
Abstract
Background
Gastric cancer (GC) is a prevalent malignancy with limited therapeutic options for advanced stages. This study aimed to identify novel therapeutic targets for GC by profiling HSP90 client kinases.
Methods
We used mass spectrometry-based activity-based protein profiling (ABPP) with a desthiobiotin-ATP probe, combined with sensitivity analysis of HSP90 inhibitors, to profile kinases in a panel of GC cell lines. We identified kinases regulated by HSP90 in inhibitor-sensitive cells and investigated the impact of MASTL knockdown on GC cell behavior. Global proteomic analysis following MASTL knockdown was performed, and bioinformatics tools were used to analyze the resulting data.
Results
Four kinases—MASTL, STK11, CHEK1, and MET—were identified as HSP90-regulated in HSP90 inhibitor-sensitive cells. Among these, microtubule-associated serine/threonine kinase-like (MASTL) was upregulated in GC and associated with poor prognosis. MASTL knockdown decreased migration, invasion, and proliferation of GC cells. Global proteomic profiling following MASTL knockdown revealed NEDD4-1 as a potential downstream mediator of MASTL in GC progression. NEDD4-1 was also upregulated in GC and associated with poor prognosis. Similar to MASTL inhibition, NEDD4-1 knockdown suppressed migration, invasion, and proliferation of GC cells.
Conclusions
Our multi-proteomic analyses suggest that targeting MASTL could be a promising therapy for advanced gastric cancer, potentially through the reduction of tumor-promoting proteins including NEDD4-1. This study enhances our understanding of kinase signaling pathways in GC and provides new insights for potential treatment strategies.
Similar content being viewed by others


Background
Gastric cancer (GC) is a prevalent malignancy with significant incidence and mortality worldwide [1]. Its aggressive nature, late-stage diagnosis, and limited treatment options all contribute to high mortality rates [2]. Therefore, innovative therapeutic strategies are urgently needed to effectively target and combat this life-threatening disease. In recent years, cancer research has been increasingly focused on identifying novel kinase targets, as they play crucial roles in regulating various cellular processes, including cell growth, proliferation, and migration [3].
Recent advances in cancer genomics, including the discovery of genetic alterations, have greatly enhanced our understanding of carcinogenesis at the molecular level [4, 5]. The application of large-scale next-generation sequencing has not only deepened our understanding of the intricate processes underlying cancer progression, but has also resulted in the development of novel genomic molecular classification systems for GC [5]. This, in turn, allowed researchers to unravel the cancer-driving mechanisms and identify diagnostic biomarkers, gene signatures, and potential anticancer targets [6,7,8]. A comparison of gene expression changes in GC and normal tumor-adjacent tissues using transcriptome sequencing revealed genetic and biochemical markers associated with tumor progression [8]. However, because protein abundance and function are not determined solely by mRNA abundance [9], these approaches can only provide limited understanding of the complex processes underlying cancer progression.
The advent of proteomics has complemented the previous omics approaches [9]. Typically, liquid chromatography (LC) is used to separate proteins and address issues related to sample complexity and dynamic range, whereas mass spectrometry (MS) is used to identify, characterize, and quantify proteins in proteomic research. Therefore, liquid chromatography–mass spectrometry (LC–MS)-based high-throughput proteomics has substantially contributed to the identification of diagnostic biomarkers and anticancer targets [10]. However, it is difficult to identify protein groups with relatively low abundance, such as kinases, which are associated with various cellular processes [11].
Activity-based protein profiling (ABPP) has emerged as a chemical proteomics approach that uses active site-directed chemical probes designed to selectively target and label the active sites of specific enzyme classes, including kinases [12]. These probes contain reactive groups that covalently bind to the active sites of the enzymes, allowing their capture and subsequent analysis. This method can be used to monitor the enzymatic activities of proteins in complex biological systems and to better understand the mechanisms of compound–target interactions [13, 14]. Moreover, by using kinase-specific activity-based probes, researchers can selectively profile the kinome in cancer cells and identify aberrantly activated or dysregulated kinases that could serve as prospective therapeutic targets [15, 16].
Heat shock protein 90 (HSP90)—a molecular chaperone involved in protein folding and stabilization—has garnered significant attention as a therapeutic target because its inhibition can lead to the destabilization and degradation of target proteins, including several kinases critical for cancer progression [17]. Numerous HSP90-client proteins play crucial roles in establishing cancer cell hallmarks [18]. This suggests that the proteome-wide profiling of HSP90 cells could be an effective strategy for identifying novel anticancer targets. Interactome analysis based on the HSP90 inhibitor PU-H71 identified oncogenic proteins and pathways, such as PI3K–AKT–mTOR, JAK–STAT, and Raf–MAPK pathways, in chronic myeloid leukemia (CML) [19]. Giulino-Roth et al. [20] used PU-H71 affinity capture and proteomics to determine the relevant HSP90 clients in Burkitt’s lymphoma, revealing that the inhibition of HSP90 targets multiple components of the PI3K/AKT/mTOR signaling pathway. Sharma et al. [21] reported that HSP90 inhibition by 17-DMAG led to the downregulation of clients involved in RAS or AKT signaling pathways, DNA repair, and sphingolipid metabolism. These reports support the notion that profiling the HSP90 client kinome may be an effective strategy for identifying novel kinase anticancer targets.
In the present study, we aimed to identify novel kinase targets in GC by investigating the HSP90 client kinome. We performed LC–MS-based ABPP to comprehensively profile the kinome under HSP90 inhibition. Among the four potential kinase targets identified, we focused on microtubule-associated serine/threonine kinase-like (MASTL)—a kinase known for its involvement in mitotic control. Our findings demonstrate that MASTL plays a crucial role in not only regulating the proliferation but also the migration and invasion of GC cells. Furthermore, global proteomic profiling revealed that knocking down MASTL led to a reduction in tumor-promoting proteins, including NEDD4-1. Similarly, silencing NEDD4-1 inhibited the proliferation, migration, and invasion of GC cells. Collectively, these results suggest that MASTL could promote GC progression through mechanisms independent of mitotic progression and could be a novel anticancer target for GC treatment.
Methods
Cell culture
All GC cells used in this study were obtained from the Korean Cell Line Bank and cultured in RPMI 1640 medium containing 10% fetal bovine serum (FBS) and 1% antibiotic–antimycotic (HyClone, Logan, UT, USA) at 37 ℃ in a humid environment with 5% CO2.
Chemicals and antibodies
AUY922 was purchased from Selleckchem (Houston, TX, USA) and dissolved in dimethyl sulfoxide (DMSO). All primary antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA), except for β-actin (Santa Cruz Biotechnology, Dallas, TX, USA), MASTL (Abgent, San Diego, CA, USA), and NEDD4 (R&D Systems, Minneapolis, MN, USA). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Western blotting
For protein extraction, the collected pellets of GC cells were lysed in NETN lysis buffer (100 mM NaCl, 20 mM Tris [pH 8.0], 0.5 mM EDTA, and 0.5% NP-40) supplemented with a protease and phosphatase inhibitor cocktail (GenDepot, Baker, TX, USA). Protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The membrane was blocked with 5% skim milk in TBST for 1 h at room temperature and immunoblotted with primary antibodies at 4 ℃ overnight. After washing with TBST, the membranes were incubated with HRP-conjugated secondary antibodies at room temperature for 1 h. An LI-COR system (LI-COR Biosciences, Lincoln, NE, USA) was used to detect chemiluminescence. Protein band intensities were measured using the ImageJ software.
