
Abstract
Ferroptosis, triggered by iron overload and excessive lipid peroxidation, plays a pivotal role in the progression of DOX-induced cardiomyopathy (DIC), and thus limits the use of doxorubicin (DOX) in clinic. Here, we further showed that cardiac ferroptosis induced by DOX in mice was attributed to up-regulation of Hmox1, as knockdown of Hmox1 effectively inhibited cardiomyocyte ferroptosis. To targeted delivery of siRNA into cardiomyocytes, siRNA-encapsulated exosomes were injected followed by ultrasound microbubble targeted destruction (UTMD) in the heart region. UTMD greatly facilitated exosome delivery into heart. Consistently, UTMD assisted exosomal delivery of siHomox1 nearly blocked the ferroptosis and the subsequent cardiotoxicity induced by doxorubicin. In summary, our findings reveal that the upregulation of HMOX1 induces ferroptosis in cardiomyocytes and UTMD-assisted exosomal delivery of siHmox1 can be used as a potential therapeutic strategy for DIC.
Graphical Abstract

Similar content being viewed by others

Introduction
Doxorubicin (DOX), which belongs to the anthracycline family, is widely used for treatment of various types of cancers. However, DOX related cardiotoxicity, which is characterized by cardiomyocyte loss, progressive cardiac enlargement, and ultimately congestive heart failure, has restricted the clinical use [1]. Approximately a quarter of patients would result in heart failure when cumulatively receiving DOX at a dose over 550 mg/m2 [2]. Elucidation of the underlying mechanism and development of potential therapeutic strategies to avoid the adverse effect is badly needed.
It has been well established that oxidative stress, mitochondrial disruption, and autophagy are involved in the DOX-induced cardiomyopathy (DIC) [3]. Very recently, ferroptosis has been revealed to play an important role in DIC [4]. Ferroptosis, is a new form of cell death characterized by the iron-dependent activation of lipoxygenase and subsequent excessive lipid peroxidation [5]. It has been demonstrated that iron-dependent ferroptosis, rather than other known cell death, has a crucial role in DOX-induced cardiomyopathy [6]. Targeting ferroptosis holds as a promising therapeutic strategy for prevention of DIC [7]. Mechanistically, DOX-induced iron accumulation and DOX-induced cardiomyopathy are at least partially mediated by upregulation of Hmox1 [7]. Further confirmation of the role of Hmox1 in DIC and therapeutical downregulation of Hmox1 would be a promising strategy.
Exosomes emerge as a promising drug carrier for nucleic acids [8, 9]. Injection of therapeutics-loaded exosomes could significantly protect heart from the cardiotoxicity [10]. However, the therapeutic efficacy was compromised by the low heart targeted delivery efficiency. Ultrasound targeted microbubble destruction (UTMD) could improve the delivery efficiency via the cavitation effect within the microvasculature of target tissues. Recently, we have found that UTMD could assist exosomal delivery into the heart [10, 11].
In this study, we further showed that cardiac ferroptosis induced by DOX in mice was attributed to up-regulation of Hmox1. UTMD assisted exosomal delivery of siHomox1 nearly blocked the ferroptosis and the subsequent cardiotoxicity induced by doxorubicin. Our findings reveal that the upregulation of HMOX1 induces ferroptosis in cardiomyocytes and UTMD-assisted exosomal delivery of siHmox1 can be used as a potential therapeutic strategy for DIC.
Results
Dox induces cardiac ferroptosis in a time and dose dependent manner
It has been revealed that ferroptosis contributes to DOX induced cardiotoxicity [7]. To further confirm whether Dox induces cardiac ferroptosis in a time dependent manner, mice were treated with 10 mg/kg Dox and then ferroptosis was examined at different time points post treatment (Fig. 1A). qPCR analysis revealed that Ptgs2 mRNA (a marker of ferroptosis [12]) expression peaked on day 2, and gradually declined since then (Fig. 1B). The increase lasted to day 5. Similarly, lipid peroxidation marker MDA, ROS indicator DHE, and cell death (Fig. 1C-G) showed similar trends. Consistently, serum LDH and CKMB increased with time, indicating obvious cardiac toxicity of Dox (Fig. 1H, I). Next, we explored whether Dox induces cardiac ferroptosis in a dose dependent manner. Mice were treated with 0, 2, 4, 6, 8 or 10 mg/kg Dox, and ferroptosis was examined on day 2. As expected, Dox dose dependently increased the ferroptosis markers (Figure S1). To further confirm whether cardiomyocyte was the main affected cell type, we performed dual immunofluorescence assay using TUNEL and cardiomyocyte marker TNNT2 in the heart. As expected, there were significant TUNEL signal colocalized with cardiomyocyte marker TNNT2 (Figure S2), suggesting that there was obvious cell death occurred in cardiomyocytes. Together, the above data suggest that doxorubicin could induce ferroptosis in the heart in a time and dose dependent manner.

