
Abstract
Background
Non-obstructive azoospermia (NOA) is the most severe form of male infertility and affects approximately 1% of men worldwide. Fanconi anemia (FA) genes were known for their essential role in DNA repair and growing evidence showed the crucial role of FA pathway in NOA. However, the underlying mechanisms for Fance deficiency lead to a serious deficit and delayed maturation of male germ cells remain unclear.
Methods
We used Fance deficiency mouse model for experiments, and collected testes or epididymides from mice at 8 weeks (8W), 17.5 days post coitum (dpc), and postnatal 11 (P11) to P23. The mice referred to three genotypes: wildtype (Fance +/+), heterozygous (Fance +/-), and homozygous (Fance −/−). Hematoxylin and eosin staining, immunofluorescence staining, and surface spread of spermatocytes were performed to explore the mechanisms for NOA of Fance −/− mice. Each experiment was conducted with a minimum of three biological replicates and Kruskal-Wallis with Dunn’s correction was used for statistical analysis.
Results
In the present study, we found that the adult male Fance −/− mice exhibited massive germ cell loss in seminiferous tubules and dramatically decreased sperms in epididymides. During the embryonic period, the number of Fance −/− prospermatogonia decreased significantly, without impacts on the proliferation (Ki-67, PCNA) and apoptosis (cleaved PARP, cleaved Caspase 3) status. The DNA double-strand breaks (γH2AX) increased at the cellular level of Fance −/− prospermatogonia, potentially associated with the increased nonhomologous end joining (53BP1) and decreased homologous recombination (RAD51) activity. Besides, Fance deficiency impeded the progression of meiotic prophase I of spermatocytes. The mechanisms entailed the reduced recruitment of the DNA end resection protein RPA2 at leptotene and recombinases RAD51 and DMC1 at zygotene. It also involved impaired removal of RPA2 at zygotene and FANCD2 foci at pachytene. And the accelerated initial formation of crossover at early pachytene, which is indicated by MLH1.
Conclusions
Fance deficiency caused massive male germ cell loss involved in the imbalance of DNA damage repair in prospermatogonia and altered dynamics of proteins in homologous recombination, DNA end resection, and crossover, providing new insights into the etiology and molecular basis of NOA.
Similar content being viewed by others


Introduction
Infertility impacts approximately 48 million couples globally, with male factors accounting for 50% of cases [1]. The most common manifestation of male infertility is spermatogenesis disorder [2], of which the majority of causative factors are unclear. Genetic factors are important causes of spermatogenesis disorder, and increasing evidence has shown the crucial role of DNA damage repair genes. Mutations of DNA damage repair genes, such as Atm, Msh4 and Msh5, lead to non-obstructive azoospermia (NOA), which is the most serious type of male infertility [3]. Deletions of DNA repair proteins, specifically RAD51, MLH1, and EXO1, are associated with spermatogenesis disorder and infertility [3, 4]. These studies underline the roles of DNA damage repair genes in the spermatogenesis.
Fanconi anemia (FA) family is known to be involved in DNA interstrand cross-links (ICLs) repair. Currently, 22 FA genes have been identified [5, 6], and growing evidence shows that the FA deficient individuals were subfertility [7]. The mutations of Fanca [8], Fancm [9], and Fancu [10] are determined to result in NOA and sertoli cell only syndrome (SCOS) in male patients. Consistently, the knockout of FA genes (Fanca, Fancc, Fancd1, Fancd2, Fancf, Fancg, Fancl, Fancm, and Fancp) in male mice leads to sterility and hypogonadism, exhibiting a notable loss of germ cells and impaired spermatogenesis [11,12,13,14,15,16,17,18,19,20,21]. The mechanisms of germ cell loss in FA males mainly involved defective proliferation and increased apoptosis of primordial germ cells (PGCs), impaired maintenance of spermatogonial stem cells (SCCs), and emerging roles in the DNA double-strand breaks (DSBs) repair at meiotic prophase [7].
Fanconi anemia complementation group E (FANCE), a component of the FA core complex (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM), is essential for the activation of the FA pathway. In the previous studies, we generated a mouse model with homozygous insertional mutagenesis of the Fance gene (OVE2364E-2a2). The Fance deficient (Fance −/−) mice displayed reduced fertility, hypogonadism, and germ cell loss in both males and females, defined as NOA and premature ovarian insufficiency (POI), respectively (Supplementary Material 1) [22, 23]. Our previous studies indicated that Fance −/− PGCs decreased evidently as early as 9.5 days post coitum (dpc) [24]. The loss of Fance −/− PGCs involved the increased apoptosis, abnormal proliferation, accumulated DNA damage and transcription-replication conflicts, and down-regulated DNA repair pathways [24, 25]. Meanwhile, we found that the mitotic-meiotic transition of Fance −/− female oocytes occurred at 13.5 dpc normally, but the progression of meiotic prophase I was arrested [26]. The male Fance −/− mice exhibited reduced meiotic cells and localized spermatogenesis [22].
