidney diseases. J Translational Med. 2019;17:264.
Smith S, Normahani P, Lane T, Hohenschurz-Schmidt D, Oliver N, Davies AH. Prevention and Management Strategies for Diabetic Neuropathy. Life. 2022;12:1185.
Poon MML, Farber DL. The whole body as the System in systems Immunology. iScience. 2020;23:101509.
Chen J, Liu Q, He J, Li Y. Immune responses in diabetic nephropathy: pathogenic mechanisms and therapeutic target. Front Immunol. 2022;13:958790.
Sun Y, Dai W, He W. Identification of key immune-related genes and immune infiltration in diabetic nephropathy based on machine learning algorithms. IET Syst Biol. 2023;17:95–106.
Zheng Z, Zheng F. Immune cells and inflammation in Diabetic Nephropathy. J Diabetes Res. 2016;2016:1841690.
Calle P, Hotter G. Macrophage phenotype and fibrosis in Diabetic Nephropathy. Int J Mol Sci. 2020;21.
Zhang X, Yang Y, Zhao Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS ONE. 2019;14:e0221991.
Su H, Lei CT, Zhang C. Interleukin-6 signaling pathway and its role in kidney disease: an update. Front Immunol. 2017;8:405.
Sun L, Kanwar YS. Relevance of TNF-α in the context of other inflammatory cytokines in the progression of diabetic nephropathy. Kidney Int. 2015;88:662–5.
Yu K, Li D, Xu F, Guo H, Feng F, Ding Y, et al. IDO1 as a new immune biomarker for diabetic nephropathy and its correlation with immune cell infiltration. Int Immunopharmacol. 2021;94:107446.
Sun HJ, Xiong SP, Cao X, Cao L, Zhu MY, Wu ZY, et al. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 2021;38:101813.
Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, Susztak K. Transcriptome analysis of human diabetic kidney disease. Diabetes. 2011;60:2354–69.
Nakagawa H, Fujita M, Fujimoto A. Genome sequencing analysis of liver cancer for precision medicine. Semin Cancer Biol. 2019;55:120–27.
Tomczak K, Czerwińska P, Wiznerowicz M. Review the Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19:A68–77.
Singh R, Zogg H, Wei L, Bartlett A, Ghoshal UC, Rajender S, et al. Gut Microbial Dysbiosis in the Pathogenesis of Gastrointestinal Dysmotility and Metabolic disorders. J Neurogastroenterol Motil. 2021;27:19–34.
Zhang B, Dong X, Hu Y, Jiang X, Li G. Classification and prediction of spinal disease based on the SMOTE-RFE-XGBoost model. PeerJ Comput Sci. 2023;9:e1280.
Zhang Y, Deng Q, Liang W, Zou X. An efficient feature selection strategy based on multiple support Vector Machine Technology with gene expression data. Biomed Res Int. 2018;2018:7538204.
Lin X, Li C, Zhang Y, Su B, Fan M, Wei H. Selecting feature subsets based on SVM-RFE and the overlapping ratio with applications in Bioinformatics. Molecules. 2017;23.
Yang J, Sui H, Jiao R, Zhang M, Zhao X, Wang L, et al. Random-Forest-Algorithm-based applications of the basic characteristics and serum and imaging biomarkers to diagnose mild cognitive impairment. Curr Alzheimer Res. 2022;19:76–83.
Acharjee A, Larkman J, Xu Y, Cardoso VR, Gkoutos GV. A random forest based biomarker discovery and power analysis framework for diagnostics research. BMC Med Genom. 2020;13:178.
Guo L, Wang Z, Du Y, Mao J, Zhang J, Yu Z, et al. Random-forest algorithm based biomarkers in predicting prognosis in the patients with hepatocellular carcinoma. Cancer Cell Int. 2020;20:251.
Wang X, Jiang L, Liu XQ, Huang YB, Zhu W, Zeng HX, et al. Identification of genes reveals the mechanism of cell ferroptosis in Diabetic Nephropathy. Front Physiol. 2022;13:890566.
Yang J, Peng L, Tian Y, Tang W, Peng L, Ning J, et al. Identification of hub genes involved in Tubulointerstitial Injury in Diabetic Nephropathy by Bioinformatics Analysis and Experiment Verification. J Immunol Res. 2022;2022:7907708.