MTT assay
Cells were seeded in 96-well plates at a density of 20–30% and allowed to adhere for 24 h. On the following day, cells were exposed to the experimental drugs. After 72 h, 10 μl of MTT reagent (5 mg/ml) was applied to the cells. After 2 h, the medium was removed, and DMSO was added to dissolve the formazan crystals. The optical absorbance was measured at 570 nm using a microplate reader (VersaMax, Molecular Devices, San Jose, CA, USA).
siRNA transfection
To suppress the expression of MASTL and NEDD4, siRNA transfections were performed using RNAiMAX reagent (Thermo Fisher Scientific, Carlsabad, CA, USA), according to the manufacturer’s instructions. siRNAs were purchased from Genolution (Seoul, South Korea). The siRNA duplex sequences used in this study are siControl (sense: 5′-CCUCGUGCCGUUCCAUCAGGUAGUU-3′; antisense: 5′-CUACCUGAUGGAACGGCACGAGGUU-3′), siMASTL (sense: 5′-GAAAGUCAGCCCUUAGAUUUU-3′; antisense: 5′-AAUCUAAGGGCUGACUUUCUU-3′), siMASTL #2 (sense: 5′-GAAUGAACUUGCAUAAUUAUU-3′; antisense: 5′-UAAUUAUGCAAGUUCAUUCUU-3′), siNEDD4 (sense: 5′-GUGCAAAUCAUCAGGUUAUUU-3′; antisense: 5′-AUAACCUGAUGAUUUGCACUU-3′), and siNEDD4 #2 (sense: 5′- UGGCGAUUUGUAAACCGAAUU-3′; antisense: 5′-UUCGGUUUACAAAUCGCCAUU-3′).
Migration and invasion assay
The cell migration assay was conducted using a transwell system in a 24-well plate (Corning, New York, NY, USA). Briefly, GC cells suspended in serum-free medium were plated into the upper chamber (pore size, 8 μm) and culture medium containing 10% FBS was added in the lower compartment. After 24 h of incubation, cells were fixed with 4% paraformaldehyde (PFA) for 20 min and stained with 0.5% crystal violet for 20 min. Subsequently, non-migrating cells were removed using a cotton swab. The migrated cells in the lower chamber were counted from three randomly chosen fields under a microscope, using the ImageJ software. For the invasion assay, the transwell filter was coated with Matrigel and cells were incubated for 48 h.
Wound healing assay
To create wounds, inserts (Ibidi, Gräfelfing, BY, Germany) were placed in a 6-well plate, and cells were seeded in each well of the insert. On the following day, the inserts were removed to create wounds and the floating cells were eliminated. Wound closure images were captured using a phase-contrast microscope (Olympus, Shinjuku, Tokyo, Japan).
Clonogenic assay
Cells were plated in 6-well plates at 3 × 103–5 × 103 cells per well and incubated in culture medium for 7 days. Colonies formed were fixed with 4% PFA for 20 min and stained with 0.5% crystal violet for 20 min.
qPCR
Total RNA was extracted using the AccuPrep® Universal RNA Extraction Kit (Bioneer, Daejeon, South Korea), following the manufacturer’s instructions. cDNA synthesis was performed using the CellScript™ cDNA Master Mix (CellSafe, Yongin, South Korea). Quantitative PCR was performed using the SYBR Green Real-Time PCR Master Mix (Toyobo, Osaka, Japan). The reaction mixture, containing cDNA, SYBR Green, forward primer, and reverse primer, underwent 35 cycles of PCR amplification with the following cycling conditions: denaturation at 95 ℃ for 5 s, annealing at 55 ℃ for 10 s, and extension at 72 ℃ for 30 s. All primer sequences used in this study are listed in Table S1.
LC–MS-based ABPP
ABPP was conducted following our previously reported methods [14] and protocols outlined in the Pierce Kinase Enrichment kit and ActivX probes (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, the collected GC cell pellets were lysed using IP lysis buffer supplemented with protease and phosphatase inhibitors. After desalting using Zeba spin desalting columns (Thermo Fisher Scientific), the desthiobiotin-ATP probe reaction was performed. Probe-labeled lysates were denatured with 5 M urea, reduced with 5 mM dithiothreitol (DTT), and alkylated with 40 mM iodoacetamide (IAA). Following buffer exchange using the Zeba spin desalting columns, samples were digested with mass spectrometry (MS)-grade trypsin. The labeled tryptic peptides were enriched using a slurry of high-capacity streptavidin-agarose resin. Captured peptides were eluted with elution buffer (50% acetonitrile (ACN), 0.1% trifluoroacetic acid (TFA)), lyophilized, and resuspended in injection buffer (50% ACN, 0.1% TFA). The eluted samples were analyzed using LC–MS (Korea Basic Science Institute, Ochang, Republic of Korea) as described previously [14, 22]. Raw MS data were processed to identify and quantify the peptides and proteins using MaxQuant software (ver. 1.5.3.8; Max Planck Institute of Biochemistry, Martinsried, Germany). The search parameters were largely set to the default values provided by MaxQuant, which are optimized for high accuracy and reproducibility across a wide range of proteomic experiments. A notable exception to the default settings was the inclusion of a custom modification: desthiobiotin-ATP at lysine residues. This modification was added to accurately reflect the specific chemical labeling used in our activity-based protein profiling (ABPP) approach. The search included common variables such as methionine oxidation as the default variable modification. Searches were performed against the Human SwissProt database.
Global proteome analysis
In-gel digestion was performed as previously described [23]. Briefly, samples were loaded onto SDS-PAGE gels, stained with Coomassie Blue, and destained overnight. The gel was sliced into fragments and washed with a destaining buffer. The gel pieces were reduced with 2 mM tris(2-carboxyethyl)phosphine (TCEP) and alkylated with 20 mM IAA. Subsequently, the samples were digested with trypsin and the resulting tryptic peptides were eluted using an elution buffer (50% ACN, 0.1% TFA). The eluted samples were analyzed by LC–MS/MS, as previously described [24]. Raw MS data were processed to identify and quantify the peptides and proteins using MaxQuant software (ver. 1.5.3.8; Max Planck Institute of Biochemistry, Martinsried, Germany) as described above.
Statistical analysis
In ABPP analysis, each condition was analyzed using three biological replicates. Proteins undetected in two or more biological replicates under the control condition were considered as "undetected." Differentially expressed proteins (DEPs) were defined as those with an average fold-change greater than twofold. In global proteome profiling, each sample was analyzed using two biological replicates and two technical LC–MS analysis replicates. A previously described method for handling missing values in replicates was followed [24]. If one of the technical replicates was missing, we used the detected value from the remaining technical replicate. If both technical replicates were missing, we added a minimal value close to zero to all missing values. DEPs were defined as those with an average fold change greater than twofold and a p-value less than 0.05. Averages were log2-transformed and p-values were log10-transformed using Microsoft Excel (Microsoft, Inc., Redmond, WA, USA) to determine the significance of the results.