Based on the previous research, we further focused on the mitotic-to-quiescent transition (17.5 dpc) and meiotic prophase I (postnatal days 11 to 23, P11-P23) of Fance −/− males. We collected the testes and epididymides from wildtype (Fance +/+), heterozygous (Fance +/-), and homozygous (Fance −/−) males to perform this study. Results revealed that the loss of embryonic germ cells in male Fance −/− mice involved impaired DNA damage repair. The delayed progression of meiotic prophase I in male Fance −/− mice resulted from the disturbed repair dynamics in homologous recombination. These findings showed that FANCE, a crucial component of the FA pathway, plays essential roles in meiosis and the embryonic development of male germ cells.
Materials and methods
Animals
All animal experiments were permitted by the Laboratory Animal Welfare and Ethical Committee of Central South University (IACUC Number: 2020sydw1041). Mice were hosted on a 12-h light/12-h dark cycle at 20-25 °C ambient temperature, with free access to food and water in the specific-pathogen-free facility. Mice were euthanized by cervical dislocation to collect the testes and epididymides. Prenatal time points were determined by following timed pregnancies, where the first day post coitum was considered 0.5 dpc. Postnatal time points were measured starting from birth, considering the first postnatal day as P1.
Fance deficient mice, bred onto an FVB/N background, were generated as previously described [22, 23]. Genotyping was performed by PCR using tail genomic DNA. Primer sequences were as follows: Fance-a:5'-TGGCATCTCCACTT CTCTATCA-3', Fance-b :(5'-AGAGCAGCCTG
GACTACTTGAG-3'), Fance-c:5'-CGTCTGTTGTGTGACTCTGGTAAC-3'), and Fance-d: 5'-CCTGGTGTGTAGCTTTGCCAATCA-3'). PCR products were separated by electrophoresis on a 1.5% (wt/vol) agarose gel (Supplementary Material 2).
Histology and immunofluorescence staining
Histology of testes and epididymides was performed on Fance +/+, Fance +/-, and Fance −/− males at 8 weeks. Testes and epididymides were dissected, fixed overnight in Bouin’s fixative solution, paraffin-embedded, sectioned (5 µm), and stained with hematoxylin and eosin using standard protocols. Sections of testes from Fance +/+, Fance +/-, and Fance −/− mice at 17.5 dpc were incubated with primary antibodies overnight at 4 ℃ and then with specified secondary antibodies for immunofluorescence staining.
Meiotic chromosome spread of spermatocytes
We collected testes of Fance +/+, Fance +/-, and Fance −/− mice at P11, P13, P17, P20, and P23 to perform the meiotic chromosome spread of spermatocytes. Briefly, seminiferous tubules were digested with 1 mg/mL type IV collagenase at 37 ℃ for 30 min. Cells were collected and placed in a buffer containing 30 mM Tris-HCl, 50 mM sucrose, and 17 mM trisodium citrate dihydrate for 10 min. Cells were then collected at 400 g for 1 min and resuspended in 100 mM sucrose for 5 min. The cell suspension was added to the same volume of buffer containing 1% PFA, 0.15% Triton X-100, and 10 mM sodium borate. After that, the suspension was spread onto slides and air-dried overnight.
Antibodies
The following antibodies were used: rabbit anti-DDX4 (Abcam, ab13840, 1:200); mouse anti-DDX4 (Abcam, ab27591, 1:200); mouse anti-Ki-67 (Santa Cruz, sc-23900, 1:100); mouse anti-PCNA (Santa Cruz, sc-56,1:150); rabbit anti-cleaved Caspase 3 (Cell Signaling, 9661, 1:400); rabbit anti-cleaved PARP (Cell Signaling Technology, 9544, 1:200); rabbit anti-γH2AX (Novus Biologicals, NB100-2280, 1:200); mouse anti-RAD51 (Novus Biologicals, NB100-148, 1:200); rabbit anti-53BP1 (Novus Biologicals, NB100-304,1:200); rabbit anti-SYCP3 (Abcam, ab15093, 1:200); mouse anti-SYCP3 (Santa Cruz, sc-74569, 1:200); rabbit anti-FANCD2 (Novus Biologicals, NB100-182, 1:100); anti-SYCP1(Abcam, ab175191, 1: 200); rabbit anti-DMC1 (Abcam, ab245217, 1:200); rat anti-RPA2 (Cell Signaling Technology, #2208, 1:100); rabbit anti-MLH1 (Abcam, ab92312, 1:200). The second antibodies used in this study were as follows: goat anti-mouse Alexa Fluor 488 (Abcam, ab150117, 1:1000); goat anti-mouse Alexa Fluor 594 (Abcam, ab150116, 1:1000); goat anti-rat Alexa Fluor 488 (Abcam, ab150157, 1:1000); goat anti-rabbit Alexa Fluor 594 (Abcam, ab150080, 1:1000). DAPI was used to counterstain cell nuclei. All images were captured using the Zeiss Axio Scope A1 microscope.
