
Abstract
Background
The roles of the transcriptional factor SIX2 have been identified in several tumors. However, its roles in gastric cancer (GC) progression have not yet been revealed. Our objective is to explore the impact and underlying mechanisms of SIX2 on the stemness of GC cells.
Methods
Lentivirus infection was employed to establish stable expression SIX2 or PFN2 in GC cells. Gain- and loss-of-function experiments were conducted to detect changes of stemness markers, flow cytometry profiles, tumor spheroid formation, and tumor-initiating ability. ChIP, RNA-sequencing, tissue microarray, and bioinformatics analysis were performed to reveal the correlation between SIX2 and PFN2. The mechanisms underlying the SIX2/PFN2 loop-mediated effects were elucidated through tissue microarray analysis, RNA stability assay, IP-MS, Co-Immunoprecipitation, and inhibition of the JNK signaling pathway.
Results
The stemness of GC cells was enhanced by SIX2. Mechanistically, SIX2 directly bound to PFN2’s promoter and promoted PFN2 activity. PFN2, in turn, promoted the mRNA stability of SIX2 by recruiting RNA binding protein YBX-1, subsequently activating the downstream MAPK/JNK pathway.
Conclusion
This study unveils the roles of SIX2 in governing GC cell stemness, defining a novel SIX2/PFN2 regulatory loop responsible for this regulation. This suggests the potential of targeting the SIX2/PFN2 loop for GC treatment (Graphical Abstracts).
Graphical Abstract

Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fifth frequent cancer and the fourth major instigator to cancer-associated fatalities all over the world [1]. While some modest improvements have been made in the treatment strategies for GC, the median survival for advanced GC remains less than 1 year [2]. Over 60% of GC patients have local or distant metastasis at the time of diagnosis, thereby, chemotherapy is the preferred selection for most patients [3, 4]. However, multidrug resistance is a critical determinant leading to cancer chemotherapy failure for GC patients [5, 6]. Chemoresistance has been linked to cancer stem cells (CSCs), which are believed to perform crucial functions in tumor metastasis, invasion, recurrence, and are the primary reason for therapy failure, including in GC [7,8,9]. Nevertheless, the mechanisms by which GC stem cells (GCSCs) maintain their stemness remain largely unknown. Thus, understanding the mechanisms contributing to the maintenance of GCSC stemness may help uncover potential targets of cancer occurrence and development, as well as aid in the development of novel cancer treatment methods.
Over the past years, abundant researches have confirmed that transcription factors are abnormally expressed in CSCs [10,11,12,13]. SIX2, which enhances metanephric mesenchyme cell proliferation and suppresses cell apoptosis during kidney development [14]. And SIX2 can define and regulate the population of multipotent self-renewing nephron progenitor cells during mammalian kidney development. [14]. Earlier researches have demonstrated that SIX2 is abnormally expressed in several cancers and closely associated with drug resistance [12, 15, 16]. As a transcription factor, SIX2 directly binds to gene promoters and regulate gene expression. We previously revealed that SIX2 facilitated the stemness of breast cancer cells by directly binding to the promoters of CYP4Z2P and CYP4Z1 [12]. Consistently, Hou et al. established that SIX2 promoted NSCLC cell stemness by stimulating E-cadherin promoter methylation [15]. However, the role of SIX2 in the stemness of GC cells remains uncertain.
Profilin (PFN), a 12–15 kDa actin-binding protein involved in actin dynamics, is ubiquitously expressed in all eukaryotes, and has a highly conservative structure (1717, 18). PFNs are engaged with numerous cellular processes, such as cell motility, metabolism, gene transcription, and signal transduction [19, 20]. Among the PFN isoforms, PFN2 has been shown to play a crucial role in tumor progression [20]. Jiang et al. established PFN2 abnormal elevation was associated with unfavorable outcome in breast cancer patients [21]. Cao et al. pointed to that PFN2 accelerated small cell lung cancer development by enhancing tumor angiogenesis [22]. Nevertheless, the biological efficacy of PFN2 in digestive tract tumors remains controversial. PFN2 instigates esophageal squamous cell carcinoma progression and metastasis [23], and significantly enhances the stemness of colorectal CSCs [24]. Nonetheless, in oral squamous cell carcinoma, PFN2 inhibits tumor growth and aggressiveness [25]. Therefore, the roles of PFN2 in various cancers require further elucidation, especially in GC, where scarce research have investigated its functions.