Results
Activity-based protein profiling revealed HSP90 client kinases in GC
Inhibition of HSP90 leads to the degradation of client proteins associated with the pathological traits of malignant cells via the proteasome pathway [25]. These traits are well-established hallmarks of cancer, including self-sufficiency of growth signals, insensitivity to growth suppression signals, evasion of apoptosis, acquisition of limitless replicative potential, sustained angiogenesis, invasion, and metastasis [18, 26,27,28,29]. Therefore, we hypothesized that client proteins that degrade as a result of HSP90 inhibition could act as potential anticancer target candidates for GC. In this study, we focused on the HSP90-regulated cancer kinome, owing to the importance of kinase signaling in GC progression [3]. First, we assessed the response of GC cells to the HSP90 inhibitor AUY922. AUY922 is a second-generation HSP90 inhibitor, specifically a synthetic radicicol-based derivative, which has been evaluated in multiple clinical trials, including those for gastric cancer [30]. It is a potent and selective inhibitor with a GI50 value in the nanomolar range, demonstrating greater antitumor efficacy compared to first-generation inhibitors such as 17-AAG [31]. Additionally, AUY922 has been shown to effectively inhibit HER2 and AKT activation in gastric cancer cells [32], supporting its relevance and suitability for our study. Our results indicated that AGS and SNU484 cells exhibited higher sensitivity to HSP90 inhibitors than other GC cells (Figure S1A and S1B). Based on these findings, we selected two AUY922-sensitive GC cell lines (AGS and SNU484) and two AUY922-resistant GC cell lines (SNU601 and MKN74) to identify the HSP90 client kinome using LC–MS-based ABPP. To enrich the kinome, we employed a commercially available desthiobiotin-ATP probe (ActivX, Thermo Scientific), which covalently labels conserved lysine residues in or near the ATP-binding pocket of enzymes, including kinases [33], and has been previously used to profile the kinome response to kinase inhibitor treatment in lung cancer [33, 34] and target profiling of dasatinib in gastric cancer [14]. The experimental approach is outlined in Fig. 1A. Briefly, cells were treated with the HSP90 inhibitor, AUY922, or vehicle control (DMSO). Subsequently, the ATP-binding proteome was labeled with a desthiobiotin-ATP probe and digested with trypsin. The labeled peptides were then enriched with streptavidin beads, followed by LC–MS analysis. Raw MS data were processed for the identification and quantification of peptides and proteins using MaxQuant software.

Identification of HSP90 client kinases through LC–MS-based activity-based protein profiling (ABPP). (A) Workflow for HSP90 client kinome profiling via desthiobiotin-ATP probe-based ABPP in GC. GC cells were incubated with vehicle control (DMSO) or 100 nM of AUY922 for 48 h and harvested (a), followed by desthiobiotin-ATP probe labeling (b). Samples were digested with trypsin (c), labeled peptides were captured by streptavidin agarose beads (d) and analyzed by LC–MS/MS (e). MaxQuant software was used to analyze LC–MS data (f). (B) HSP90 client kinases in AUY922-sensitive GC cells were illustrated in the kinome tree. (C) Common HSP90 client kinases across GC cell lines were illustrated in a Venn diagram
We identified a total of 117 peptides that corresponded to 83 kinases in AGS, 86 peptides that corresponded to 64 kinases in SNU484, 106 peptides that corresponded to 68 kinases in SNU601 and 108 peptides that corresponded to 67 kinases in MKN74. We defined kinases as HSP90-regulated if their intensities were reduced by more than 50% compared with the vehicle control. A list of the HSP90-regulated kinases in each cell line is provided in Table S2. We identified seven common HSP90 client kinases, MASTL, STK11, CHEK1, MET, PKM, and CDK1, in AUY922-sensitive GC cells (AGS and SNU484) and eight common HSP90 client kinases, PDPK1, MAP4K5, PKM, STK38, TAOK3, EIF2AK2, and DTYMK, in AUY922-resistant GC cells (SNU601 and MKN74; Fig. 1B and Figure S2). We hypothesized that the HSP90 client kinases exclusively identified in both sensitive cell types (AGS & SNU484; Fig. 1C)–MASTL, MET, CHEK1 and STK11 – could be important for GC progression. Previous studies have reported that MET, CHEK1, and STK11 are HSP90 client proteins [35,36,37,38,39].
Among these four kinases, we focused on microtubule-associated serine/threonine kinase-like (MASTL) due to its potential as a novel tumor promoting protein in GC. MASTL is reportedly a master regulator that promotes mitotic progression by inhibiting PP2A via ENSA [33, 34]. High expression of MASTL has been observed in a variety of human cancers and is associated with poor clinical outcomes in breast, gastric, and colon cancer [40,41,42] as well as recurrence after treatment in various cancer [42,43,44]. Notably, MASTL inhibition selectively killed breast cancer cells, but not normal cells, through the induction of mitotic catastrophe [44] and radiosensitization in NSCLC cells, but not in primary human fibroblasts [45]. Treatment with AUY922 significantly reduced MASTL expression specifically in AUY922-sensitive GC cells, validating MASTL as a potential novel HSP90 client kinase (Figure S3). Collectively, these findings indicate that MASTL is a promising candidate for anticancer treatment of GC.
Knockdown of MASTL impaired GC proliferation, migration, and invasion
Based on TCGA database, a high mutation frequency of MASTL was observed in GC across a wide range of human tumors (Figure S4). MASTL expression in tumor tissues was significantly higher than that in normal tissues and correlated with the GC tumor grade (Figs. 2A and B, and Figure S5). In addition, it has been reported that MASTL expression was associated with epithelial-to-mesenchymal transition (EMT) status and survival, serving as an independent prognostic factor for patients with GC [41]. Based on these results, MASTL can be considered a potential anticancer target for the treatment of GC.

MASTL is highly expressed in GC and knockdown of MASTL inhibited proliferation, migration and invasion of GC cells. A Differential MASTL mRNA expression was analyzed in gastric tumors (T) and normal tissues (N) using the GEPIA. B Expression analysis of MASTL in different stages of GC using UALCAN database. Statistical analyses are shown in Table S3. C GC cells were transfected with siControl or siMASTL, and the effect of MASTL knockdown on proliferation of GC cells was analyzed by clonogenic assay. D GC cells were transfected with siControl or siMASTL #2, and the effect of MASTL knockdown on GC migration was analyzed by wound healing assay. E AGS cells were transfected with siControl and siMASTL #2, and the effect of MASTL knockdown on AGS migration was analyzed by Transwell assay. F SNU484 cells were transfected with siControl and siMASTL #1, and the effect of MASTL knockdown on SNU484 invasion was analyzed by invasion assay. Representative images of migrated or invaded cells on the membrane (magnification: 200x) are shown. G Effect of MASTL knockdown on MMP-1 expression was analyzed by qPCR. Error bars indicate the standard deviation of representative triplicates from at least three experiments, which showed similar results. NS: not significant, *: p < 0.05, **: p < 0.01, ***: p < 0.001
To evaluate the potential of targeting MASTL as a novel anticancer approach, we silenced MASTL expression (Figure S6) and examined its effects on GC cell proliferation. The clonogenic assay showed that MASTL silencing inhibited the proliferation of GC cells (Fig. 2C). To further explore the role of MASTL in GC metastasis, we tested whether MASTL inhibition impaired GC migration/invasion and found that MASTL-deficient GC cell migration was reduced (Figs. 2D and E). Similarly, MASTL knockdown significantly decreased GC cell invasion (Fig. 2F). Given the observed effect of MASTL inhibition on GC migration and invasion, we hypothesized that MASTL regulates MMPs associated with tumor growth, differentiation, migration, and invasion at various cancer stages [46,47,48]. To examine this, we tested whether MASTL inhibition could impair MMP expression and found that the MASTL knockdown reduced MMP-1 mRNA expression in GC (Fig. 2G).