Statistical analysis
At least three biological replicates were conducted for each experiment. Data were presented as mean ± S.D. Kruskal-Wallis with Dunn’s correction was employed for multiple comparisons between groups. P < 0.05 was considered statistically significant. Statistical analyses were performed using Prism Software (GraphPad). Statistical parameters, including statistical tests and P-values, were reported in the figure legends.
Results
Fance deficiency leads to germ cell loss in adult male mice
A general characteristic of FA-null mice was profound germline defects, including hypogonadism and germ cell depletion [7]. Fance −/− mice have been reported that the weights and sizes of the testes were significantly lower than the control mice from 1 to 8 weeks of age [22]. Consistently, we detected the sizes of testes and epididymides from Fance −/− mice at 8 weeks were smaller than those of their wild-type and heterozygous siblings (Fig. 1A). We further compared sections of testes and epididymides from Fance +/+, Fance +/-, and Fance −/− mice using H&E staining (Fig. 1B). Seminiferous tubules from Fance +/+ and Fance +/- mice had normal architecture and contained germ cells at various maturation stages. The majority of seminiferous tubules in Fance −/− testes were severely degenerated and the number of germ cells was sharply reduced. Consistent with the histological analysis of testes, sperms in epididymides of Fance −/− mice were much fewer than controls. Concerned the testes of Fance −/− mice displayed a mosaic pattern, we further classified the seminiferous tubules (Fig. 1C). In the testes of Fance −/− mice, only 15% of seminiferous tubules had relatively normal morphology, 52% of seminiferous tubules only contained Sertoli cells, 19% of the seminiferous tubules manifested massive germ cell loss, and 14% of Fance −/− tubules had round spermatids as the most advanced spermatogenic cells (Fig. 1D). Collectively, our results indicated that Fance deficiency results in massive germ cell loss and displays a mosaic pattern in the testes of adult male mice.

Fance deficiency results in gonadal hypoplasia and germ cell loss in adult male mice
Fance deficiency reduces prospermatogonia without impact on the proliferation and apoptosis
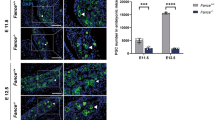
To determine the germ cell defects that occurred during embryonic development, we conducted further study on the Fance −/− mice at 17.5 dpc. At 17.5 dpc, the male germ cells were at the mitotic-to-quiescent transition and were called prospermatogonia. Immunofluorescence staining was conducted on testes sections using the germ cell marker DDX4 (Fig. 2A). In Fance −/− testes at 17.5 dpc, 78% of seminiferous tubules only contained sertoli cells, and only 22% of tubules had germ cells (Fig. 2B). And there was a significant decrease in the number of germ cells per germ cell-containing tubule in Fance −/− testes compared with the controls (Fig. 2C). It implied that the number of Fance −/− prospermatogonia has declined significantly at 17.5 dpc.

Fance deficiency decreases number of prospermatogonia
We performed immunofluorescence on Fance +/+, Fance +/-, and Fance −/− testis sections to detect DDX4 and Ki-67 (a marker of cell proliferation) at 17.5 dpc (Fig. 3A). The proportions of Ki-67 positive prospermatogonia were similar among Fance −/− mice and controls (Fig. 3B). We further found ki-67 foci number per ki-67 positive prospermatogonia did not differ significantly between controls and Fance −/− mice (Fig. 3C). Meanwhile, we performed immunostaining for the proliferating cell nuclear antigen (PCNA) (Fig. 3D), which is a DNA replication and repair protein. The percentage of PCNA positive germ cells did not differ significantly between controls and Fance −/− germ cells (Fig. 3E). The percentages of Ki-67 or PCNA-positive germ cells were very low to negligible among the three genotypes. These results indicated that the proliferation status of Fance −/− prospermatogonia was normal.

Fance is dispensable for mitotic-to-quiescent transition and apoptosis of prospermatogonia
Immunofluorescence stainings of cleaved PARP and cleaved Caspase 3 were utilized to estimate the apoptosis of germ cells (Fig. 3F, H). Results showed that the apoptosis was comparable among Fance −/− mice and controls (Fig. 3G, I). It means the deficiency of Fance did not increase the apoptosis of germ cells at 17.5 dpc. Collectively, these results reflect that the proliferation and apoptosis of Fance −/−prospermatogonia are unaffected at the mitotic-to-quiescent transition.