Dox induces ferroptosis in a time dependent manner. A, Schematic representation of the experimental procedure. B, qPCR analysis of Ptgs2 mRNA expression in the hearts at indicate time points from mice receiving Dox treatment. C, MDA level in the hearts at indicate time points from mice treated same as above. D, Representative DHE staining in the heart tissues from mice treated same as above. E, Representative TUNEL staining in the heart tissues from mice treated same as above. F, Quantification data of Panel D. G, Quantification data of Panel E. H, Serum LDH level at indicate time points from mice treated same as above. I, Serum CKMB level at indicate time points from mice treated same as above. Scale bar: 50 μm. Data are presented as the mean ± SEM. *, p\(\:<\)0.05 by one-way ANOVA
Repeated treatments with ferrostatin-1 have systemic toxicity
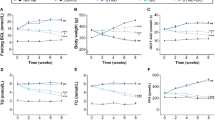
Ferrostatin-1 (Fer-1) was identified as a potent inhibitor of ferroptosis [13]. Mouse cardiomyocyte HL-1 were treated with Dox with or without Fer-1 (Figure S3A). As expected, Dox significantly up-regulated Ptgs2 mRNA level and lipid peroxidation, while Fer-1 blocked the increase (Figure S3B, C). Consistently, in vivo treatment of Fer-1 nearly blocked the increase of Ptgs2 expression(Figure S4A, B), lipid peroxidation marker MDA(Figure S3C), ROS (Figure S3D, E), and cell death (Figure S4F, G) in the heart induced by Dox in mouse model. With the inhibition of ferroptosis, cardiac structure and function were also restored. HE staining and Sirius red staining revealed that Fer-1 treatment reduced the fibrosis (collagen content) in the heart (Figure S5A-D). Moreover, the heart weight was also increased and the serum LDH (lactate dehydrogenase [14]) was decreased to nearly the normal level (Figure S5E, F). Consistently, echocardiography examination revealed that Fer-1 treatment significantly blocked the impairment of cardiac systolic and diastolic function induced by Dox (Figure S5G-L). However, repeated treatments of Fer-1 reduced the body weight and survival of mice (Fig. 2A-C), suggesting the systemic toxic effects of Fer-1. The apparent discrepancy might be explained by that one dose of Fer-1 treatment could attenuate cellular signal and ferroptosis, while multiple dosing could also cause systemic side-effects. Notably, Ferroptosis lasted during Dox treatment and other strategies to inhibit ferroptosis persistently and safely are thus needed.

Repeated treatment of Fer-1 causes severe side-effects. A, Schematic representation of the experimental procedure. Mice were treated with Dox and Fer-1 at indicated time and body weight and survival were monitored. B, Body weight change in mice treated as indicated. Data are presented as the mean ± SEM. *, p\(\:<\)0.05 by one-way ANOVA. C, Kaplan-Meier survival curves of mice treated as indicated
DOX-induced ferroptosis is mainly attributed to up-regulation of Hmox1
Further identification of the molecular mechanism involved in DOX-induced ferroptosis would provide novel specific target for cardiotoxicity therapy. We thus probed the expression of genes associated with Ferroptosis. Among the gene candidates, Hmox1 was found significantly upregulated in the heart both at mRNA and protein levels from mice treated with Dox (Fig. 3A-D). Consistently, Dox also increased Hmox1 expression in Dox treated HL-1 cells (Fig. 3E). With Hmox1 knockdown by siRNA (Figure S6A, B, Fig. 3F), Dox induced ferroptosis was also inhibited significantly (Fig. 3G).