Global proteomics analysis identified MASTL-regulated proteome in GC
The molecular mechanism of MASTL as an oncogene remains to be fully elucidated. Overexpression of MASTL reportedly induces the dephosphorylation of glycogen synthase kinase-3 (GSK-3) at its inhibitory sites (S9/S21) and promotes hyperphosphorylation of AKT (S473) by degrading PHLPP, a phosphatase responsible for AKT dephosphorylation [49]. Additionally, the previous study demonstrated that inhibiting MASTL significantly impairs the phosphorylation of GSK-3β at its inhibitory site (S9) and the nuclear accumulation and activity of β-catenin and expression of Bcl-xL and Survivin, anti-apoptotic proteins [42]. However, we found that inhibition of MASTL failed to affect GSK-3β phosphorylation in GC cells (Figure S7), suggesting that MASTL could promote GC via unknown mechanisms.
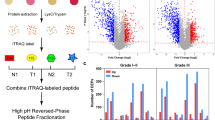
To elucidate the tumor promoting mechanisms of MASTL in GC, we profiled global proteome alteration occurring in MASTL-deficient SNU484 cells. The workflow for identifying the MASTL-regulated proteome is shown in Fig. 3A. Briefly, SNU484 cells were transfected with siControl or two different siRNAs targeting MASTL (Figure S8). The proteins were then in-gel digested, and the extracted peptides were analyzed using LC–MS/MS, followed by data processing with MaxQuant software. We identified a total of 4181 proteins (Table S4). Differentially expressed proteins (DEPs) were defined as those with an average fold change greater than twofold and a p-value less than 0.05, as illustrated in volcano plots and the Venn diagram (Figs. 3B and C). We identified 136 downregulated proteins with siMASTL #1 and 122 with siMASTL #2. Similarly, 83 upregulated proteins were identified with siMASTL #1 and 110 with siMASTL #2. Ultimately, 33 proteins were found to be downregulated and 18 were found to be upregulated by both siRNAs (Fig. 3C), which were used for subsequent bioinformatic analyses.

Analysis of global proteome in MASTL-deficient GC cells revealed MAST-regulated protein candidates. (A) Workflow for MASTL-regulated proteome profiling via global proteome analysis in GC. SNU484 cells were transfected with two different MASTL targeting siRNAs or control siRNA for 48 h (a), followed by in-gel digestion (b). The resulting tryptic peptides were analyzed by LC–MS/MS (c). MaxQuant software was used to analyze LC–MS data (d). (B) Differentially expressed proteins (DEPs) in MASTL-deficient GC cells were visualized by volcano plot. Non-axial vertical and horizontal lines denote twofold change and p = 0.05, respectively. (C) Commonly decreased and increased DEPs were illustrated using Venn diagrams. (D) Metascape analysis of down-regulated proteins. (E) Metascape analysis of up-regulated proteins
To gain biological insights into MASTL-regulated proteome, we performed comprehensive pathway enrichment analyses using the Metascape database, which integrates GO function and KEGG pathway analyses [50] (Figs. 3D and E). The downregulated proteins were mainly enriched in the mitotic cell cycle, meiotic cell cycle, regulation of nucleocytoplasmic transport, response to salt, and establishment of organelle localization. Upregulated proteins were mainly enriched in amino acid catabolic processes and autophagy. Additionally, KEGG pathway enrichment analyses revealed that the downregulated proteins were primarily enriched in the cell cycle, mitotic, PIP3 activates AKT signaling, and deubiquitination pathways. Conversely, upregulated proteins were mainly enriched in cargo recognition for clathrin-mediated endocytosis. Given prior research linking MASTL to the AKT signaling pathway in different cancers [49, 51], we focused on proteins related to “PIP3 activates AKT signaling” in the KEGG pathway annotations. Among the down-regulated proteins, NEDD4-1, TNRC6B, RAP2C, USP34, STRN, and PPP2R1B were enriched in this pathway (Table S5). In particular, we focused on NEDD4-1 because of its association with cancer progression. NEDD4-1, E3 ubiquitin ligases, preferentially induces ubiquitination of substrates through K63 conjugation involved in DNA repair, protein trafficking, autophagy, immunity, and inflammation compared with K48 conjugation [52, 53]. NEDD4-1 regulates multiple substrates involved in cancer cell proliferation, migration, invasion, and drug sensitivity [52]. Notably, it was reported that NEDD4-1 overexpression in p53−/− primary MEFs reduced PTEN expression, antagonizing the activation of the PI3K-AKT-mTOR signaling pathway [54]. Likewise, inhibition of NEDD4-1 induces dephosphorylation of AKT, increases the expression of PTEN, and reduces the growth and migration of hepatocellular carcinoma cells [55]. Importantly, NEDD4-1 has been reported to be overexpressed in gastric cardia adenocarcinoma (GCA) compared to adjacent normal tissues and highlighted as a significant prognostic biomarker for gastric cardia adenocarcinoma [56]. This indicates that NEDD4-1 may be important for GC progression as a novel downstream signaling molecule of MASTL.
Knockdown of NEDD4 impaired the viability, migration and invasion of GC cells
Using bioinformatic databases, we assessed the increased expression correlated with the survival rate of patients with GC (Fig. 4A).We validated the mass spectrometry results using Western blotting and RT-qPCR, which indicated that MASTL knockdown significantly reduced NEDD4-1 expression in GC cells at both the protein and mRNA levels (Figs. 4B and S9). Inhibition of MASTL failed to impair AKT phosphorylation (Fig. 4B), suggesting that NEDD4-1 could promote GC progression beyond its potential role in regulating PTEN-AKT pathway. To confirm that the reduction of NEDD4-1, as a result of MASTL knockdown, inhibits GC phenotypes, we silenced NEDD4-1 (Figure S10) and tested its impact on GC cell proliferation, migration, and invasion. Similar to MASTL inhibition, silencing NEDD4-1 impaired the viability of GC cells (Fig. 4C). Furthermore, NEDD4-1 knockdown significantly reduced the migration and invasion of GC cells (Figs. 4D–F). These results suggest that NEDD4-1 could be one of the major downstream effectors of MASTL contributing to GC progression.