Li Y, Li Y, Yang T, Wang M. Dioscin attenuates oxLDL uptake and the inflammatory reaction of dendritic cells under high glucose conditions by blocking p38 MAPK. Mol Med Rep. 2020;21:304–10.
Harris RC. The role of the epidermal growth factor receptor in Diabetic kidney disease. Cells. 2022;11.
Huang CC, Lee CC, Lin HH, Chang JY. Cathepsin S attenuates endosomal EGFR signalling: a mechanical rationale for the combination of cathepsin S and EGFR tyrosine kinase inhibitors. Sci Rep. 2016;6:29256.
Sun Y, Deng M, Ke X, Lei X, Ju H, Liu Z, et al. Epidermal Growth Factor Protects against High Glucose-Induced Podocyte Injury Possibly via modulation of Autophagy and PI3K/AKT/mTOR signaling pathway through DNA methylation. Diabetes Metab Syndr Obes. 2021;14:2255–68.
Tso PH, Wang Y, Yung LY, Tong Y, Lee MM, Wong YH. RGS19 inhibits Ras signaling through Nm23H1/2-mediated phosphorylation of the kinase suppressor of Ras. Cell Signal. 2013;25:1064–74.
Zhang Y, Xu L, Lu Y, Zhang J, Yang M, Tian Y, et al. Protective effect of Cordyceps sinensis against diabetic kidney disease through promoting proliferation and inhibiting apoptosis of renal proximal tubular cells. BMC Complement Med Ther. 2023;23:109.
Satirapoj B, Dispan R, Radinahamed P, Kitiyakara C. Urinary epidermal growth factor, monocyte chemoattractant protein-1 or their ratio as predictors for rapid loss of renal function in type 2 diabetic patients with diabetic kidney disease. BMC Nephrol. 2018;19:246.
Gipson DS, Trachtman H, Waldo A, Gibson KL, Eddy S, Dell KM, et al. Urinary epidermal growth factor as a marker of Disease Progression in Children with Nephrotic Syndrome. Kidney Int Rep. 2020;5:414–25.
Li D, Wang N, Zhang L, Hanyu Z, Xueyuan B, Fu B, et al. Mesenchymal stem cells protect podocytes from apoptosis induced by high glucose via secretion of epithelial growth factor. Stem Cell Res Ther. 2013;4:103.
Morin NJ, Laurent G, Nonclercq D, Toubeau G, Heuson-Stiennon JA, Bergeron MG, et al. Epidermal growth factor accelerates renal tissue repair in a model of gentamicin nephrotoxicity in rats. Am J Physiol. 1992;263:F806–11.
Harskamp LR, Gansevoort RT, van Goor H, Meijer E. The epidermal growth factor receptor pathway in chronic kidney diseases. Nat Rev Nephrol. 2016;12:496–506.
Vuletic S, Dong W, Wolfbauer G, Day JR, Albers JJ. PLTP is present in the nucleus, and its nuclear export is CRM1-dependent. Biochim Biophys Acta. 2009;1793:584–91.
Schlitt A, Liu J, Yan D, Mondragon-Escorpizo M, Norin AJ, Jiang XC. Anti-inflammatory effects of phospholipid transfer protein (PLTP) deficiency in mice. Biochim Biophys Acta. 2005;1733:187–91.
Tan KC, Shiu SW, Wong Y, Tam S. Plasma phospholipid transfer protein activity and subclinical inflammation in type 2 diabetes mellitus. Atherosclerosis. 2005;178:365–70.
Cho J, Min HY, Lee HJ, Hyun SY, Sim JY, Noh M et al. RGS2-mediated translational control mediates cancer cell dormancy and tumor relapse. J Clin Invest. 2023;133.
Gurley SB, Griffiths RC, Mendelsohn ME, Karas RH, Coffman TM. Renal actions of RGS2 control blood pressure. J Am Soc Nephrol. 2010;21:1847–51.
Jang H-S, Kim JI, Noh M, Rhee MH, Park KM. Regulator of G protein signaling 2 (RGS2) deficiency accelerates the progression of kidney fibrosis. Biochimica et Biophysica Acta (BBA) -. Mol Basis Disease. 2014;1842:1733–41.
