
Abstract
Background
Respiratory microbiota is closely related to tuberculosis (TB) initiation and progression. However, the dynamic changes of respiratory microbiota during treatment and its association with TB progression remains unclear.
Methods
A total of 16 healthy individuals and 16 TB patients (10 drug-sensitive TB (DS-TB) and 6 drug-resistant TB (DR-TB)) were recruited. Sputum samples were collected at baseline for all anticipants and after anti-TB treatment at Month-6 for TB patients. High throughput 16 S RNA sequencing was used to characterize the respiratory microbiota composition.
Results
Compared to the healthy individuals, TB patients exhibited lower respiratory microbiota diversity (p < 0.05). This disruption was alleviated after anti-TB treatment, especially for DS-TB patients. Parvimonas spp. numbers significantly increased after six months of anti-TB treatment in both DS-TB and DR-TB patients (p < 0.05). Rothia spp. increase during treatment was associated with longer sputum-culture conversion time and worse pulmonary lesion absorption (p < 0.05). Besides, Moraxella spp. prevalence was associated with longer sputum-culture conversion time, while Gemella spp. increase was associated with worsening resolving of pulmonary lesions (p < 0.05).
Conclusion
Dynamic changes of respiratory microbiota during anti-TB treatment is closely related to TB progression. The involvement of critical microorganisms, such as Parvimonas spp., Rothia spp., Moraxella, and Gemella spp., appears to be associated with pulmonary inflammatory conditions, particularly among DR-TB. These microorganisms could potentially serve as biomarkers or even as targets for therapeutic intervention to enhance the prognosis of tuberculosis patients.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is the second leading infectious killer after COVID-19, leading to a total of 1.4 million deaths in 2022 [1]. Treatment failure is a major contributor to TB patients’ death, especially for drug-resistant TB. To date, treatment failure is frequent, currently at 12% for drug-susceptible TB and 37% for multidrug- or rifampicin-resistant tuberculosis (MDR/RR-TB) [2]. While many factors such as inadequate compliance and drug resistance are involved, they do not explain all the treatment failures. Exploring the factors related to anti-TB treatment response is critical for the improvement of treatment outcome, particularly for drug-resistant TB patients.
Respiratory microbiota is a potential critical factor related to anti-TB response since it is involved in the TB initiation and immune regulation [3,4,5]. In healthy individuals, the lower respiratory tract is predominantly colonized by Prevotella spp., Veillonella spp., and Streptococcus spp. [6]. The decrease of Prevotella spp. was reported as the characteristic of sputum smear-positive TB patients whereas the genus Streptococcus spp. dominates the lung microbiome of Mycobacterium tuberculosis (MTB) negative individuals [7]. The dysbacteriosis has been the main characteristic of TB patients.
Previous study also attempted to connect the respiratory microbiota and TB recurrence and treatment response, which mostly focused on pre-treatment microbiota characteristics [8]. Yet, the prolonged and intensive use of anti-TB antibiotics may lead to significant changes in respiratory microbiota [9, 10], which may have more implications for anti-TB response. Furthermore, the dynamic changes in respiratory microbiota may be more prominent in drug-resistant TB patients as the antibiotics recommended for drug-resistant TB, such as levofloxacin or moxifloxacin, linezolid, amikacin, imipenem/cilastatin and meropenem [11], have broad-spectrum antibacterial activity. However, the associations between the dynamic changes of respiratory microbiota and the treatment response in TB patients have not been previously reported.
In the current study, we investigated the associations between changes in respiratory microbiota and anti-TB treatment outcome among drug-sensitive TB (DS-TB) and drug-resistant TB (DR-TB) patients. These results will provide certain guiding significance for anti-TB response prediction and clinical medication of TB.
Materials and methods
Participants
A total of 16 TB patients (10 drug-sensitive and 6 drug-resistant TB patients) and 16 healthy individuals were recruited between January to March 2019 in Guangzhou Chest Hospital.