Upregulation of Hmox1 contributes to Dox induced ferroptosis. A, Schematic illustration of the gene network regulating ferroptosis. B, Schematic representation of the experimental procedure. Mice were treated with Dox and the gene expression were analyzed on day 2. C, qPCR analysis of gene candidates in control and Dox-treated mice. Data are presented as the mean ± SEM. *, p\(\:<\)0.05 by t test. D, Western blot analyses of cardiac Hmox1 protein in the heart from control and Dox-treated mice. Gapdh was included as a loading control. E, Western blot analyses of cardiac Hmox1 protein in the HL-1 cells treated with or without Dox. F, Knockdown efficiency of siHmox1 as determined by western blot. G, Lipid peroxidation as analyzed by flow cytometry. Cells of indicated treatments were stained with the fluorescent probe C11-BODIPY before flow cytometry analysis
UTMD assisted exosomal delivery of siHmox1 reduces ferroptosis and cardiotoxicity
In view of the above data, we assumed that in vivo delivery of siHmox1 would reduce ferroptosis and cardiotoxicity. Exosomes are emerging as a novel siRNA carrier and promising in clinical translation. Exosomes from HEK293 cells were isolated and characterized (Figure S7A). TEM, Western blot and size distribution analysis confirmed the identity of the exosomes (Figure S7B-D). Accordingly, exosomes were efficiently loaded with siRNA via electroporation (Figure S8A-B), which in turn reduced the expression of Hmox1 in HL1 cells when cells receiving the exosome treatment (Figure S8C, D).
Next, we aimed to deliver exosomes into the heart. Exosomes were labeled with fluorescent dye, followed by injection in vivo (Figure S9A). As expected, both ex vivo fluorescent imaging and confocal microscope analysis showed that the liver, spleen, and lung were the dominant targets of exosomes, while there were few exosomes delivered into the heart (Figure S9B-C). Our previous study revealed that UTMD could enhance the local delivery of exosomes via the cavitation effects. Consistent with previous findings, prior UTMD treatment in the heart region facilitated the exosome delivery in the heart region (Fig. 4A-C). The promoting effects last at least for 6 min, as exosome delivery could be similarly enhanced for exosomes injected even 6 min after UTMD. To further confirm whether cardiomyocyte was the main target of the delivery strategy, we have examined the distribution of FAM-labeled siRNA in the heart. As expected, there are significant FAM signal colocalized with cardiomyocyte marker TNNT2, suggesting that these siRNAs were at least partially distributed in these cardiomyocytes (Figure S10). Notably, there are also FAM in TNNT2 negative cells, suggesting that these siRNA could be also delivered into other cells like endothelial cells and or fibroblasts. The optimized frequency of UTMD to facilitate exosome uptake by heart was further explored by monitoring the knockdown efficiency of Gapdh. siGapdh encapsulated by exosomes were delivered, followed by indicated UTMD treatment as depicted in Figure S11A. Notably, repeated UTMD with 2 h intervals could increase the delivery and knockdown efficiency slightly (Figure S11A, B). However, repeated UTMD with intervals also caused the cell damage and inflammation, as manifested by increased expression of VCAM-1 (Figure S11C). Thus, we chose to use UTMD only once in the following study.
In the following experiments, we aimed to explore the efficacy of UTMD assisted exosomal delivery of siHmox1 on ferroptosis and cardiotoxicity. Hereafter, the exosomes with Hmox1 siRNA loaded was thus termed as ExosiHmox1. Followed injection via tail vein, ExosiHmox1 alone had no effects on the expression of Hmox1 in the heart. In contrast, prior treatment of UTMD significantly enhance the siRNA delivery and thus reduced the expression of Hmox1 (Fig. 4F-H). Collectively, these data indicate that the current strategy could functionally deliver siRNA into the heart.

UTMD promotes the delivery of exosomes into the heart. A, Schematic representation of the experiment procedure. Mice were injected with or without DiO or DiR labelled exosomes, followed by UTMD radiation in the heart region in the UTMD group. B, Ex vivo fluorescence images showing the distribution of exosomes in the hearts from control or UTMD treated mice. C, Representative images of localization of the exosomes in the TNNT-2 positive cardiomyocytes. D, Ex vivo fluorescence images showing the distribution of exosomes in the hearts from mice with indicated treatments. For UTMD treatment groups, mice were additionally injected with exosomes at indicate time post UTMD. E, Quantification data of panel D. F, Schematic representation of the experiment procedure. G, qPCR analysis of the in vivo knockdown efficiency of siHmox1 in hearts from mice treated as indicated. Data are presented as the mean ± SEM. *, p\(\:<\)0.05 by one-way ANOVA. H, In vivo knockdown efficiency of siHmox1 in mice with different treatments, as determined by qPCR
We next investigated whether this strategy can prevent the ferroptosis induced by Doxorubicin in vivo (Fig. 5A). qPCR analysis revealed that UTMD + ExosiHmox1 treatment reduced Ptgs2 mRNA expression significantly (Fig. 5B). Similarly, lipid peroxidation marker MDA (Fig. 5C), ROS indicator DHE (Fig. 5D-E), and cell death (Fig. 5F-G) were all significantly inhibited in the UTMD + ExosiHmox1 treatment group. Together, the above data suggest that UTMD + ExosiHmox1 treatment could inhibit the ferroptosis induced by Dox.