Fance deficiency impacts the DSBs level and the activity of HR and NHEJ repair in prospermatogonia
Based on the important roles of Fanconi anemia genes in DNA damage response and repair [5], we explored the DNA damage and repair status of Fance −/− germ cells at 17.5 dpc. The phosphorylation of H2AX at serine 139 (γH2AX) was used to measure the presence of DSBs in germ cells, which is the most severe type of DNA damage (Fig. 4A). No significant difference was seen in the proportions of γH2AX-positive germ cells among Fance +/+, Fance +/-, and Fance −/− mice (Fig. 4B). However, in Fance −/− mice, the number of γH2AX foci per γH2AX-positive germ cell significantly increased, suggesting an increase in DSBs at the cellular level. (Fig. 4C).

Fance is required for DSBs repair of prospermatogonia
Homologous recombination (HR) and non-homologous end joining (NHEJ) are the main methods responsible for repairing DSBs and preserving the integrity of the genome [27]. 53BP1, which mediated the NHEJ pathway, was detected by immunostaining [28] (Fig. 4D). The expression of 53BP1 was significantly elevated in Fance −/− germ cells compared with Fance +/- (Fig. 4E). Despite differences, the majority of Fance +/+, Fance +/-, and Fance −/− germ cells were positive for 53BP1, indicating that the NHEJ is the predominant DSBs repair way of male germ cells at 17.5 dpc. We performed immunostaining for the HR factor RAD51(Fig. 4F). The results revealed a significant reduction in the proportion of RAD51-positive germ cells in Fance −/− mice compared to the control groups (Fig. 4G). The increased DSBs observed in Fance −/− germ cells at 17.5 dpc may be linked with the imbalance between NHEJ and HR mechanisms.
Fance deficiency delays the progression of meiotic prophase I
To investigate the roles of FANCE in meiotic prophase I, we performed meiotic chromosome spread and immunofluorescence staining in Fance +/+, Fance +/-, and Fance −/− spermatocytes from P11 to P23. Firstly, we examined the meiotic progression by SYCP3 (a marker for the meiotic chromosome axis) and γH2AX immunostaining of spermatocytes (Fig. 5A). At P11, only leptotene spermatocytes were observed in Fance −/− mice. And the proportion of Fance −/− leptotene spermatocytes was much more than the Fance +/+ and Fance +/- mice (Supplementary Material 3A). At P13, Fance −/− spermatocytes developed to mid pachytene. Compared with the controls, Fance −/− spermatocytes exhibited a reduced proportion of mid pachytene and an increased proportion of leptotene (Supplementary Material 3B). At P17, all stages of spermatocytes in meiotic prophase were identified in controls. Whereas, the diplotene spermatocytes couldn’t be detected in Fance −/− spermatocytes, the proportion of late pachytene spermatocytes decreased and the proportion of leptotene spermatocytes increased (Fig. 5B). We further detected all the stages of the meiotic prophase I in Fance −/− spermatocytes at P20 and P23, proportions of late meiotic spermatocytes (late pachytene and diplotene) dramatically reduced and the proportion of leptotene spermatocytes significantly increased (Supplementary Material 3C, D). These results indicated that Fance deficiency slowed the meiotic prophase I progression of spermatocytes and hindered most spermatocytes at the leptotene stage.

Meiotic prophase I progression is delayed in Fance −/− spermatocytes
The pattern of γH2AX signals was similar in Fance +/+, Fance +/-, and Fance −/− spermatocytes at the leptotene and zygotene stages. However, at pachytene, 49% of the Fance −/− spermatocytes showed a persistent presence of γH2AX signals on autosomes (Fig. 5C, D). In contrast, the abnormal γH2AX signals were detected in only 11% and 11% of the Fance +/+ and Fance +/- pachytene spermatocytes, respectively (Fig. 5D). Collectively, Fance deficiency led to defective DSBs repair in autosomes of pachytene spermatocytes. Additionally, Immunostaining of central components of synaptonemal complex SYCP1 and SYCP3 revealed that the chromosome synapsis was relatively normal at various meiotic stages in Fance +/+, Fance +/-, and Fance −/− spermatocytes (Fig. 6).

The chromosome synapsis of Fance −/− spermatocytes is relatively normal
Fance is essential for meiotic recombination and the resolution of FANCD2 foci from autosomes during meiosis
Next, we explored the underlying mechanisms of the disturbed meiotic progression and DSBs repair in Fance-deficient mice. We investigated the HR repair of programmed DSBs by immunostaining of RAD51 and DMC1 (two recombinases of meiotic HR repair) (Fig. 7A, E). We observed the numbers of RAD51 and DMC1 foci on chromosome axes in leptotene spermatocytes were comparable in the three genotypes (Fig. 7B, F). At the zygotene stage, the numbers of RAD51 and DMC1 foci recruited at chromosome axes were significantly decreased in Fance −/− spermatocytes compared with controls (Fig. 7C, G). The number of RAD51 and DMC1 foci returned to the normal level at the pachytene Fance −/− spermatocytes (Fig. 7D, H).