Considering the promoting role of SIX2 in the stemness of breast cancer cells and NSCLC cells, and the pivotal role of PFN2 in tumor progression, we hypothesize that both SIX2 and PFN2 play crucial roles in the regulation of gastric cancer stemness and may drive the enhancement of stemness in GC cells. In this study, we report a regulatory loop between SIX2 and PFN2 that enhanced the stemness of GC cells. Specifically, SIX2 enhances the stemness by directly binding to the promoter of PFN2, while PFN2 regulates SIX2 promoter activity and mRNA stability through the recruitment of RNA binding protein Y-box binding protein 1 (YBX-1). Moreover, the MAPK/JNK signaling pathway is required for the regulation of SIX2/PFN2 regulatory loop in GC cell stemness. Importantly, SIX2 and PFN2 expression are negatively associated with overall survival (OS), post-progression survival (PPS), and first progression (FP). Meanwhile SIX2 and PFN2 expression are positively correlated, emphasizing the essential roles of this regulatory loop in GC progression. Consequently, our research provides a novel perspective on the relationship between transcription factors and stemness maintenance in gastric cells.
Material and method
Cell lines and reagents
AGS and MKN-45 were purchased from Procell Life Science (Wuhan, China). HEK293T was obtained from the Jiangsu Key Laboratory of Tumorigenesis and Intervention. AGS cells were cultured in a special medium for AGS cell (Procell Life Science, Wuhan, China). MKN-45 cells were cultured in RPMI-1640 with 20% FBS. HEK293T cells were in high glucose DMEM medium (KeyGen BioTECH, Nanjing, China) with 10% fetal bovine serum (OmnimAbs, Shanghai, China). Cells were validated annually by short tandem repeat (STR) DNA analysis. B27 (Cat. No. abs9120), EGF (Cat. No. abs04176) and bFGF (Cat. No. abs45152529) were purchased from absin, (Shanghai, China). Actinomycin D (Sigma-Aldrich, USA) were used for RNA stability assay. The MEK signaling pathway inhibitor PD0325901 (1 µM) and JNK signaling pathway inhibitor SP600125 (10 µM) were purchased from Selleck (Texas, USA).
Plasmid, siRNA and cell transfection
The SIX2 coding sequence was amplified via polymerase chain reaction (PCR) technology from a human cDNA template. Next, it was ligated into the pCDNA4 HisMaxB vector or p3xflag-cmv-10 vector(especially for co-IP) and verified by DNA sequencing. Similarly, the coding sequences of PFN2, NONO, and YBX-1 were synthesized by GENEWIZ (suzhou, China), and then inserted into the p3xflag-cmv-10 vector or pCDNA3.1-HA and then verified by DNA sequencing. The siRNA concentration utilized in this study was 50 nM, and all siRNA sequences specific to the target were synthesized in Genepharma (Shanghai, China). Sequences of siRNA were listed as follows: si-SIX2 (sense 5’ – GCGAGCUCUACAAGAUCCUTT − 3’); and si-PFN2 (sense 5’ – GCUACUGCGACGCCAAAUATT − 3’). Transfection of plasmids or siRNA were performed using Jet-PRIME. Primer sequences were registered at Supplementary Table 1.
Lentivirus and stable cell lines
The SIX2 coding area, SIX2 shRNA sequences, and PFN2 coding area were sub-cloned into pGLV3/H1/GFP + Puro Vector, and GenePharma had completed lentivirus packaging. Western blot analysis was used to verify expression levels.