Efficient siHmox1 delivery reduces ferroptosis induced by Dox. A, Schematic representation of the experimental procedure. B, qPCR analysis of Ptgs2 mRNA expression in the hearts at indicate time points from mice receiving indicated treatments. C, MDA level in the hearts at indicate time points from mice treated same as above. D, Representative DHE staining in the heart tissues from mice treated same as above. E, Quantification data of Panel D. F, Representative TUNEL staining in the heart tissues from mice treated same as above. G, Quantification data of Panel F. Data are presented as the mean ± SEM. *, p\(\:<\)0.05 by one-way ANOVA
Consistent with the inhibition of ferroptosis, cardiac structure and function were also restored. Mice were treated as indicated in Fig. 6A, and at the end of experiment mice were subjected to echocardiography and histology analysis. HE staining and Sirius red staining revealed that UTMD + ExosiHmox1 treatment reduced the fibrosis (collagen content) in the hearts challenged with Dox, in comparison to other groups (Fig. 6B-D). Moreover, the heart weight in the UTMD + ExosiHmox1 treatment group was nearly same as the basic level (Fig. 6E). Heart weight is determined by both cell loss due to cardiotoxicity and subsequent fibrogenic repair. In other words, the heart weight change in our study was dominant by the Dox treatment induced cell loss, rather than the subsequent fibrosis.

Efficient siHmox1 delivery alleviates cardiac injury in Dox treated mice. A, Schematic representation of the experimental procedure. B, Representative images of myocardial H&E staining from the mice of indicated groups. C, Representative images of myocardial Sirius red staining from the mice of indicated treatments. D, Quantification of the collagen content. E, The heart/body weight ratio measured in mice same as above
Consistently, echocardiography examination revealed that UTMD + ExosiHmox1 significantly blocked the impairment of cardiac systolic and diastolic function induced by Dox (Fig. 7A-C). No death and significant body weight decrease were observed among the groups, suggesting that the strategy should be safe.

Echocardiography analysis of cardiac function in siHmox1 treated mice. A, Representative images of echocardiography. B, Quantification of systolic function as determined by EF value. C, Diastolic function as determined by E/A value. Data are presented as the mean ± SEM. n = 6 per group. *P < 0.05. EF, ejection fraction; FS, fractional shortening; MV, mitral valve
Methods
Mice husbandry
The male C57Bl6 mice were obtained from the experimental animal center of the Fourth Military Medical University (Xi’an, China). C57Bl/6 mice were housed at 22 ± 2 °C with a 12-h light/dark cycle. All animal experiments complied with the ARRIVE guidelines and were carried out in accordance with National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978).
Doxorubicin treatment in mice
To construct the doxorubicin induced cardiotoxicity mouse model, mice were injected intraperitoneally with doxorubicin (Yuanye biotechnology, China) diluted in PBS at 10 mg/kg, together with other indicated treatments. At the end of the experiment, the mice were subjected to echocardiography examination and/or histology analysis.
Histology
Hearts from treated mice were fixed with 4% paraformaldehyde (pH 7.4), embedded in paraffin, and then sliced into 5-µm thickness section. The sections were stained with Hematoxylin and Eosin (H&E) for routine histological examination. To measure collagen deposits, sections were stained with Sirius red. Quantification was performed using ImageJ software (National Institutes of Health).
Flow cytometry analysis of lipid peroxidation
Cells with indicated treatments were incubated in 2 µM C11-BODIPY581/591 (Invitrogen) for 20 min at 37 °C. The whole process was kept in the dark. Fluorescent signal was analyzed by flow cytometry.