Fance is required for HR repair at meiotic prophase
Given that the FANCD2 functioned in repairing programmed DSBs, promoting chromosome synapsis, and regulating crossover formation during meiosis in spermatogenesis [29], we examined the expression of FANCD2, which acted downstream of FANCE. At the mid pachytene, we found FANCD2 foci accumulated on autosomes persistently in Fance −/−spermatocytes but disappeared on autosomes in Fance +/+ and Fance +/− spermatocytes (Fig. 8). These observations reflected that Fance plays a crucial role in the proper recruitment of DNA repair factors during meiotic recombination.

Fance is required for the resolution of FANCD2 foci from autosomes at meiotic prophase
Fance plays a crucial role in DNA end resection and the initiation of crossover formation during meiosis
DNA end resection of programmed DSBs generates single-stranded DNA (ssDNA) to load RAD51 and DMC1 recombinases. We performed investigations on the process of DNA end excision by examining the ssDNA-binding protein RPA2, which can reflect the amount of ssDNA generated (Fig. 9A). It was found that leptotene Fance −/− spermatocytes had a lot fewer RPA2 foci at the chromosome axes than the controls (Fig. 9B). At the zygotene stage, Fance −/− spermatocytes had far more RPA2 foci than Fance +/+ and Fance +/- spermatocytes (Fig. 9C). At pachytene, there was no significant difference in the number of RPA2 foci in spermatocytes among the three groups (Fig. 9D). These observations proved that FANCE plays a crucial role in the DNA end resection of programmed DSBs.

Fance is required for DNA end resection and corssover at meiotic prophase
We evaluated the effect of FANCE on crossover formation by measuring MLH1, a mismatch repair factor that accumulates at the meiotic crossover site (Fig. 9E). At early pachytene, an apparent increase in MLH1 foci numbers was detected in Fance −/− spermatocytes compared with controls (Fig. 9F). At the mid and late pachytene stages, the numbers of MLH1 foci were similar among Fance −/− mice and their wildtype and heterozygous counterparts (Fig. 9G, H). These data indicated that although Fance −/− spermatocytes can largely complete crossover, their initial formation was accelerated.
Discussion
In the present study, we found that Fance deficiency results in germline defects of males, including prospermatogonia loss at the embryonic period and meiotic defects of spermatocytes at meiotic prophase I. Moreover, we further elucidated that the disruption of the DNA damage repair of prospermatogonia and defects in meiotic recombination, DNA end resection, and crossover formation in spermatocytes were the underlying mechanisms for the germline defects. Collectively, our results clarify the involvement and mechanism of FANCE during the germ cell development of males.
Firstly, we revealed that Fance −/− mice displayed germline defects, including germ cell loss in testes and sperms decreased in epididymides, which is consistent with FA patients and other mouse models [8,9,10,11,12,13,14,15,16,17,18,19,20,21]. Previous studies have revealed that FA proteins regulate the fertility of FA males by influencing the PGCs proliferation and maintenance of SCCs [7]. We conducted further exploration to investigate the precise mechanism by which FANCE regulates spermatogenesis.
Prospermatogonia links the embryonic development of male PGCs to the postnatal development of spermatogonia and the establishment of SCCs [30]. The proper timing of a quiescence period in prospermatogonia is important for the successful establishment of SCCs pool [31]. Although the number of Fance −/− prospermatogonia reduced significantly at 17.5 dpc, the proliferation of the remaining prospermatogonia was normal, which means Fance −/− prospermatogonia entered quiescence on a normal timeline. Meanwhile, the Fance −/− prospermatogonia underwent a relatively normal apoptotic status.
Our outcomes found persistent DSBs in Fance −/− prospermatogonia, which were also detected in Fancd1-mutant mice [32]. Additionally, we found the main repair pathway of prospermatogonia was NHEJ. When compared to controls, the NHEJ activity of Fance −/− prospermatogonia was higher and the HR activity was lower. Consistent with our previous finding that loss of Fance in the oocytes stimulates NHEJ repair and inhibits HR repair [26].
Meiosis is necessary for the formation of functional gametes from diploid germ cells during gametogenesis in mammals. The Fance −/− male germ cells showed a delay in entering meiotic prophase I at P11 and the progression of meiotic prophase I was hindered during the subsequent periods. Thus, we hypothesize that the absence of Fance resulted in a delay in the initiation and progression of meiotic prophase I.