To be eligible for the study, TB patients were required to meet following criteria: (1) age > 18 years old; (2) sputum culture identified as MTB; (3) without respiratory infection induced by non-TB pathogen based on the infection indicators in blood (monocyte-to-lyphocyte ratio, procalcitonin, C-reactive protein) [12, 13], imaging examination (chest CT) [14], and pathogen detection (sputum culture for common non-TB bacterial and fungal, antibody detection for common virus and chlamydia, and polymerase chain reaction for virus if necessary) [15]; (4) without severe systemic diseases (malignant hypertension, myocardial infarction, etc.), diabetes, or malignant tumors.
During the same period, 16 healthy medical staff working in the same hospital and frequency-matched to the cases (± 5 years), were recruited. For healthy medical workers, detailed inclusion criteria were as follow: (1) aged > 18 years old; (2) physical examination and chest X-ray showing no abnormalities; (3) without respiratory infection based on the pneumonia-related symptoms (including fever, cough, expectoration, or chest pain), the infection indicators in blood (procalcitonin, C-reactive protein), imaging examination (chest CT), and pathogen detection (sputum culture for TB and non-TB pathogens, antibody detection for common virus and chlamydia, and polymerase chain reaction for virus if necessary) [15, 16]; (4) without history of chronic respiratory diseases, coronary heart disease, malignant tumors, diabetes, hyperthyroidism, malnutrition, and obesity; (5) without history of smoking.
The study was approved by the ethics committee of Guangzhou Chest Hospital and each participant provided their informed consent before inclusion in the study.
Sputum collection
We adopted the method of ultrasonic atomization-induced deep sputum collection to collect the samples for 16 S rRNA sequencing. This method was non-invasive and barely collected the samples from upper respiratory tract [17]. Furthermore, researches have demonstrated that there was no statistically significant difference in the sensitivity and specificity of pathogen detection between deep sputum samples and bronchoalveolar lavage (BAL) samples [18]. For the TB patients who could provide sputum samples spontaneously expectorated, we also performed ultrasonic atomization-induced deep sputum collection on them to ensure the samples were comparable by exclude the bias caused by different sputum collection procedures.
Briefly, sputum were induced using a hypertonic saline solution with an ultrasonic nebulizer. For healthy individuals who should not have any sputum at all, bronchial secretion was collected. The minimal volume of each collection was 2-5 ml. In cases where this volume was not achievable after the induction procedure, we will repeated the induction and collected the best sample for analysis. The ultrasonic nebulizer was disinfected after each use according to standard hospital protocols to prevent cross-contamination. Saline solutions were changed for each patient to maintain sterility.
Drug susceptibility definition
In vitro experiments using either molecular or genotypic techniques to detect resistance-conferring mutations, or phenotypic methods (by BACTEC MGIT 960 System) were used to determine the drug susceptibility of MTB, as previously described [19]. MTB isolates susceptible to all four of the first-line drugs were identified as drug-sensitive MTB (DS-MTB). MTB isolates resistant to any anti-TB medicine were identified as drug-resistant MTB (DR-MTB).
Treatment outcome definition
The TB patients receive anti-TB treatments according to the principles outlined in the WHO consolidated guidelines on tuberculosis [11, 20]. The treatment outcomes were defined based on the sputum culture results and chest X-rays/CT evaluation [21]. Patients with markedly effective treatment outcome were defined as the ones who had two consecutive sputum culture negative results in the sixth month of treatment at leats 30 days apart, significant absorption of pulmonary lesions (absorbed ≥ 50%), and cavities closure. Patients with effective treatment outcome were defined as the ones who had two consecutive sputum culture negative results in the month 6 of treatment at least 30 days apart, but the chest X-rays/CT evaluation results did not meet the definition of markedly effective. Patients with treatment failure were defined as the ones whose sputum culture was positive in the fifth month or later during treatment.
DNA extraction
Sputum samples were liquefied with 4% NaOH and buffered in pH 6.8 phosphate buffer. Centrifugation was performed and the sediment was retained for DNA extraction. The NEB next microbiome DNA enrichment Kit (New England Biolabs, Ipswich, MA, US) was used to extract microbial community DNA according to the manufacturer’s instructions. The extracted DNA was quantified using the Qubit® dsDNA BR Assay Kit (Invitrogen, USA) and its quality was checked by running equal aliquots of the sample on 1% agarose gel.