NEDD4-1 is upregulated in GC and associated with poor prognosis of patients with GC; inhibition of NEDD4-1 reduced proliferation, migration and invasion of GC cells. A Kaplan–Meier survival curves were generated to compare overall survival (OS) of stomach adenocarcinoma (STAD) patients with high and low NEDD4-1 expression using Kaplan–Meier Plotter (https://kmplot.com/analysis/). NEDD4-1 expression was categorized based on microarray data with an expression range of 32–1084, using a cutoff value of 327. Patients with NEDD4-1 expression ranging from 32 to 327 were classified as the low expression group, while those with expression from 327 to 1084 were classified as the high expression group. B GC cells were transfected with control or siRNA targeting MASTL for 48 h and expressions of MASTL, NEDD4-1, p-AKT (Ser 473) were analyzed by western blotting. C GC cells were transfected with siControl and siNEDD4-1 and the effect of NEDD4-1 knockdown on proliferation in GC cells was analyzed by clonogenic assay. D GC cells were transfected with siControl and siNEDD4-1 #1 and the effect of NEDD4-1 knockdown on GC migration was analyzed by wound healing assay. E AGS cells were transfected with siControl and siNEDD4-1 #1 and the effect of NEDD4-1 knockdown on AGS migration was analyzed by transwell migration assay. F SNU484 cells were transfected with siControl and siNEDD4-1 #1, and transwell invasion assay was performed. Representative images of migrated or invaded cells on the membrane (magnification: 200x) are shown. Error bars indicate the standard deviation of representative triplicates from at least three experiments, which showed similar results. NS: not significant, *: p < 0.05, **: p < 0.01, ***: p < 0.001
Discussion
Large-scale molecular profiling and classification of GC based on actionable genetic alterations have facilitated the development of personalized treatments for advanced GC [57]. For example, trastuzumab combined with chemotherapy is recommended as the first-line treatment for HER-2 overexpressing GC patients, in accordance with the approved standard of care in the USA [57, 58]. Additionally, ramucirumab, a monoclonal antibody against the vascular endothelial growth factor receptor 2, and pembrolizumab, a programmed death-1 (PD-1) inhibitor, are available as second- or third-line therapies. TAS-102, also known as tipiracil hydrochloride, is another available option for third-line therapy for GC. However, strategies for treating patients with HER2-negative GC tumors are relatively limited, emphasizing the urgent need for novel anticancer targets to improve the outcomes of patients with advanced GC.
The molecular chaperone HSP90 plays a crucial role in regulating the activity and stability of client proteins [59]. By regulating these proteins, HSP90 is involved in the pathological hallmarks of malignant cells, including self-sufficiency of growth signals, insensitivity to growth suppression signals, evasion of apoptosis, acquisition of limitless replicative potential, sustained angiogenesis, invasion and metastasis [18, 26,27,28,29]. Notably, HSP90 is more abundantly expressed in gastric cancer tissues than in normal mucosa [60] and is linked to tumor size, metastasis, and survival of patients with GC [61]. It has been reported that inhibition of HSP90 impairs GC proliferation and degrades target proteins including HER-2, Akt, and thymidylate synthase [62]. However, owing to concerns about toxicity [25], HSP90 has not been considered a target candidate for anticancer treatment. This concern has led to profiling of HSP90 client proteins to identify novel anticancer targets.
In this study, a desthiobiotin-ATP probe was used to profile HSP90 client kinases expressed at low levels in the samples. Unfortunately, we could not completely confirm whether all kinases in the sample were labeled with the desthiobiotin-ATP probe or whether loss occurred during purification. However, we identified 83 kinases in AGS, 64 kinases in SNU484, 68 kinases in SNU601, and 67 kinases in MKN74. Additionally, we identified seven common HSP90 client kinases in AUY922-sensitive GC cells and eight common HSP90 client kinases in AUY922-resistant GC cells. Recently, fluorophosphonate probes for the serine hydrolase class were synthesized and reported to selectively label active enzymes, but not their inactive precursor (zymogen) or inhibitor-bound forms [63,64,65]. However, desthiobiotin-ATP probes label both active and inactive forms of kinases [34, 66]. Considering the traits of these probes, AUY922 may have potential effects on both the active and inactive forms of HSP90 client kinases.
Using the ABPP approach, we identified four candidate proteins: MASTL, MET, CHEK1, and STK11 (Fig. 1C). Consistent with our results, Consistent with our results, MET and CHEK1 have been previously reported as HSP90 client proteins [35, 67, 68]. Activation of Met by Hepatocyte Growth Factor (HGF) can confer resistance to lapatinib in HER2-positive gastric cancer cells, there by compromising its therapeutic efficacy [69]. Knockdown of c-Met reduced peritoneal dissemination as well as tumor size in a gastric cancer xenograft model [70]. CHEK1, a critical regulator of cell cycle transition in DNA damage response, plays a significant role in promoting survival and growth of gastric cancer cells. CHK1 inhibitor, LY2606368, reduced tumor volume and weight in a gastric cancer PDX model. Particularly, the combination of LY2606368 with BMN673, a PARP inhibitor, demonstrated a synergistic anticancer effect [71]. Notably, MASTL expression correlates with epithelial-mesenchymal transition (EMT) status, tumor recurrence, and poor survival outcomes in GC [41]. These studies indicate a relationship between these kinases and the clinicopathology of GC. Unlike other candidates, STK11 is a tumor suppressor and considered an HSP90 client protein [39]. Previous studies indicated that STK11 degradation due to HSP90 inhibition can lead to tumor development [39]. However, our experiments demonstrated that HSP90 inhibitors significantly impaired GC proliferation (Figures S1A and S1B). These findings suggest that, among other client proteins, there may be potential anticancer targets in GC with mutations or loss of STK11.
In this study, we emphasize the importance of MASTL as a potential anticancer target for GC treatment. We demonstrated that MASTL inhibition impaired GC proliferation, migration, and invasion. Although the effect of MASTL knockdown on GC proliferation can be explained by its role as a key regulator of cell cycle progression, its impact on GC migration and invasion is less intuitive. Global proteomic analysis revealed NEDD4-1 as one of the downstream of MASTL contributing these phenotypes (Figs. 4C–F). Previous studies have reported that NEDD4-1 regulates multiple substrates via ubiquitination [52, 53]. PTEN, a negative regulator of the PI3K-Akt pathway, is a well-known substrate of NEDD4-1 [54, 55]. However, NEDD4-1 and PTEN appeared to be uncorrelated in GC tumor samples from GEPIA (Figure S11). In addition, inhibition of MASTL failed to reduce AKT phosphorylation (Fig. 4B), indicating that NEDD4-1 may not be associated with the stability of PTEN in GC. Accordingly, we hypothesized that previous studies on NEDD4-1 substrates would aid in understanding the role of the MASTL in GC progression. Wang et al. reported that recombinant NEDD4-1 contributes to the stability of MDM2, a negative regulator of p53, by promoting K63-type polyubiquitination. [72, 73]. They demonstrated that the inhibition of NEDD4-1 enhances p53 expression and activity, which leads to enhanced responses to DNA damage and growth inhibition [72]. Considering the results of this study, in which the inhibition of MASTL and NEDD4-1 reduced cell proliferation, MDM2 may be associated with the role of MASTL and NEDD4-1 in GC progression. MMP14 has been reported to have strong ECM-degrading capabilities and is highly expressed in GC tissues compared to the non-cancerous mucosa [74]. Notably, it has been reported that MMP14, which is involved in pericellular proteolysis and invasion, is monoubiquitinated at K581 by NEDD4-1 [75]. Similarly, we showed that the inhibition of MASTL and NEDD4 reduced GC cell invasion. In addition, high levels of MMP14 in tissues and serum are associated with worse survival in patients [74, 76]. Collectively, MASTL may promote GC invasion through NEDD4-1-mediated mono-ubiquitination of MMP14.