The repair of programmed DSBs is an essential event during meiosis. Multiple studies have observed the presence of persistent γH2AX foci in spermatocytes of Fancd1, Fancn, and Fanco mouse models [32,33,34]. These suggested that the mutations of FA genes result in unrepaired DNA and unsynapsed regions on homologous chromosomes. In line with previous research, our findings demonstrated the presence of abnormal γH2AX foci on autosomes of Fance −/− pachytene spermatocytes.
FANCD2 is the key element of the FA pathway and maintains genome stability in various DNA damage response pathways. Recent studies reported that FANCD2 foci on chromosome axes were abolished in mutant spermatocytes deficient for FANCA, FANCB, FANCC, and FANCI [35, 36], indicating their essential function for FANCD2 foci formation during meiosis. We noticed FANCD2 foci accumulated on autosomes at the leptotene and zygotene stages and disappeared at the pachytene stage in controls. Deficiency of FANCE resulted in the FANCD2 foci accumulating on autosomes at the pachytene stage persistently. FANCD2 is required for HR during meiotic DSBs repair in Caenorhabditis elegans [37] and promotes DSBs repair through HR in human somatic cells [38]. We speculate that the Fance deficiency affects the HR repair of FANCD2, which participates in forming the unrepaired DSBs on autosomes at Fance −/− pachytene spermatocytes.
Meiotic recombination, mediated by the recombinases RAD51 and DMC1, is essential for chromosome synapsis to repair the DSBs produced at leptotene. We observed the decreased recruitment of RAD51 and DMC1 at zygotene Fance −/− spermatocytes. These might be the reason for the delayed repair of DSBs and the impeded progression of meiotic prophase I. Consistent with our study, the number of RAD51 foci declined in Fancb [39], Fancd1 [14, 32], Fanco [34], and Fancs [32] mutant mice. Similarly, DMC1 foci, a meiotic specific paralog of RAD51, was decreased in Fancd1 mutant mice [14]. Whereas, Kurzbauer et al. indicated that DMC1 foci manifested an increase in Fancd2 mutant Arabidopsis [33]. And the DMC1 foci in Fancs mutant spermatocytes remain unaltered [32]. These results imply that FA genes play important but not exactly the same roles in the recruitment of RAD51 and DMC1. Collectively, FANCE plays a role in ensuring proper meiotic progression by influencing the functions of RAD51, DMC1, and FANCD2 to repair the programmed DSBs of meiotic prophase I.
DNA end resection of DSBs generates ssDNA for loading RAD51 and DMC1. Shi et al. reported that when the ssDNA-binding protein RPA is lost, RAD51/DMC1 recombinases can’t be recruited to the programmed DSBs sites [38]. We observed that RPA2 foci reduced at leptotene Fance −/− spermatocytes, earlier than the decrease of DMC1 and RAD51. We infer that the lack of FANCE in spermatocytes decreased the recruitment of RAD51/DMC1 at zygotene owing to the reduction of RPA2 at leptotene. Additionally, we observed an increased number of RPA2 foci in Fance −/−spermatocytes at the subsequent zygotene stage, preceding the increase of FANCD2 foci. Feeney et al. reported that removal of RPA from ICLs sites is perturbed in FANCW-deficient cells [40]. Previous research indicated that FANCW facilitates HR by promoting the unloading of RPA and RAD51 from DNA damage sites [41]. These findings suggest that Fance may facilitate HR by promoting the removal of RPA2 and FANCD2 during the meiosis.
Furthermore, we observed an elevated number of MLH1 foci in Fance −/− spermatocytes at early pachytene and normal numbers of foci at mid and late pachytene. These indicated that the defect in Fance accelerates the formation of crossovers but has no impact on the completion of crossovers. Different expression patterns of MLH1 were observed in FA mouse models, including an increased number of MLH1 foci in FANCJ [42] and FANCP [43], reduced MLH1 foci in FNACO [34] and FANCR [44], and a lack of MLH1 foci in FANCS [32].
Conclusions
In summary, we have elucidated the important role and mechanism of FANCE in spermatogenesis. Our study demonstrated that Fance deficiency results in prospermatogonia loss at the embryonic stage and meiotic defects of spermatocytes at meiotic prophase I. We further detected the increased DSBs and the imbalance of the NHEJ and HR repair in Fance −/− prospermatogonia. During meiosis, FANCE deficiency caused reduced recruitment of RPA2, DMC1, and RAD51, and delayed disassociation of RPA2 and FANCD2 from chromosomes. Meanwhile, an accelerated formation of crossover was detected (Supplementary Material 4).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- FANCE:
-
Fanconi anemia complementation group E
- NOA:
-
Nonobstructive azoospermia
- FA:
-
Fanconi anemia
- Dpc:
-
Days post coitum
- P:
-
Postnatal
- ICLs:
-
Interstrand crosslinks
- SCOS:
-
Sertoli cell only syndrome
- PGCs:
-
Primordial germ cells
- SCCs:
-
Spermatogonial stem cells
- DSBs:
-
Double-strand breaks
- POI:
-
Premature ovarian insufficiency
- PCNA:
-
Proliferating cell nuclear antigen
- HR:
-
Homologous recombination
- NHEJ:
-
Non-homologous end joining
- ssDNA:
-
Single-stranded DNA
References
Mascarenhas MN, Flaxman SR, Boerma T, Vanderpoel S, Stevens GA. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9(12): e1001356.