Library construction
The variable region V1-V3 of bacterial 16 S rRNA gene was amplified using degenerate PCR primers 8 F (5’-AGAGTTTTGATYMTGGCTCAG-3’) and 518R (5’-ATTACCGCGGCTGCTCG-3’). Both forward and reverse primers were tagged with Illumina adapter, pad and linker sequences. PCR enrichment was performed in a 50 μL reaction containing 30ng template, polymerase and PCR master mix. PCR cycling conditions were: 94℃ for 3 min, 30 cycles of 94℃ for 30s, 50℃ for 45s, 72℃ for 45s and final extension at 72℃ for 10 min. The PCR products were purified with AmpureXP beads and eluted in Elution buffer. Libraries were qualified by the Agilent 2100 bioanalyzer (Agilent, USA). The validated libraries were used for sequencing on Illumina HiSeq platform (BGI, Shenzhen, China) following the standard pipelines of Illumina, and generating 2 × 300 bp paired-end reads.
Sequencing and bioinformatics analysis
Raw reads were filtered to remove adapters and low-quality and ambiguous bases. Then paired-end reads were added to tags by the Fast Length Adjustment of Short reads program (FLASH, v1.2.11) to get the tags. All sequences were aligned and clustered into operational taxonomic units (OTUs) with 97% similarity using UPARSE software (v7.0.0.1090), and chimeric sequences were compared with the Gold database using UCHIME (v4.2.40) to detect. Then, the OTU representative sequences were taxonomically classified using the Ribosomal Database Project (RDP) classifier v.2.2, with a minimum confidence threshold of 0.6, and processed using QIIME (v1.8.0) on the Greengenes database v201305.
The optimized sequences were mapped back to the OTU representative sequences using the USEARCH_global method, and the abundance of OTU sequences in each sample was obtained. Alpha diversity were estimated by MOTHUR (v1.31.2) at the OTU level.
Statistical analyses
Statistical analysis comparing non-categorical or ordinal variables between groups was performed with Mann-Whitney U test, while Chi-square test was used to compare categorical variables between groups. To explore the difference in microbiota before and after anti-TB treatment, paired. T test was performed. The foldchanges of microbiota during anti-TB treatment were calculated, which were used to evaluate the associations between dynamic changes in microbiota and treatment outcomes. All analyses were conducted with a two-sided p-value of 0.05.
Results
Participant characteristics
A total of 16 healthy individuals and 16 TB patients (10 drug-sensitive TB patients (DS) and 6 drug-resistant TB patients (DR) were included in the current study. As shown in Table 1, the median age of healthy individuals, DS-TB patients, and DR-TB patients were 37.5, 33, and 39, and no significant difference was observed. The patient cohorts and the healthy control group are consist of 11 and 3 males, 5 and 13 females, respectively. The patient cohorts and the healthy control group are consist of 4 and 0 current smokers, 2 and 0 ex-smokers, 10 and 16 no history of smoking, respectively. All the TB patients included in the current study achieved effective treatment. However, compared to DS-TB patients, the treatment outcome of DR-TB patients were less effective, with poorer resolving of pulmonary lesions and less cavity closure. Also, the markedly effective rate of DS-TB patients (50%) was higher than that of DR-TB patients (14.29%).
Association between respiratory microbiota diversity and anti-TB treatment outcome
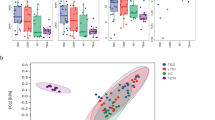
We analyzed the diversity of respiratory microbiota in healthy individuals, pre-treated TB patients and post-treated TB patients. Compared to the healthy individuals, TB patients exhibited lower respiratory tract microbiota diversity (Fig. 1A), and the disruption was alleviated after anti-TB treatment (Fig. 1B). Notably, the recovery of respiratory microbiota diversity was more significant in DS-TB patients but not in DR-TB patients (Fig. 1B). Such results is consistent with the clinical evidence that DR-TB results in severe destruction of lung tissues and intense inflammation, leading to more pronounced clinical symptoms. Such results suggests that the dysbiosis of the respiratory microbiota is a crucial factor influencing the clinical outcomes of patients with pulmonary tuberculosis.