DHE and TUNEL staining
Fresh heart tissues were sectioned to 10 μm thicknesses and incubated with superoxide anion fluorescent probe dihydroethidium (Cat HY-D0079, MCE) or TUNEL as instructed. The slices were counterstained with Hoechst and then washed three times with PBST for 5 min before examination under fluorescence microscope.
Cell culture and treatment
Mouse cardiomyocyte HL-1 was cultured in DMEM supplemented with 10% FBS, 1% penicillin and streptomycin, and incubated at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Cells were incubated with 0.5 µM doxorubicin for 48 h. For Hmox1 knockdown, negative control or siRNA (20 nM) was transfected into HL-1 cells using Lipofectamine 2000, followed by doxorubicin treatment one day later.
qPCR analysis
Total RNA was isolated using Trizol (Invitrogen) and reverse-transcription was conducted using PrimeScript First-Strand cDNA Synthesis Kit (Takara, China). Gene expression was analyzed using PrimeScript RT Master Mix (Roche, Switzerland) as instructed. PCR was performed at least in triplicate and relative expression was calculated by normalizing to the control samples using 2-ddCt method. The primer sequences are listed in Table S1.
Western blot
Protein samples were prepared using RIPA lysis buffer and separated in 12% SDS-PAGE gel (120 V for stacking gel and 160 V for separation gel). After transferred to nitrocellulose membrane, the membrane was blocked with 5% bovine serum albumin for 1 h, followed by incubation overnight with primary antibodies anti-GM130 (66662-1-Ig, Proteintech), anti-CD9 (60232-1-Ig, Proteintech), HMOX1 (66743-1-Ig, Proteintech) or anti-GAPDH (60004-1-Ig, Proteintech) at 4 °C. Then, the membrane was washed and incubated with corresponding secondary antibodies at room temperature for 1 h, and visualized by the ECL Prime Western Blotting Detection Reagent.
Measurement of MDA content
Malondialdehyde (MDA) levels were measured using Lipid Peroxidation MDA Assay Kit (Cat#S0131, Beyotime) as instructed.
Exosome isolation, loading, labeling and tracking
Cell culture supernatant was collected and centrifuged at 13,200 g and 4 °C for 30 min to remove large microvesicles. The supernatant was then filtered twice through 0.22 μm filters and then exosomes were pelleted with ultracentrifugation at 120,000 g for 4 h. Pellets were washed once with PBS and centrifuged at 120,000 g, 4 °C for 1 h again. The exosome morphology was confirmed by electron microscopy. Briefly, the exosomes were added onto the grid and stained with 2% uranyl acetate, followed by imaging with the transmission electron microscope (JEM-2000EX TEM, JEOL Ltd., Tokyo, Japan). Size distribution of the isolated exosomes was also analyzed by nanoparticle tracking assay.
To load siRNA (negative control, siHmox1, or siGapdh) into exosomes, about 10^10 exosomes in 400 µl were electroporated with 1 OD siRNAs of interest (GenePharma, Shanghai) at 700 V/150 mF in 0.4 cm wide electroporation cuvettes. Free nucleic acids outside the exosomes were removed by RNase, followed by additional exosome isolation with ultracentrifugation as described above. The exosome suspension was dissociated to avoid possible aggregates before use.
For in vivo tracking, purified exosomes were first incubated with fluorescent dye DiR/DiO/DiI (at the final concentration of 10 mM, Invitrogen, Carlsbad, CA) and then free dye was removed by another round of centrifugation. Mice were injected with labelled exosomes (100 µg at protein concentration in 100 µL PBS) with or without the aid of ultrasound targeted microbubble destruction. Different organs were harvested and processed for ex vivo image or microscope analysis. Cardiomyocytes were stained with anti-TNNT-2 to visualize the cellular location of exosomes/FAM-labeled siRNA.
Ultrasound targeted microbubble destruction and exosome delivery into the heart
UTMD and UTMD aided exosome in vivo delivery were conducted similar as describe before. Briefly, mice were prepared with the irradiated region hair shaved, and then anaesthetized with 2% isoflurane. The mixture composed of 100 µL SonoVue™ microbubble (Bracco imaging, SA) and 100 µL exosomes (100 µg at protein concentration in 100 µL PBS) was injected slowly via tail vein. Two minutes after injection, ultrasound irradiation was performed in the heart region through the thorax. Ultrasound pulses were applied to the targeted region for 1 min in total at a duty cycle of 50% by a 0.66 MHz US instrument (Gift from Chongqing Medical University) armed with the probe area of 4.5 cm2. For therapeutic intervention, the mice were treated with exosomes encapsulated with control or Hmox1 siRNA (100 mg at protein concentration in 100 µL PBS) with or without the aid of UTMD one day before doxorubicin treatment. Groups treated with doxorubicin, UTMD, or exosome alone served as controls.