Schlegel PN, Sigman M, Collura B, De Jonge CJ, Eisenberg ML, Lamb DJ, et al. Diagnosis and Treatment of Infertility in Men: AUA/ASRM Guideline PART II. J Urol. 2021Jan;205(1):44–51.
Gunes S, Al-Sadaan M, Agarwal A. Spermatogenesis, DNA damage and DNA repair mechanisms in male infertility. Reprod Biomed Online. 2015Sep;31(3):309–19.
Qin J, Huang T, Wang J, Xu L, Dang Q, Xu X, et al. RAD51 is essential for spermatogenesis and male fertility in mice. Cell Death Discov. 2022Mar 15;8(1):118.
Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013Jan 17;493(7432):356–63.
Semlow DR, Walter JC. Mechanisms of Vertebrate DNA Interstrand Cross-Link Repair. Annu Rev Biochem. 2021;20(90):107–35.
Tsui V, Crismani W. The Fanconi Anemia Pathway and Fertility. Trends Genet. 2019;35(3):199–214. https://doi.org/10.1016/j.tig.2018.12.007.
Krausz C, Riera-Escamilla A, Chianese C, Moreno-Mendoza D, Ars E, Rajmil O, Pujol R, et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med. 2019Jan;21(1):189–94.
Yin H, Ma H, Hussain S, Zhang H, Xie X, Jiang L, et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med. 2019Jan;21(1):62–70.
Yang Y, Guo J, Dai L, Zhu Y, Hu H, Tan L, et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet. 2018Sep;55(9):628–36.
Cheng NC, van de Vrugt HJ, van der Valk MA, Oostra AB, Krimpenfort P, de Vries Y, et al. Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum Mol Genet. 2000Jul 22;9(12):1805–11.
Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. 2003Aug 15;12(16):2063–76.
Nadler JJ, Braun RE. Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. Genesis. 2000Jul;27(3):117–23.
Sharan SK, Pyle A, Coppola V, Babus J, Swaminathan S, Benedict J, et al. BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development. 2004Jan;131(1):131–42.
Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, et al. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003Aug 15;17(16):2021–35.
Whitney MA, Royle G, Low MJ, Kelly MA, Axthelm MK, Reifsteck C, et al. Germ cell defects and hematopoietic hypersensitivity to gamma-interferon in mice with a targeted disruption of the Fanconi anemia C gene. Blood. 1996Jul 1;88(1):49–58.
Bakker ST, van de Vrugt HJ, Visser JA, Delzenne-Goette E, van der Wal A, Berns MA, et al. Fancf-deficient mice are prone to develop ovarian tumours. J Pathol. 2012Jan;226(1):28–39.
Koomen M, Cheng NC, van de Vrugt HJ, Godthelp BC, van der Valk MA, Oostra AB, et al. Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum Mol Genet. 2002Feb 1;11(3):273–81.
Agoulnik AI, Lu B, Zhu Q, Truong C, Ty MT, Arango N, et al. A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum Mol Genet. 2002Nov 15;11(24):3047–53.
Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, et al. Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet. 2009Sep 15;18(18):3484–95.
Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PHL, McIntyre RE, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011Feb;43(2):147–52.
Fu C, Begum K, Jordan PW, He Y, Overbeek PA. Dearth and Delayed Maturation of Testicular Germ Cells in Fanconi Anemia E Mutant Male Mice. PLoS ONE. 2016Aug 3;11(8): e0159800.
Fu C, Begum K, Overbeek PA. Primary Ovarian Insufficiency Induced by Fanconi Anemia E Mutation in a Mouse Model. PLoS ONE. 2016Mar 3;11(3): e0144285.
Suye S, Yin H, Zhou Z, Zheng C, Ren Z, Shi L, et al. Histological and transcriptomic analysis of Fance-deficient PGCs reveal the possible mechanisms of their depletion. Reproduction. 2023Jun 12;166(1):65–75.
Zhou Z, Yin H, Suye S, Ren Z, Yan L, Shi L, et al. Fance deficiency inhibits primordial germ cell proliferation associated with transcription-replication conflicts accumulate and DNA repair defects. J Ovarian Res. 2023Aug 10;16(1):160.