Association between respiratory microbiota diversity and anti-TB treatment outcome. (A) Boxplot showing difference in respiratory microbiota diversity between healthy individuals, DS-TB patients, and DR-TB patients. (B) Dynamic changes of microbiota diversity for TB patients, DS-TB patients, and DR-TB patients. (C) Heatmap visualization of bacterial generas abundance in TB patients before and after anti-TB treatments. (D) Venn diagram showing the number of microbiota generas that significantly changed after anti-TB treatment. (E) Dynamic changes of Parvimonas spp., Megasphaera spp., Streptococcus spp., and Prevotella spp. before and after anti-TB treatment
Building upon these results, we further explored the critical bacterial generas that significantly changed during anti-TB treatment and found that Parvimonas spp. numbers significantly increased after 6 months of anti-TB treatment among both DS-TB and DR-TB patients (Fig. 1C and D, and 1E). We also found that Megasphaera spp. pevalence significantly increased while Streptococcus spp. significantly decreased after anti-TB treatment among DS-TB patients. Prevotella spp. occurrence marginally increased after anti-TB treatment among DR-TB patients (Fig. 1E). Given that all the TB patients included in the current study achieved favorable treatment outcome, our results suggested that these genus were indicators of favorable treatment outcome.
Association between dynamic changes of respiratory microbiota and sputum-culture conversion
We further explored the roles of respiratory microbiota in anti-TB treatment efficacy. Conversion of sputum cultures is one of the key indicators for anti-TB treatment efficacy and therefore, its association with the respiratory microbiota changes was investigated (Fig. 2A). We found that the elevation of Moraxella spp. and Rothia spp. were associated with longer sputum-culture conversion time, while elevation of Kingella spp. and Peptostreptococcus spp. were associated with shorter period for sputum-culture conversion (Fig. 2B and C). Specific to DS-TB patients, only the marginally significant association between Moraxella spp. elevation and sputum-culture conversion time was retained (Fig. 2C). Significant association between dynamic changes of microbiota and sputum-culture conversion time among DR-TB patients was not observed.

Association between dynamic changes of respiratory microbiota and sputum-culture conversion. (A) Heatmap visualization of foldchanges in bacterial generas abundance in TB patients with long and short sputum-culture conversion time. (B) Venn diagram showing the number of microbiota generas that were associated with sputum-culture conversion. (C) Boxplot showing the foldchanges in Moraxella spp., Rothia spp., Kingella spp., and Peptostreptococcus spp. among TB patients with long and short sputum-culture conversion time
Association between dynamic changes of respiratory microbiota and resolving of pulmonary lesions
Pulmonary lesion absorption is another key indicators for anti-TB treatment efficacy and the association between dynamic changes of respiratory microbiota and resolving of pulmonary lesions (Fig. 3A). We found that the elevation of Rothia spp., Gemella spp., Alloprevotella spp., Saccharibacteria spp., and Corynebacterium spp. were associated with worse pulmonary lesion absorption, while Peptostreptococcaceae_incertae_sedis spp. was associated with better pulmonary lesion absorption (Fig. 3B). Among the DS-TB patients, the elevation of Rothia spp. and Gemella spp. was associated with worse resolving of pulmonary lesions. No bacterial genus was found to be associated with resolving of pulmonary lesions among DR-TB patients.

Association between dynamic changes of respiratory microbiota and resolving of pulmonary lesions. (A) Heatmap visualization of foldchanges in bacterial generas abundance in TB patients with complete and incomplete resolving of pulmonary lesions. (B) Venn diagram showing the number of microbiota generas that were associated with resolving of pulmonary lesions. (C) Boxplot showing the foldchanges in Rothia spp., Gemella spp., Alloprevotella spp., Saccharibacteria spp., Corynebacterium spp., and Peptostreptococcaceae_incertae_sedis spp. among TB patients with complete and incomplete resolving of pulmonary lesions
Discussion
Multiple studies have suggested associations between dysbiosis and TB occurrence and progression [22,23,24]. The extensive use of antibiotics during anti-TB treatment can significantly alter the lung microbiota [25]. However, the relationship between these microbial changes and the progression of tuberculosis, as well as the efficacy of anti-TB therapy, remains unclear. In this study, we investigated the associations between microbial changes and clinical outcomes of TB patients, providing new evidence that the lung microbiota may be a key factor influencing TB progression. We further revealed significant associations between various microorganisms, including Parvimonas spp., Rothia spp., Moraxella spp. and Gemella spp., and clinical outcomes of TB patients. These microorganisms may serve as biomarkers or even drug targets to improve the prognosis of patients with pulmonary TB.