RNA sequencing
MKN-45 cells were seeded into a six-well plate, and RNA was extracted after transfection Flag-six2 or Flag plasmid for 48 h. Novogene (Beijing, China) completed RNA sequencing and data analysis. Sequencing data was uploaded in GEO as GSE234584.
Tissue microarray analysis
This chip was purchased from Outdo Biotech Company (Shanghai, China), which contains human cancer tissues and normal adjacent tissue collected from 75 GC patients with informed consent (HStmA150CS02). The supplier also provided relevant clinical and pathological information. The collection of human tissue was approved by the Shanghai Medical Institution Ethics Committee in accordance with ethical guidelines. Ethics approval number is SHYJS-CP-1,807,020.
Servicebio (Wuhan, China) completed immunohistochemical analysis, and the quantification was executed using Quantity-one Software. The staining sections was scored, and the scoring criteria refer to the literature [26]. Furthermore, Boster Biological Technology completed the RNA-fluorescence in situ hybridization assays.
qPCR
Details were provided in our previous research. [12]. Amplification specificity was routinely verified by performing a melting curve analysis. Primer sequences were given in Supplementary Table 1.
Western blot analysis
Experimental details were described previously [12]. Information about the antibodies were given in Supplementary Table 2.
Flow cytometry
Spread cells in a six-well plate, and harvested 72 h after transfection with plasmid or siRNA. ALDH1 enzyme activity was detected using ALDEFLUOR™ Kit (STEMCELL Technologies, Cat. No. 01700, USA). Finally, enzymatic activity was measured using a FACSC alibur flow cytometer (BD Biosciences).
Mammosphere formation assay
Details were presented in our previous research [27].The information on the reagents used in this assay are at the front. Images were captured with microscope.
Chromatin immunoprecipitation assay (ChIP)
ChIP-IT Express (Active Motif, Cat. No. 53008, USA) was used to perform the ChIP assays following the standard protocols.
Dual luciferase reporter assay
PFN2, SIX2, and their promoter sequences were inserted into the pGL3-promoter vector. Cells were harvested 72 h after co-transfection with plasmids. Using Dual Luciferase Reporter Assay Kit (Cat. No. DL101-01, vazyme, Nanjin, China) following the manufacturer’s standard protocols to detect luciferase activity.
Co-immunoprecipitation (Co-IP)
Using Co-IP kit (Thermo Scientific, Cat. No. 88804, USA) following the manufacturer’s standard protocols, followed by detection of the protein lysates via western blot analysis.
RNA stability assay
Details were in our previous research [28].
RNA-Fluorescence in situ hybridization (RNA-FISH)
Frozen slices were utilized to perform fluorescence in situ hybridization for SIX2 and PFN2 using a detailed procedure previously described [29].
In vivo tumorigenesis study
6 weeks old Nude BALB/c mice were purchased from GemPharmatech (Nanjing, China) and raised under standard pathogen-free conditions. MKN-45 cells infected with the different virus were ortho-topically implanted into the inguinal mammary gland of nude mice. For the tumor-limiting dilution assay, MKN-45 cells with SIX2 overexpression and vector were at the concentration of 1 × 107,1 × 106, and 1 × 105, while MKN-45 cells with SIX2 knockdown and control were at the concentration of 5 × 107,5 × 106, and 5 × 105 cells. 14 days later, collected tumors and determined tumor formation rate after mice were euthanized. Animal operating procedures were approved by the Animal Ethics Committee of China Pharmaceutical University (2022-05-040). to Evaluate stem cell frequency using ELDA method [30].
Statistical analysis
Statistical analysis was utilized GraphPad Prism 9. Data are expressed as mean ± SD from three independent experiments. Statistical significance between groups was analyzed using t-test. P < 0.05 or less was considered statistically significant.