Hwang IY, Park C, Harrison K, Boularan C, Galés C, Kehrl JH. An essential role for RGS protein/Gαi2 interactions in B lymphocyte-directed cell migration and trafficking. J Immunol. 2015;194:2128–39.
Hu S, Ren S, Cai Y, Liu J, Han Y, Zhao Y, et al. Glycoprotein PTGDS promotes tumorigenesis of diffuse large B-cell lymphoma by MYH9-mediated regulation of Wnt–β-catenin–STAT3 signaling. Cell Death Differ. 2022;29:642–56.
Nagata N, Fujimori K, Okazaki I, Oda H, Eguchi N, Uehara Y, et al. De novo synthesis, uptake and proteolytic processing of lipocalin-type prostaglandin D synthase, beta-trace, in the kidneys. FEBS J. 2009;276:7146–58.
Liu Y-R, Tang Z-S, Duan J-A, Chen L, Sun J, Zhou R, et al. ER-depletion lowering the ‘hypothalamus-uterus-kidney’ axis functions by perturbing the renal ERβ/Ptgds signalling pathway. Aging. 2019;11:9500–29.
De Gregorio C, Contador D, Campero M, Ezquer M, Ezquer F. Characterization of diabetic neuropathy progression in a mouse model of type 2 diabetes mellitus. Biol Open. 2018;7.
Chen J, Harris RC. Interaction of the EGF receptor and the Hippo Pathway in the Diabetic kidney. J Am Soc Nephrol. 2016;27:1689–700.
Swamy M, Jamora C, Havran W, Hayday A. Epithelial decision makers: in search of the ‘epimmunome’. Nat Immunol. 2010;11:656–65.
Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell–cell interactions and communication from gene expression. Nat Rev Genet. 2021;22:71–88.
Zhou W, Liu Y, Hu Q, Zhou J, Lin H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: evidence based on bioinformatics. BMC Nephrol. 2022;23:303.
Zhao Y, Guo Y, Jiang Y, Zhu X, Liu Y, Zhang X. Mitophagy regulates macrophage phenotype in diabetic nephropathy rats. Biochem Biophys Res Commun. 2017;494:42–50.
Yu Y, Lin Q, Ye D, Wang Y, He B, Li Y, et al. Neutrophil count as a reliable marker for diabetic kidney disease in autoimmune diabetes. BMC Endocr Disorders. 2020;20:158.
Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. 2006;17:368–77.
Uddin M, Lau LC, Seumois G, Vijayanand P, Staples KJ, Bagmane D, et al. EGF-induced bronchial epithelial cells drive neutrophil chemotactic and anti-apoptotic activity in asthma. PLoS ONE. 2013;8:e72502.
Ochieng P, Nath S, Macarulay R, Eden E, Dabo A, Campos M, et al. Phospholipid transfer protein and alpha-1 antitrypsin regulate hck kinase activity during neutrophil degranulation. Sci Rep. 2018;8:15394.
Chen D, Liu Y, Chen J, Lin H, Guo H, Wu Y, et al. JAK/STAT pathway promotes the progression of diabetic kidney disease via autophagy in podocytes. Eur J Pharmacol. 2021;902:174121.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Ozaki T, Kaibori M, Matsui K, Tokuhara K, Tanaka H, Kamiyama Y, et al. Effect of thiol-containing molecule cysteamine on the induction of inducible nitric oxide synthase in hepatocytes. JPEN J Parenter Enter Nutr. 2007;31:366–71. discussion 71-2.
Sun H, Tsai Y, Nowak I, Liesveld J, Chen Y. Eltrombopag, a thrombopoietin receptor agonist, enhances human umbilical cord blood hematopoietic stem/primitive progenitor cell expansion and promotes multi-lineage hematopoiesis. Stem Cell Res. 2012;9:77–86.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
CG and YD carried out the studies, participated in collecting data, and drafted the manuscript. CG and YH performed the statistical analysis and participated in its design. CG and WW participated in acquisition, analysis, or interpretation of data and draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Guo, C., Wang, W., Dong, Y. et al. Identification of key immune-related genes and potential therapeutic drugs in diabetic nephropathy based on machine learning algorithms. BMC Med Genomics 17, 220 (2024). https://doi.org/10.1186/s12920-024-01995-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01995-4