A global proteomics approach successfully profiled proteins altered by MASTL knockdown. While our experimental validation was focused on NEDD4-1, we could identify potential oncogenic proteins whose expression was reduced by knocking down MASTL. Among them, Anillin (ANLN), an actin-binding protein, possesses a multidomain structure and interacts with cytoskeletal components [77, 78]. Anillin, known as a regulator of cytokinesis, is reported as a marker of poor prognosis and is involved in cancer progression and metastasis in multiple types of cancer [79,80,81,82,83]. Recent study has reported that Anillin is highly expressed and associated with progression in GC [84]. Notably, the Cancer Genome Atlas (TCGA) database showed a high correlation (r = 0.69, P = 9.1e-59) between the expression of ANLN and MASTL in GC tissues (Figure S12). A comprehensive pan-cancer analysis revealed that high ANLN expression is associated with poor survival outcomes in more than ten cancer types; however, gastric cancer was not specifically highlighted as one of those types [85]. ANLN depletion has been reported to inhibit gastric cancer cell proliferation, migration, and invasion in vitro, and to contribute to tumor growth of gastric cancer cells in vivo [86]. Conversely, another study indicated that high ANLN expression is associated with better prognosis in gastric cancer, suggesting that this result may be related to the unique pathological features of GC [87]. This accumulated evidence suggests that, along with NEDD4-1, Anillin could be one of the major downstream effectors of MASTL contributing to GC progression. However, our proteomics approach has limitations in identifying the direct substrates of MASTL. To address this concern, additional phosphoproteomic analysis is required to identify MASTL substrates. We speculate that combining these approaches will provide a comprehensive understanding of role of MASTL in GC progression.
Conclusions
LC–MS-based ABPP revealed four HSP90 client kinases—MASTL, STK11, CHEK1 and MET—as potential targets for GC treatment. Among these proteins, we focused on MASTL, whose inhibition impairs GC proliferation, migration, and invasion. Global proteome profiling revealed NEDD4-1 as a potential downstream mediator of MASTL involved in GC cell proliferation, migration, and invasion. The results of this study deepen our understanding of GC and contribute to its treatment approaches by providing insights on novel therapeutic targets.
Availability of data and materials
Raw proteomics data obtained from ABPP analysis have been deposited on MassIVE database with the accession number MSV000093754 (https://doi.org/10.25345/C52805900). FTP for reviewers: ftp://MSV000093754@massive.ucsd.edu, Password: “cancer450$”. Raw proteomics data obtained from global proteome profiling have been deposited on MassIVE database with the accession number MSV000093757 (https://doi.org/10.25345/C5NZ8115C). FTP for reviewers: ftp://MSV000093757@massive.ucsd.edu, Password: “cancer450$”.
Abbreviations
- ABPP:
-
Activity-based protein profiling
- CAN:
-
Acetonitrile
- CML:
-
Chronic myeloid leukemia
- DEPs:
-
Differentially expressed proteins
- DMSO:
-
Dimethyl sulfoxide
- DTT:
-
Dithiothreitol
- EMT:
-
Epithelial-to-mesenchymal transition
- FBS:
-
Fetal bovine serum
- GC:
-
Gastric cancer
- GSK-3:
-
Glycogen synthase kinase-3
- HRP:
-
Horseradish peroxidase
- HSP90:
-
Heat shock protein 90
- IAA:
-
Iodoacetamide
- MASTL:
-
Microtubule-associated serine/threonine kinase-like
- PFA:
-
Paraformaldehyde
- SDS-PAGE:
-
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TCEP:
-
Tris(2-carboxyethyl)phosphine
- TFA:
-
Trifluoroacetic acid
References
Rawla P, Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz Gastroenterol. 2019;14(1):26–38.
Machlowska J, Baj J, Sitarz M, Maciejewski R, Sitarz R. Gastric cancer: epidemiology, risk factors, classification, genomic characteristics and treatment strategies. Int J Mol Sci. 2020;21(11):4012.
Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP, et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer. 2018;17(1):48.
Lin W-C, Kao H-W, Robinson D, Kung H-J, Wu C-W, Chen H-C. Tyrosine kinases and gastric cancer. Oncogene. 2000;19(49):5680–9.
Katona BW, Rustgi AK. Gastric cancer genomics: advances and future directions. Cell Mol Gastroenterol Hepatol. 2017;3(2):211–7.
Tsimberidou AM, Fountzilas E, Bleris L, Kurzrock R. Transcriptomics and solid tumors: The next frontier in precision cancer medicine. Semin Cancer Biol. 2022;84:50–9.
Supplitt S, Karpinski P, Sasiadek M, Laczmanska I. Current achievements and applications of transcriptomics in personalized cancer medicine. Int J Mol Sci. 2021;22(3):1422.
Zhang W, Liu S, Zhan H, Yan Z, Zhang G. Transcriptome sequencing identifies key pathways and genes involved in gastric adenocarcinoma. Mol Med Rep. 2018;18(4):3673–82.
Zhang Z, Wu S, Stenoien DL, Pasa-Tolic L. High-throughput proteomics. Annu Rev Anal Chem (Palo Alto Calif). 2014;7:427–54.
Kwon YW, Jo HS, Bae S, Seo Y, Song P, Song M, et al. Application of proteomics in cancer: recent trends and approaches for biomarkers discovery. Front Med (Lausanne). 2021;8:747333.
Wissing J, Jansch L, Nimtz M, Dieterich G, Hornberger R, Keri G, et al. Proteomics analysis of protein kinases by target class-selective prefractionation and tandem mass spectrometry. Mol Cell Proteomics. 2007;6(3):537–47.
Barglow KT, Cravatt BF. Activity-based protein profiling for the functional annotation of enzymes. Nat Methods. 2007;4(10):822–7.
Wang S, Tian Y, Wang M, Wang M, Sun GB, Sun XB. Advanced activity-based protein profiling application strategies for drug development. Front Pharmacol. 2018;9:353.
Choi KM, Cho E, Bang G, Lee SJ, Kim B, Kim JH, et al. Activity-based protein profiling reveals potential dasatinib targets in gastric cancer. Int J Mol Sci. 2020;21(23):9276.
Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, et al. Functional interrogation of the kinome using nucleotide acyl phosphates. Biochemistry. 2007;46(2):350–8.
Kurimchak AM, Kumar V, Herrera-Montavez C, Johnson KJ, Srivastava N, Davarajan K, et al. Kinome profiling of primary endometrial tumors using multiplexed inhibitor beads and mass spectrometry identifies SRPK1 as candidate therapeutic target. Mol Cell Proteomics. 2020;19(12):2068–90.