Echocardiography
Mice were anaesthetized with inhaled isoflurane via a circuit anesthesia apparatus. Visual Sonics Vevo 2100 ultrasound instrument (Visual Sonics Inc., Toronto, ON, Canada) armed with 40 MHz linear transducer was employed for cardiac function analysis. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were obtained from M-mode, and E/A value was obtained by pulsed-wave Doppler echocardiography. The heart rate was monitored continuously during the whole process [15]. The identities of animals were kept blinded to the echocardiography investigators.
Statistics
The data were expressed as mean ± SEM or otherwise indicated. One-way ANOVA analysis was used for multiple group comparison or t-test for comparison between two groups by GraphPad Prism8. Significant differences were considered at p < 0.05.
Discussion
In the present study, we have revealed that cardiac ferroptosis induced by DOX in mice was attributed to up-regulation of Hmox1. UTMD greatly facilitated exosome delivery into heart. Consistently, UTMD assisted exosomal delivery of siHomox1 nearly blocked the ferroptosis and the subsequent cardiotoxicity induced by doxorubicin.These findings reveal that the upregulation of HMOX1 induces ferroptosis in cardiomyocytes and UTMD-assisted exosomal delivery of siHmox1 can be used as a potential therapeutic strategy for DIC.
DOX, a widely used chemotherapeutic drug, has dosage-dependent cardiac toxicity, which often decreases the quality of life and even causes life-threatening disorders. Various mechanisms how DOX exerts its toxic effects on cardiomyocytes have been identified. The most accepted notion was that excessive DOX accumulated in the mitochondria results in excessive ROS generation and oxidative stress-induced cell death subsequently [16]. Death of cardiomyocytes is crucial in the development of cardiotoxicity. DOX has also been reported to induce various forms of cell death, including apoptosis, autophagy, necroptosis, ferroptosis and pyroptosis [17]. Different from the caspase-dependent apoptosis, ferroptosis is a novel form of regulated cell death characterized by the iron-dependent accumulation of lipid peroxidation. Ferroptosis is morphologically, biochemically, and genetically different from apoptosis, and other forms of death, such as necroptosis, and autophagy. Inhibition of either apoptosis or necroptosis only partially improved the survival of DOX-treated cardiac cells [18], suggesting that other forms of cell death are likely involved in DOX-induced cardiotoxicity. A recent publication found that both apoptosis and ferroptosis were involved in the DIC model and combined usage of zVAD (apoptosis inhibitor) and Fer-1 (ferroptosis inhibitor) almost completely abolished DOX-induced cardiomyocytes death [19]. Consistent with previous studies, we here further confirmed that ferroptosis is a big player in DIC. Notably, a recent study showed that ferroptosis inhibition rather than apoptosis inhibition prolongs the survival rate in DIC model, suggesting that ferroptosis accounts for a higher ratio than apoptosis in the DIC model [4]. Notably, apoptosis and ferroptosis might accounts for the major cell death in a time dependent manner, with apoptosis mainly peaked in the early phase (10 h), while ferroptosis dominant beyond 30 h following DOX treatment [4, 7, 20].
Up to now, the exact molecular network involved in DOX-induced ferroptosis remains elusive. Previous studies presented that DOX treatment led to conceivable changes in ferroptosis-related markers, including NCOA4, GPX4, HMOX1, GSH, and MDA levels [4, 7, 20]. Consistent with previous study, we found that up-regulation of Hmox1 accounts for the obvious ferroptosis, which might be explained by the fact that Hmox1 causes heme degradation and leads to release of free iron, which accumulates in mitochondria and triggers lipid peroxidation in heart. The ferroptosis promoting effects of Hmox1 is paradoxical to its widely considered cardioprotective role in cardiac ischemia/reperfusion injury and permanent coronary ligation-induced heart failure [21, 22]. One explanation is that Hmox1 functions in a context dependent manner. Further studies to reveal the underlying mechanisms would be helpful in developing Hmox1-based therapy.