Results
Elevated SIX2 expression predicts a worse survival rate in GC patients
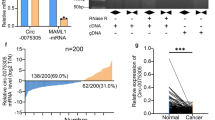
We initially assessed the survival data in GC patients expressing SIX2 through Kaplan Meier plotter database (http://kmplot.com) [31]. As presented in Fig. 1A and C, the higher the level of SIX2 expressed in GC patients, the worse their OS, PPS and FP probability. Consistently, KM-plotter analysis by R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl) exhibited that patients with high SIX2 expression have worse overall survival (Fig. 1D). Additionally, we estimated the relationship between stemness markers (ALDH1A1, SOX2) expression and SIX2 levels using R2. Remarkably, our analysis demonstrated that SIX2 expression and the stemness markers are positively correlated in GC tissues (Fig. 1E and F). These above-described results prove that SIX2 might have a significant impact in GC progression. Interestingly, the tissue microarray analysis exhibited that SIX2 was highly expressed in adjacent tissue in comparison with that in GC tissues (Fig. 1G). Furthermore, we detected the protein level of SIX2 in human gastric adenocarcinoma cell lines (MKN-45 and AGS) and human gastric mucosal epithelial cells lines (GES-1), and identified that SIX2 expressed at higher levels in GES-1 than in MKN-45 and AGS (Fig. 1H and I). This unexpected result prompts us to continue exploring the relationship between SIX2 and GC cells.

The SIX2 expression in GC patients. (A) Correlation between SIX2 expression and OS probability. (B) Correlation between SIX2 expression and PPS probability. (C) Correlation between SIX2 expression and FP probability. (D) Correlation between SIX2 expression and OS probability using R2. (E) Correlation between SIX2 and ALDH1A1 expression of in GC tissues via R2. (F) Correlation between SIX2 and SOX2 expression of in GC tissues via R2. (G) IHC detection and scores of SIX2 protein levels in tissue chip. (H and I) Western blot detection and the quantitative results of SIX2 levels in different cells
SIX2 promotes the stemness of GC cells
SIX2 was stably overexpressed and knocked down in two GC cell lines (MKN-45, AGS) via lentivirus infection. We found that overexpression of SIX2 upregulated the expression of stemness markers (ALDH1A1, SOX2, OCT3/4), while knockdown of SIX2 exhibited an opposite effect (Fig. 2A and B, Figure S1A and S1B). Flow cytometry analysis revealed that overexpression of SIX2 increased the percentage of Aldefluor-positive cells, known for their self-renewal properties, while knockdown of SIX2 decreased this percentage (Fig. 2C and D and Figure S1C). These results suggest that SIX2 can positively enrich Aldefluor-positive cells known to possess self-renewal properties [32]. Besides, we observed an increase in the CD44 + sub-population in GC cells upon SIX2 overexpression, while SIX2 knockdown had opposite effects (Figure S1D). Owing to the higher concentration of CSC-like cells in non-adherent tumor cell spheres rather than adherent tumor cells [33], we determined the effects of SIX2 on sphere-formation ability and found that SIX2 overexpression increased the capacity of cell spheroid formation, as manifested by an increase in spheroid size and number, while SIX2 knockdown reversed the effects (Fig. 2E and F). Surprisingly, AGS cells with or without overexpressing SIX2 could hardly form spheres (data not shown). Moreover, we observed the enhanced tumorigenic ability in GC cells with SIX2 overexpression, evidence of which was confirmed in the tumor-limiting dilution assay with increased tumor formation rate and stem cell frequency, while SIX2 knockdown exhibited opposite effects (Fig. 2G and H). Therefore, these results indicate that SIX2 could exacerbate the stemness of GC cells both in vivo and in vitro.