Weidenauer L, Wang T, Joshi S, Chiosis G, Quadroni MR. Proteomic interrogation of HSP90 and insights for medical research. Expert Rev Proteomics. 2017;14(12):1105–17.
Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347–65.
Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–26.
Giulino-Roth L, van Besien HJ, Dalton T, Totonchy JE, Rodina A, Taldone T, et al. Inhibition of Hsp90 suppresses PI3K/AKT/mTOR signaling and has antitumor activity in burkitt lymphoma. Mol Cancer Ther. 2017;16(9):1779–90.
Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, et al. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cellular Proteom. 2012;11(3):M111.014654.
Lee S-Y, Lee H, Yun SH, Park EC, Seo G, Kim H-Y, et al. Proteomics-based diagnostic peptide discovery for severe fever with thrombocytopenia syndrome virus in patients. Clin Proteomics. 2022;19(1):28.
Lee SJ, Choi KM, Bang G, Park SG, Kim EB, Choi JW, et al. Identification of nucleolin as a novel AEG-1-interacting protein in breast cancer via interactome profiling. Cancers (Basel). 2021;13(11):2842.
Park SG, Ji MJ, Ham IH, Shin YH, Lee SM, Lee CH, et al. Secretome analysis reveals reduced expression of COL4A2 in hypoxic cancer-associated fibroblasts with a tumor-promoting function in gastric cancer. J Cancer Res Clin Oncol. 2023;149(8):4477–87.
Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–16.
Workman P. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone. Cancer Lett. 2004;206(2):149–57.
Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berl). 2004;82(8):488–99.
Chiosis G, Vilenchik M, Kim J, Solit D. Hsp90: the vulnerable chaperone. Drug Discov Today. 2004;9(20):881–8.
Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–10.
Magyar CTJ, Vashist YK, Stroka D, Kim-Fuchs C, Berger MD, Banz VM. Heat shock protein 90 (HSP90) inhibitors in gastrointestinal cancer: where do we currently stand?-A systematic review. J Cancer Res Clin Oncol. 2023;149(10):8039–50.
Jensen MR, Schoepfer J, Radimerski T, Massey A, Guy CT, Brueggen J, et al. NVP-AUY922: a small molecule HSP90 inhibitor with potent antitumor activity in preclinical breast cancer models. Breast Cancer Res. 2008;10(2):R33.
Park KS, Hong YS, Choi J, Yoon S, Kang J, Kim D, et al. HSP90 inhibitor, AUY922, debilitates intrinsic and acquired lapatinib-resistant HER2-positive gastric cancer cells. BMB Rep. 2018;51(12):660–5.
Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18(6):699–710.
Kim JY, Stewart PA, Borne AL, Fang B, Welsh EA, Chen YA, et al. Activity-based proteomics reveals heterogeneous kinome and ATP-binding proteome responses to MEK inhibition in KRAS mutant lung cancer. Proteomes. 2016;4(2):16.
Miyajima N, Tsutsumi S, Sourbier C, Beebe K, Mollapour M, Rivas C, et al. The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and-resistant MET-driven tumor models. Can Res. 2013;73(23):7022–33.
Rice MA, Kumar V, Tailor D, Garcia-Marques FJ, Hsu E-C, Liu S, et al. SU086, an inhibitor of HSP90, impairs glycolysis and represents a treatment strategy for advanced prostate cancer. Cell Rep Med. 2022;3(2):100502.
Arlander SJ, Felts SJ, Wagner JM, Stensgard B, Toft DO, Karnitz LM. Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J Biol Chem. 2006;281(5):2989–98.
Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003;278(52):52572–7.
Boudeau J, Deak M, Lawlor MA, Morrice NA, Alessi DR. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem J. 2003;370(3):849–57.
Rogers S, McCloy RA, Parker BL, Gallego-Ortega D, Law AMK, Chin VT, et al. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene. 2018;37(33):4518–33.
Sun XJ, Li YL, Wang LG, Liu LQ, Ma H, Hou WH, et al. Mastl overexpression is associated with epithelial to mesenchymal transition and predicts a poor clinical outcome in gastric cancer. Oncol Lett. 2017;14(6):7283–7.
Uppada SB, Gowrikumar S, Ahmad R, Kumar B, Szeglin B, Chen X, et al. MASTL induces colon cancer progression and chemoresistance by promoting Wnt/beta-catenin signaling. Mol Cancer. 2018;17(1):111.
Wang L, Luong VQ, Giannini PJ, Peng A. Mastl kinase, a promising therapeutic target, promotes cancer recurrence. Oncotarget. 2014;5(22):11479.
Yoon YN, Choe MH, Jung KY, Hwang SG, Oh JS, Kim JS. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer. 2018;18(1):716.
Nagel R, Stigter-van Walsum M, Buijze M, van den Berg J, van der Meulen IH, Hodzic J, et al. Genome-wide siRNA screen identifies the radiosensitizing effect of downregulation of MASTL and FOXM1 in NSCLC. Mol Cancer Ther. 2015;14(6):1434–44.
Yadav L, Puri N, Rastogi V, Satpute P, Ahmad R, Kaur G. Matrix metalloproteinases and cancer - roles in threat and therapy. Asian Pac J Cancer Prev. 2014;15(3):1085–91.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74.
Zhang W, Huang X, Huang R, Zhu H, Ye P, Lin X, et al. MMP1 overexpression promotes cancer progression and associates with poor outcome in head and neck carcinoma. Comput Math Methods Med. 2022;2022:3058342.
Vera J, Lartigue L, Vigneron S, Gadea G, Gire V, Del Rio M, et al. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Elife. 2015;4:e10115.
Alzeeb G, Arzur D, Trichet V, Talagas M, Corcos L, Le Jossic-Corcos C. Gastric cancer cell death analyzed by live cell imaging of spheroids. Sci Rep. 2022;12(1):1488.
Reshi I, Nisa MU, Farooq U, Gillani SQ, Bhat SA, Sarwar Z, et al. AKT regulates mitotic progression of mammalian cells by phosphorylating MASTL, leading to protein phosphatase 2A inactivation. Mol Cell Biol. 2020;40(10):e00366–18.
Huang X, Chen J, Cao W, Yang L, Chen Q, He J, et al. The many substrates and functions of NEDD4-1. Cell Death Dis. 2019;10(12):904.
Boase NA, Kumar S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene. 2015;557(2):113–22.
Wang X, Trotman LC, Koppie T, Alimonti A, Chen Z, Gao Z, et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell. 2007;128(1):129–39.
Huang ZJ, Zhu JJ, Yang XY, Biskup E. NEDD4 promotes cell growth and migration via PTEN/PI3K/AKT signaling in hepatocellular carcinoma. Oncol Lett. 2017;14(3):2649–56.
Sun A, Yu G, Dou X, Yan X, Yang W, Lin Q. Nedd4-1 is an exceptional prognostic biomarker for gastric cardia adenocarcinoma and functionally associated with metastasis. Mol Cancer. 2014;13:1–10.