In our previous research [26], as well as studies conducted by other scholars [22], it has been consistently observed that TB patients have lower microbial diversity compared to healthy individuals, indicating that dysbiosis may be a key factor influencing the occurrence and progression of TB. In this study, we further showed that the microbial diversity of patients significantly increases during the successful anti-TB treatment, suggesting that improving dysbiosis may be a clinically feasible strategy to improve the prognosis of TB patients. Notably, the recovery of microbial diversity in DR-TB patients is not as pronounced as in DR-TB patients. This may be attributed to the more severe dysbiosis and the use of broad-spectrum antibiotics in DR-TB patients. We also supposed that insufficient recovery of microbial diversity may contribute to the higher risk of recurrence in DR-TB patients. Overall, such findings collectively suggest that dysbiosis is a key factor influencing the progression of TB, and alleviating the dysbiosis may be a clinically feasible approach to improve the prognosis of TB patients.
Exploring the key microbial that influence the prognosis and treatment efficacy of TB patients will provide clues for developing new biomarkers or even therapeutic approaches to improve clinical outcomes. To achieve this, we investigated the associations between microbial changes and clinical outcomes in TB patients. Our results suggest that Parvimonas may be a key microbial influencing TB progression, as its abundance significantly increases after successful anti-TB treatment in both DS- and DR-TB patients. Previous studies have suggested the potential role of Parvimonas spp. in promoting TB progression, as its abundance in TB patients is significantly higher than in healthy individuals [27]. These findings are inconsistent with our results, and the reasons for this discrepancy are still unclear. It has been shown that Parvimonas spp. is negatively associated with IL-15 and the latter one is a critical cytokine in maintaining T cell immunity [28]. We supposed that the elevation of Parvimonas spp. reflects the negative feedback of the anti-TB immunity. Suppressing the excessive anti-TB immune response can alleviate tissue damage and is a critical factor for the recovery of TB patients.
Rothia spp. was another microbiota that might be critical for TB patients, since its elevation during treatment was associated with longer sputum-culture conversion time and worse pulmonary lesion absorption. Previous studies showed that Rothia spp. was enriched in TB patients [29], which are consistent with our results and suggest the potential role of Rothia spp. in promoting TB progression. Rothia spp. is emerging as opportunistic pathogens associated with various infections in immunocompromised and immunocompetent individuals. Previous studies also found that Rothia spp. was positively associated with IL-10 [28], a cytokine that inhibits the immune response. Such results suggest that the elevation of Rothia spp. during treatment might contribute to the disruption in anti-TB immunity. Additionally, Rothia spp. was reported to produce the siderophore enterobactin in the human oral niche and the siderophore in lung microenvironment may be helpful for TB to acquire iron from the host and grow [30]. Moreover, Rothia spp. was found to be unique to TB patients [31], suggesting that targeting Rothia spp. may be an attractive therapy strategy for TB patients with low toxicity.
In addition to Parvimonas spp. and Rothia spp., we also identified Moraxella spp. and Gemella spp. as critical microbial that affect sputum-culture conversion and pulmonary lesion absorption in TB patients, respectively. Previous studies have suggested that Moraxella spp. in the pulmonary microenvironment downregulates the expression of epidermal defense genes and thereby increasing the risk of tuberculosis and promoting the progression of other respiratory diseases [32]. As for Gemella spp., it is supposed to influence TB progression by modulating the IFN-gamma pathway [28]. Considering that Moraxella spp. is specifically associated with sputum conversion while Gemella spp. is specifically related to pulmonary lesion absorption, we supposed that there may be more specific mechanisms underlying the association of Moraxella spp. with sputum conversion and the relationship of Gemella spp. with pulmonary lesion absorption, respectively.