Yin H, Suye S, Zhou Z, Cai H, Fu C. The reduction of oocytes and disruption of the meiotic prophase I in Fanconi anemia E-deficient mice. Reproduction. 2022Jul 14;164(3):71–82.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016Jan;26(1):52–64.
Lei T, Du S, Peng Z, Chen L. Multifaceted regulation and functions of 53BP1 in NHEJ-mediated DSB repair (Review). Int J Mol Med. 2022Jul;50(1):90.
Zhao S, Huang C, Yang Y, Xu W, Yu Y, Wen C, et al. DNA repair protein FANCD2 has both ubiquitination-dependent and ubiquitination-independent functions during germ cell development. J Biol Chem. 2023Mar;299(3): 102905.
Gura MA, Bartholomew MA, Abt KM, Relovská S, Seymour KA, Freiman RN. Transcription and chromatin regulation by TAF4b during cellular quiescence of developing prospermatogonia. Front Cell Dev Biol. 2023Oct;12(11):1270408.
Du G, Oatley MJ, Law NC, Robbins C, Wu X, Oatley JM. Proper timing of a quiescence period in precursor prospermatogonia is required for stem cell pool establishment in the male germline. Development. 2021;148(9):dev194571.
Hartford SA, Chittela R, Ding X, Vyas A, Martin B, Burkett S, et al. Interaction with PALB2 Is Essential for Maintenance of Genomic Integrity by BRCA2. PLoS Genet. 2016Aug 4;12(8): e1006236.
Simhadri S, Peterson S, Patel DS, Huo Y, Cai H, Bowman-Colin C, et al. Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. J Biol Chem. 2014Aug 29;289(35):24617–29.
Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, Martin BK, et al. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J Cell Biol. 2007;176(5):581–92.
Alavattam KG, Kato Y, Sin HS, Maezawa S, Kowalski IJ, Zhang F, et al. Elucidation of the Fanconi Anemia Protein Network in Meiosis and Its Function in the Regulation of Histone Modifications. Cell Rep. 2016Oct 18;17(4):1141–57.
Xu L, Xu W, Li D, Yu X, Gao F, Qin Y, Yang Y, Zhao S. FANCI plays an essential role in spermatogenesis and regulates meiotic histone methylation. Cell Death Dis. 2021Aug 9;12(8):780.
Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010Jul 9;39(1):25–35.
Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010Jul 9;329(5988):219–23.
Kato Y, Alavattam KG, Sin HS, Meetei AR, Pang Q, Andreassen PR, et al. FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum Mol Genet. 2015Sep 15;24(18):5234–49.
Feeney L, Muñoz IM, Lachaud C, Toth R, Appleton PL, Schindler D, et al. RPA-Mediated Recruitment of the E3 Ligase RFWD3 Is Vital for Interstrand Crosslink Repair and Human Health. Mol Cell. 2017;66(5):610–621.e4.
Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol Cell. 2017;66(5):622–634.e8.
Sun X, Brieño-Enríquez MA, Cornelius A, Modzelewski AJ, Maley TT, Campbell-Peterson KM, et al. FancJ (Brip1) loss-of-function allele results in spermatogonial cell depletion during embryogenesis and altered processing of crossover sites during meiotic prophase I in mice. Chromosoma. 2016Jun;125(2):237–52.
Holloway JK, Mohan S, Balmus G, Sun X, Modzelewski A, Borst PL, et al. Mammalian BTBD12 (SLX4) protects against genomic instability during mammalian spermatogenesis. PLoS Genet. 2011Jun;7(6): e1002094.
Dai J, Voloshin O, Potapova S, Camerini-Otero RD. Meiotic Knockdown and Complementation Reveals Essential Role of RAD51 in Mouse Spermatogenesis. Cell Rep. 2017Feb 7;18(6):1383–94.
Acknowledgements
The author thanks the people who were involved in the study.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82271674), Hunan Provincial Innovation Foundation for Postgraduate [grant number QL20230061], and Postgraduate Innovative Project of Central South University [grant number 2022XQLH152].
Author information
Authors and Affiliations
Contributions
The work presented here was conducted in collaboration with all authors. Chun Fu and Huan Yin designed the research. Huan Yin performed the research. Huan Yin and Zhixian Zhou analyzed the data and prepared figures, and Huan Yin wrote the paper. All authors contributed to writing the paper, and all approved the final version for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Laboratory Animal Welfare and Ethical Committee of Central South University (IACUC Number: 2020sydw1041).
Consent for publication
All patients agreed to participate in the study and signed the informed consent form.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yin, H., Zhou, Z. & Fu, C. Fance deficiency impaired DNA damage repair of prospermatogonia and altered the repair dynamics of spermatocytes. Reprod Biol Endocrinol 22, 113 (2024). https://doi.org/10.1186/s12958-024-01284-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-024-01284-w