Preventing cardiac cell death is essential to develop an effective cardioprotective strategy. Ferroptosis can be prevented or even reversed using iron chelation and ROS scavenger [23]. Interestingly, although the potent iron chelator, such as deferoxamine, is used to treat iron overload diseases such as thalassemia and sickle cell disease [24], its protective effect with respect to DIC is unclear and worth to be explored. We here revealed that repeated injection of Ferrostatin itself is toxic and had no persistent protective effects.
Cardiovascular diseases remain one of the leading causes of death worldwide. Although we only studied the role of ferroptosis, a recently discovered iron-dependent form of cell death, in cardiac injury induced by doxorubicin. Upregulation of heme oxygenase Hmox1 and subsequent ferroptosis should be also involved in other cardiovascular diseases [25,26,27]. Knockdown of Hmox1 and inhibiting ferroptosis via UTMD assisted exosomal delivery strategy significantly reduced DIC, suggesting a potential therapeutic approach for treating and/or preventing various heart diseases associated with ferroptosis.
Conclusion
In summary, we here have further confirmed that ferroptosis mediates the pathogenesis of DOX-induced cardiotoxicity via upregulation of Hmox1. Our findings suggest that selectively knockdown of Hmox1 in cardiomyocytes via UTMD-assisted exosome delivery may represent a practical therapeutic approach for management of DOX-induced cardiotoxicity without compromising the drug’s anticancer properties. Future studies are needed to test the potential clinical implications of this proposed therapeutic strategy.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ALOX4:
-
Alx homeobox 4
- CK:
-
Creatine kinase
- DHE:
-
Dihydroergotamine
- DFP:
-
Deferiprone、
- DIC:
-
Dox-induced cardiomyopathy
- DiI:
-
1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine
- DiO:
-
3,3′-Dioctadecyloxacarbocyanine perchlorate
- DiR:
-
1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyan ineiodide
- DOX:
-
Doxorubicin
- ER:
-
Endoplasmic reticulum
- Exo:
-
Exosome
- Fer:
-
Ferroptosis
- FTH1:
-
Ferritin heavy chain 1
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GPX4:
-
Glutathione peroxidase 4.
- GSH:
-
Glutathione
- HE:
-
Hematoxylin and eosin
- Hmox1:
-
Heme oxygenase-1
- LDH:
-
Lactate dehydrogenases
- LPCAT3:
-
Lysophosphatidylcholine acyltransferase 3
- LVEF:
-
Left ventricular ejection fraction
- LVFS:
-
Left ventricular fractional shortening
- MDA:
-
Malondialdehyde
- mRNA:
-
Message RNA
- NFE2L2:
-
Nuclear factor erythroid factor-2
- NOX1:
-
NADPH oxidase 1
- PLOOH:
-
Phospholipid hydroperoxides
- PUFA:
-
Polyunsaturated fats
- qPCR:
-
Real-time quantitative polymerase chain reaction
- ROS:
-
Reactive oxygen species
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- TEM:
-
Transmission electron microscopy
- US:
-
Ultrasound
- UTMD:
-
Ultrasound-targeted microbubble destruction
References
Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother. 2021;139:111708.
Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97(11):2869–79.
Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox Biol. 2020;29:101394.
Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, Hu L, Huang K, He J. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022;29(10):1982–95.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Ta N, Qu C, Wu H, Zhang D, Sun T, Li Y, Wang J, Wang X, Tang T, Chen Q, Liu L. Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy. Proc Natl Acad Sci U S A. 2022;119(36):e2117396119.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–80.
Ke W, Afonin KA. Exosomes as natural delivery carriers for programmable therapeutic nucleic acid nanoparticles (NANPs). Adv Drug Deliv Rev. 2021;176:113835.
Afonin KA, Dobrovolskaia MA, Church G, Bathe M. Opportunities, barriers, and a strategy for overcoming Translational challenges to Therapeutic Nucleic Acid Nanotechnology. ACS Nano. 2020;14(8):9221–7.
Sun W, Zhao P, Zhou Y, Xing C, Zhao L, Li Z, Yuan L. Ultrasound targeted microbubble destruction assisted exosomal delivery of miR-21 protects the heart from chemotherapy associated cardiotoxicity. Biochem Biophys Res Commun. 2020;532(1):60–7.