Notably, the microbiota dysbiosis may increase the risk of secondary pneumonia for TB patients, especially for the DR-TB patients, though such comorbidities were not observed in the current study due to small sample size. Secondary pneumonia is a critical comorbidity, which affects the curated efficacy of anti-TB treatment and prognosis [33]. The current analyses about the dynamic changes of microbiota showed that some TB patients might experience anti-pathogen immunity disruption and opportunistic pathogens elevation, which might be the biological basis for secondary infections. Whether targeting the microbiota can prevent or cure secondary pneumonia is a clinical issue that is warranted to be addressed.
There are some inevitable limitations in this study. First, the sample size was relatively small, which restricted us from conducting further stratified analysis. In addition, the small sample size inevitably led to unbalanced distribution of sex among healthy individuals, DS-TB, and DR-TB patients. Although previous studies showed no evidence that the microbiota of the respiratory tract differed significantly between the sexes [34], further studies with larger sample size and more balanced gender distribution should be conducted to validate our findings. Second, the long-term follow-up data was absent, which prevented us from investigating the association between microbial changes and long-term clinical outcomes of TB patients. Future studies will require larger sample sizes and longer follow-up data to validate the findings of this study. Third, the latent TB infection in healthy individuals was not tested in the current study. Although the primary objective of the current study was to explore the associations between the dynamic changes of respiratory microbiota and the treatment response in TB patients and the information of latent TB infection in healthy individuals might not influence the main conclusion, further studies that excluding the healthy individuals with latent TB infection should be conducted to figure out the actual microbiota differences between healthy individuals and TB patients. These microbiota differences may be involved in the TB progression as well.
In conclusion, the respiratory microbiota diversity increased after anti-TB treatment, with certain bacterial species becoming enriched or reduced. The findings provided by this study will help us better understand the role of respiratory microbiota and its association with treatment outcomes, which could be used to guide and personalize anti-TB treatment in the future. The respiratory microbiome may serve as a potential biomarker for predicting TB progression, distinguishing drug resistance, and early treatment evaluation. However, due to the limited number of cases included in this study, further sample size expansion and large-scale multicenter cohort studies are needed to verify these findings.
Data availability
All data generated or analyzed during this study are included in this pubilshed article. Data can be accessed from NCBI Sequence Read Archive (SRA) and the number was PRJNA1080014 (www.ncbi.nlm.nih.gov/sra/PRJNA1080014).
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- MTB:
-
Mycobacterium tuberculosis
- PTB:
-
Pulmonary tuberculosis
- RNA:
-
Ribonucleic acid
- TB:
-
Tuberculosis
- WHO:
-
World Health Organization
References
Bagcchi S, WHO’s global tuberculosis report. 2022. The Lancet Microbe. 2023;4(1):e20.
World Health Organization. Global tuberculosis report 2023. Geneva:World Health Qrganization; 2023.
Namasivayam S, Sher A, Glickman MS, Wipperman MF. The microbiome and tuberculosis: early evidence for cross talk. mBio. 2018;9(5):e01420–18.
Osei Sekyere J, Maningi NE, Fourie PB. Mycobacterium tuberculosis, antimicrobials, immunity, and lung-gut microbiota crosstalk: current updates and emerging advances. Ann N Y Acad Sci. 2020;1467(1):21–47.
Naidoo CC, Nyawo GR, Wu BG, Walzl G, Warren RM, Segal LN, Theron G. The microbiome and tuberculosis: state of the art, potential applications, and defining the clinical research agenda. Lancet Respiratory Med. 2019;7(10):892–906.
Salisbury ML, Han MK, Dickson RP, Molyneaux PL. Microbiome in interstitial lung disease: from pathogenesis to treatment target. Curr opin pulm med. 2017;23(5):404–10.