Selim JH, Shaheen S, Sheu WC, Hsueh CT. Targeted and novel therapy in advanced gastric cancer. Exp Hematol Oncol. 2019;8:25.
Orditura M, Galizia G, Sforza V, Gambardella V, Fabozzi A, Laterza MM, et al. Treatment of gastric cancer. World J Gastroenterol. 2014;20(7):1635–49.
Kamal A, Boehm MF, Burrows FJ. Therapeutic and diagnostic implications of Hsp90 activation. Trends Mol Med. 2004;10(6):283–90.
Zuo D-S, Dai J, Bo A-H, Fan J, Xiao X-Y. Significance of expression of heat shock protein90α in human gastric cancer. World J Gastroenterol. 2003;9(11):2616.
Giaginis C, Daskalopoulou SS, Vgenopoulou S, Sfiniadakis I, Kouraklis G, Theocharis SE. Heat Shock Protein-27, -60 and -90 expression in gastric cancer: association with clinicopathological variables and patient survival. BMC Gastroenterol. 2009;9:14.
Lee KH, Lee JH, Han SW, Im SA, Kim TY, Oh DY, et al. Antitumor activity of NVP-AUY922, a novel heat shock protein 90 inhibitor, in human gastric cancer cells is mediated through proteasomal degradation of client proteins. Cancer Sci. 2011;102(7):1388–95.
Cravatt BF, Wright AT, Kozarich JW. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414.
Kidd D, Liu Y, Cravatt BF. Profiling serine hydrolase activities in complex proteomes. Biochemistry. 2001;40(13):4005–15.
Jessani N, Liu Y, Humphrey M, Cravatt BF. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci. 2002;99(16):10335–40.
McAllister FE, Niepel M, Haas W, Huttlin E, Sorger PK, Gygi SP. Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Anal Chem. 2013;85(9):4666–74.
Arlander SJ, Felts SJ, Wagner JM, Stensgard B, Toft DO, Karnitz LM. Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J Biol Chem. 2006;281(5):2989–98.
Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J Biol Chem. 2003;278(52):52572–7.
Chen CT, Kim H, Liska D, Gao S, Christensen JG, Weiser MR. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11(3):660–9.
Wang XL, Chen XM, Fang JP, Yang CQ. Lentivirus-mediated RNA silencing of c-Met markedly suppresses peritoneal dissemination of gastric cancer in vitro and in vivo. Acta Pharmacol Sin. 2012;33(4):513–22.
Yin Y, Shen Q, Zhang P, Tao R, Chang W, Li R, et al. Chk1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer. Am J Cancer Res. 2017;7(3):473.
Xu C, Fan CD, Wang X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene. 2015;34(3):281–9.
Zhao Y, Yu H, Hu W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin (Shanghai). 2014;46(3):180–9.
Kasurinen A, Gramolelli S, Hagstrom J, Laitinen A, Kokkola A, Miki Y, et al. High tissue MMP14 expression predicts worse survival in gastric cancer, particularly with a low PROX1. Cancer Med. 2019;8(16):6995–7005.
Eisenach PA, de Sampaio PC, Murphy G, Roghi C. Membrane type 1 matrix metalloproteinase (MT1-MMP) ubiquitination at Lys581 increases cellular invasion through type I collagen. J Biol Chem. 2012;287(14):11533–45.
Kasurinen A, Tervahartiala T, Laitinen A, Kokkola A, Sorsa T, Bockelman C, et al. High serum MMP-14 predicts worse survival in gastric cancer. PLoS ONE. 2018;13(12):e0208800.
Piekny AJ, Glotzer M. Anillin is a scaffold protein that links RhoA, actin, and myosin during cytokinesis. Curr Biol. 2008;18(1):30–6.
Naydenov NG, Koblinski JE, Ivanov AI. Anillin is an emerging regulator of tumorigenesis, acting as a cortical cytoskeletal scaffold and a nuclear modulator of cancer cell differentiation. Cell Mol Life Sci. 2021;78:621–33.
Wang G, Shen W, Cui L, Chen W, Hu X, Fu J. Overexpression of Anillin (ANLN) is correlated with colorectal cancer progression and poor prognosis. Cancer Biomark. 2016;16(3):459–65.
Zhang L-H, Wang D, Li Z, Wang G, Chen D-B, Cheng Q, et al. Overexpression of anillin is related to poor prognosis in patients with hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2021;20(4):337–44.
Magnusson K, Gremel G, Rydén L, Pontén V, Uhlén M, Dimberg A, et al. ANLN is a prognostic biomarker independent of Ki-67 and essential for cell cycle progression in primary breast cancer. BMC Cancer. 2016;16(1):1–13.
Zhou W, Wang Z, Shen N, Pi W, Jiang W, Huang J, et al. Knockdown of ANLN by lentivirus inhibits cell growth and migration in human breast cancer. Mol Cell Biochem. 2015;398:11–9.
Idichi T, Seki N, Kurahara H, Yonemori K, Osako Y, Arai T, et al. Regulation of actin-binding protein ANLN by antitumor miR-217 inhibits cancer cell aggressiveness in pancreatic ductal adenocarcinoma. Oncotarget. 2017;8(32):53180.
Jia H, Yu F, Li B, Gao Z. Actin-binding protein Anillin promotes the progression of gastric cancer in vitro and in mice. J Clin Lab Anal. 2021;35(2):e23635.
Cui Z, Mo J, Song P, Wang L, Wang R, Cheng F, et al. Comprehensive bioinformatics analysis reveals the prognostic value, predictive value, and immunological roles of ANLN in human cancers. Front Genet. 2022;13:1000339.
Zhang L, Wei Y, He Y, Wang X, Huang Z, Sun L, et al. Clinical implication and immunological landscape analyses of ANLN in pan-cancer: A new target for cancer research. Cancer Med. 2023;12(4):4907–20.
Shi Y, Ma X, Wang M, Lan S, Jian H, Wang Y, et al. Comprehensive analyses reveal the carcinogenic and immunological roles of ANLN in human cancers. Cancer Cell Int. 2022;22(1):188.
Funding
This work was supported by the National Research Foundation of Korea [grant numbers RS-2023–00248021, 2020R1A6A3A13073061, RS-2024–00344028], the Commercialization Promotion Agency for R&D Outcomes (COMPA) funded by the Ministry of Science and ICT (MSIT) [grant number 2024–24020010-11r, R&D Equipment Engineering Education Program], and the Korea Institute of Science and Technology Research Project [grant number 2V09701].
Author information
Authors and Affiliations
Contributions
K.-M. Choi and J.-Y. Kim designed the research. K.-M. Choi, S.-J. Kim and M.-J. Ji performed experiments. K.-M. Choi, S.-J. Kim, M.-J. Ji, E. Kim, J.-S. Kim, H.‑M. Park and J.-Y. Kim analyzed data. K.-M. Choi, S.-J., Kim and J.-Y. Kim wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choi, KM., Kim, SJ., Ji, MJ. et al. Activity-based protein profiling and global proteome analysis reveal MASTL as a potential therapeutic target in gastric cancer. Cell Commun Signal 22, 397 (2024). https://doi.org/10.1186/s12964-024-01783-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01783-8