Sun W, Xing C, Zhao L, Zhao P, Yang G, Yuan L. Ultrasound assisted Exosomal Delivery of tissue responsive mRNA for enhanced efficacy and minimized off-Target effects. Mol Ther Nucleic Acids. 2020;20:558–67.
Chen X, Comish PB, Tang D, Kang R. Characteristics and biomarkers of Ferroptosis. Front Cell Dev Biol. 2021;9:637162.
Du Y, Guo Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discov. 2022;8(1):501.
Wu Y, Lu C, Pan N, Zhang M, An Y, Xu M, Zhang L, Guo Y, Tan L. Serum lactate dehydrogenase activities as systems biomarkers for 48 types of human diseases. Sci Rep. 2021;11(1):12997.
Yuan L, Wang T, Liu F, Cohen ED, Patel VV. An evaluation of transmitral and pulmonary venous doppler indices for assessing murine left ventricular diastolic function. J Am Soc Echocardiogr. 2010;23(8):887–97.
Shi S, Chen Y, Luo Z, Nie G, Dai Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal. 2023;21(1):61.
Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021;12(4):339.
Kitakata H, Endo J, Ikura H, Moriyama H, Shirakawa K, Katsumata Y, Sano M. Therapeutic targets for DOX-Induced Cardiomyopathy: role of apoptosis vs. Ferroptosis. Int J Mol Sci 23(3) (2022).
Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI, Imai H, Tsutsui H. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 8(6) (2023).
Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11(7):3052–9.
Dunn LL, Kong SMY, Tumanov S, Chen W, Cantley J, Ayer A, Maghzal GJ, Midwinter RG, Chan KH, Ng MKC, Stocker R. Hmox1 (Heme Oxygenase-1) protects against ischemia-mediated Injury via stabilization of HIF-1alpha (hypoxia-Inducible Factor-1alpha). Arterioscler Thromb Vasc Biol. 2021;41(1):317–30.
Jazwa A, Stepniewski J, Zamykal M, Jagodzinska J, Meloni M, Emanueli C, Jozkowicz A, Dulak J. Pre-emptive hypoxia-regulated HO-1 gene therapy improves post-ischaemic limb perfusion and tissue regeneration in mice. Cardiovasc Res. 2013;97(1):115–24.
Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, Volpe JJ, Rosenberg PA. 12-Lipoxygenase plays a key role in cell death caused by glutathione depletion and arachidonic acid in rat oligodendrocytes. Eur J Neurosci. 2004;20(8):2049–58.
Qadah T. Deferasirox versus deferoxamine in managing iron overload in patients with Sickle Cell Anaemia: a systematic review and meta-analysis. J Int Med Res. 2022;50(12):3000605221143290.
Wang K, Chen XZ, Wang YH, Cheng XL, Zhao Y, Zhou LY, Wang K. Emerging roles of ferroptosis in cardiovascular diseases. Cell Death Discov. 2022;8(1):394.
Hu H, Chen Y, Jing L, Zhai C, Shen L. The Link between Ferroptosis and Cardiovascular diseases: a novel target for treatment. Front Cardiovasc Med. 2021;8:710963.
Ayer A, Zarjou A, Agarwal A, Stocker R. Heme Oxygenases in Cardiovascular Health and Disease. Physiol Rev. 2016;96(4):1449–508.
Acknowledgements
We are grateful to the technical help to Jing Zhang from the Department of Ultrasound in Tangdu Hospital, Fourth Military Medical University.
Funding
This study was supported by the National Natural Science Foundation of China 82302223 and 82202167.
Author information
Authors and Affiliations
Contributions
JC, SQ and YL: Design, Experiment and Writing-original draft. WQ and TZ: Cell and Animal experiments. LZ, ZL and YD: Conceptualization, Methodology, Writing—review and editing, Supervision. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Protocols of animal experiments included in this study were approved by the policies and guidelines of the Animal Ethics Committee of Fourth Military Medical University.
Consent for publication
All authors confirmed that this work has not been published before, and was not under consideration for publication elsewhere. All authors have approved the submission of this manuscript to Journal of Nanobiotechnology.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, J., Qiu, S., Liu, Y. et al. Ultrasound targeted microbubble destruction assisted exosomal delivery of siHmox1 effectively inhibits doxorubicin-induced cardiomyocyte ferroptosis. J Nanobiotechnol 22, 531 (2024). https://doi.org/10.1186/s12951-024-02794-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-024-02794-w