Hu Y, Cheng M, Liu B, Dong J, Sun L, Yang J, Yang F, Chen X, Jin Q. Metagenomic analysis of the lung microbiome in pulmonary tuberculosis - a pilot study. Emerg Microbes Infections. 2020;9(1):1444–52.
Wu J, Liu W, He L, Huang F, Chen J, Cui P, Shen Y, Zhao J, Wang W, Zhang Y, et al. Sputum microbiota associated with new, recurrent and treatment failure tuberculosis. PLoS ONE. 2013;8(12):e83445.
Xiao G, Cai Z, Guo Q, Ye T, Tang Y, Guan P, Zhang J, Ou M, Fu X, Ren L, et al. Insights into the unique lung microbiota profile of pulmonary tuberculosis patients using metagenomic next-generation sequencing. Microbiol Spectr. 2022;10(1):e0190121.
Zhang M, Shen L, Zhou X, Chen H. The microbiota of human lung of pulmonary tuberculosis and the alteration caused by anti-tuberculosis drugs. Curr Microbiol. 2022;79(11):321.
WHO consolidated guidelines on tuberculosis. Module 4: treatment-drug-resistant tuberculosis treatment, 2022 update. Geneva:World Health Qrganization; 2022.
Malik AA, Gandhi NR, Marcy O, Walters E, Tejiokem M, Chau GD, Omer SB, Lash TL, Becerra MC, Njuguna IN, et al. Development of a clinical prediction score including monocyte-to-lymphocyte ratio to inform tuberculosis treatment among children with HIV: a multicountry study. Open Forum Infect Dis. 2022;9(11):ofac548.
Zenner D, Brals D, Nederby-Ohd J, Menezes D, Aldridge R, Anderson SR, de Vries G, Erkens C, Marchese V, Matteelli A et al. Drivers determining tuberculosis disease screening yield in four European screening programmes: a comparative analysis. Eur Respir J. 2023;62(4).
Nambu A, Ozawa K, Kobayashi N, Tago M. Imaging of community-acquired pneumonia: roles of imaging examinations, imaging diagnosis of specific pathogens and discrimination from noninfectious diseases. World J Radiol. 2014;6(10):779–93.
Torres A, Cilloniz C, Niederman MS, Menendez R, Chalmers JD, van der Wunderink RG. Poll T: pneumonia. Nat Rev Dis Primers. 2021;7(1):25.
Metlay JP, Waterer GW, Long AC, Anzueto A, Brozek J, Crothers K, Cooley LA, Dean NC, Fine MJ, Flanders SA, et al. Diagnosis and treatment of adults with community-acquired pneumonia. An official clinical practice guideline of the American Thoracic Society and Infectious Diseases Society of America. Am J Respir Crit Care Med. 2019;200(7):e45–67.
Park JS. Efficacy of induced sputum for the diagnosis of pulmonary tuberculosis in adults unable to expectorate sputum. Tuberc Respir dis. 2015;78(3):203–9.
Shi W, Zhu S. The application of metagenomic next-generation sequencing in detection of pathogen in bronchoalveolar lavage fluid and sputum samples of patients with pulmonary infection. Comput math Method m. 2021;2021(1):7238495.
WHO consolidated guidelines on tuberculosis. Module 3: diagnosis –Rapid diagnostics for tuberculosis detection. Geneva:World Health Organization; 2024.
WHO consolidated guidelines on tuberculosis. Module 4: treatment - drug-susceptible tuberculosis treatment. Geneva: World Health Organization; 2022.
Zhang SY, Fu JY, Guo XY, Wu DZ, Zhang T, Li C, Qiu L, Shao CR, Xiao HP, Chu NH, et al. Improvement cues of lesion absorption using the adjuvant therapy of traditional Chinese medicine Qinbudan tablet for retreatment pulmonary tuberculosis with standard anti-tuberculosis regimen. Infect dis Poverty. 2020;9(1):50.
Krishna P, Jain A, Bisen PS. Microbiome diversity in the sputum of patients with pulmonary tuberculosis. Eur J Clin Microbiol Infect Dis. 2016;35(7):1205–10.
Adami AJ, Cervantes JL. The microbiome at the pulmonary alveolar niche and its role in mycobacterium tuberculosis infection. Tuberculosis. 2015;95(6):651–8.
Lin D, Wang X, Li Y, Wang W, Li Y, Yu X, Lin B, Chen Y, Lei C, Zhang X, et al. Sputum microbiota as a potential diagnostic marker for multidrug-resistant tuberculosis. Int J Med Sci. 2021;18(9):1935–45.
Luies L, Reenen MV, Ronacher K, Walzl G, Loots DT. Predicting tuberculosis treatment outcome using metabolomics. Biomark Med. 2017;11(12):1057–67.
Cai X, Luo Y, Zhang Y, Lin Y, Wu B, Cao Z, Hu Z, Wu X, Tan S. Airway microecology in rifampicin-resistant and rifampicin-sensitive pulmonary tuberculosis patients. BMC Microbiol. 2022;22(1):286.
Cheung MK, Lam WY, Fung WY, Law PT, Au CH, Nong W, Kam KM, Kwan HS, Tsui SK. Sputum microbiota in tuberculosis as revealed by 16S rRNA pyrosequencing. PLoS ONE. 2013;8(1):e54574.
Xia X, Chen J, Cheng Y, Chen F, Lu H, Liu J, Wang L, Pu F, Wang Y, Liu H, et al. Comparative analysis of the lung microbiota in patients with respiratory infections, tuberculosis, and lung cancer: a preliminary study. Front Cell Infect Microbiol. 2022;12:1024867.
HaileMariam M, Yu Y, Singh H, Teklu T, Wondale B, Worku A, Zewude A, Mounaud S, Tsitrin T, Legesse M, et al. Protein and microbial biomarkers in sputum discern acute and latent tuberculosis in investigation of pastoral Ethiopian cohort. Front Cell Infect Microbiol. 2021;11:595554.
Uranga CC, Arroyo P, Duggan BM, Gerwick WH, Edlund A. Commensal oral Rothia mucilaginosa produces enterobactin, a metal-chelating siderophore. mSystems. 2020;5(2):e00161–20.
Eshetie S, van Soolingen D. The respiratory microbiota: new insights into pulmonary tuberculosis. BMC Infect Dis. 2019;19(1):92.
Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Müller-Quernheim J, Greulich T, Nowinski A, Barta I, Stendardo M, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis. Lancet Microbe. 2021;2(7):e300–10.
Tan SY, Liang ZZ, Chiwala G, Kuang HB, Huang ZP, Qin HJ, Li Y, Li YQ, Adnan Hameed HM, Zhang TY. The effects of secondary pneumonia on the curative efficacy of multidrug-resistant tuberculosis: a retrospective cohort study. Biomed Environ Sci. 2018;31(12):908–12.
Ma Z, Li W. How and why men and women differ in their microbiomes: medical ecology and network analyses of the microgenderome. Adv Sci. 2019;6(23):1902054.
Acknowledgements
We sincerely thank the Guangdong Provincial Health Commission and Guangzhou Municipal Health Commission for their funding support and the Key Laboratory of Respiratory Diseases of Guangzhou Chest Hospital for its basic support.
Funding
The work was supported by the Guangzhou Science and Technology Bureau Municipal-University(Institute) Joint Funding Project[grant numbers: 2024A03J0585] and Guangdong Provincial Health Commission Funding Project[grant numbers: A2019342]. The funders provided fnancial and technical support for the study.
Author information
Authors and Affiliations
Contributions
YL and ZL progressed experimental design, data collection, molecular experiments, and manuscript writing. XC and YL did microbiology experiments, data collection and analysis. BW, YF and ZC made patient selection and analysis. ST made protocol and project development, served as scientifc advisor of experimental design, and manuscript editing. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee of Guangzhou Chest Hospital and each participant provided their informed consent before inclusion in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lin, Y., Liang, Z., Cai, X. et al. Dynamic changes of respiratory microbiota associated with treatment outcome in drug-sensitive and drug-resistant pulmonary tuberculosis. Ann Clin Microbiol Antimicrob 23, 83 (2024). https://doi.org/10.1186/s12941-024-00742-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-024-00742-y