SIX2 enhances the stemness of GC cells. (A) Western blot detection and the quantitative results of stemness markers protein expression levels in MKN-45 and AGS cells with or without SIX2 overexpression. (B) Western blot detection and the quantitative results of stemness markers protein expression levels in MKN-45 and AGS cells with or without SIX2 knockdown. (C) The percentage of Aldefluor-positive cells were detected via Flow cytometry analysis in MKN-45cells depicted in (A). (D) Flow cytometry analysis of the percentage of Aldefluor-positive cells in MKN-45 cells depicted in (B). (E) Spheroid formation (10x) of MKN-45cells depicted in (A). (F) Spheroid formation (10x) of MKN-45cells depicted in (B). (G and H) Tumors size and tumors number of MKN-45 cells with different treatment (Vector, SIX2, si-NC, si-SIX2), respectively. The tumor formation rate and the ELDA analysis was used to analyze the frequency of stem cells. Data are presented as mean ± SD, n = 3, *P < 0.05 vs. vector or si-NC
SIX2 regulates PFN2 expression by binding to PFN2 promoter
We next explored the mechanisms how SIX2 promotes the stemness of GC cells. Through transcriptome sequencing analysis, we screened for possible downstream targets of SIX2 through and identified the top ten genes that significantly increased with SIX2 overexpression (Fig. 3A). As SIX2 is a transcription factor, we analyzed the binding sites of SIX2 on the promoter of above genes by JASPAR database [34]. We sought out two top-scoring putative binding sites on the PFN2 promoter (Supplementary Table 3). The two binding sites on the antisense strand have duplicate regions, so we combine these two sites into one, and described as binding sites (A) The binding sites on the sense strand with a binding score of 13.42 and relative score 0.86, indicating a strong possibility of binding were designated as binding sites (B) As shown in Fig. 3B and D, SIX2 overexpression upregulated PFN2 levels, while knockdown of SIX2 downregulated it. The WT or MUT binding sites were displayed in Fig. 3E. Luciferase reporter gene analysis illustrated that SIX2-overexpressed HEK293T cells had a higher up-regulation (about 22.77 times) of luciferase activity at WT binding sites A and B, while at mutant binding sites A or B only had a moderate up-regulation (about 14.87 times or 12.53 times). Interestingly, the activity of MUT A and B was also upregulated about 7.35 times (Fig. 3F). These above results confirmed that SIX2 bind directly to PFN2 promoter through binding sites A and B, and suggested the possibility of other binding sites. The docking results of SIX2 binding to the PFN2 promoter were illustrated in Fig. 3G. Chromatin immunoprecipitation (ChIP) analysis further validated that the region containing the binding site A and B was prominently significantly augmented in DNA pulled down by anti-SIX2 (Fig. 3H). In summary, our results validated SIX2 could directly to bind to PFN2 promoter and activate its transcription.

SIX2 regulates PFN2 expression by binding to its promoter. (A) Top 20 changed mRNA in MKN-45 cells with or without SIX2 overexpression. (B) PFN2 protein expression levels detected by western blot. (C and D) PFN2 mRNA levels were detected via qRT-PCR. (E) Potential binding sites for SIX2 on PFN2 promoter. (F) Luciferase report experiment for detecting relative luciferase activity. (G) Docking results of SIX2 binding to the PFN2 promoter. (H) ChIP assay detection of the abundance of PFN2 promoter with anti-SIX2 and IgG. Data are presented as mean ± SD, n = 3, *P < 0.05 vs. vector, si-NC or Control
PFN2 is accountable for the stemness regulation of GC cells mediated by six2
To further investigate whether SIX2 promotes the stemness of GC cells through PFN2, MNK-45 and AGS cells with SIX2 overexpression or MKN-45 cells with SIX2 knockdown were transfected with si-PFN2 or PFN2 expression plasmid, respectively. The results showed that si-PFN2 attenuated the stemness-promoting effect via SIX2 overexpression, characterized by the decrease percentage of Aldefluor-positive cells (Fig. 4A), cell spheroid formation (Fig. 4C), and stemness marker expression (Figure S2A and S2B). In contrast, the effect of overexpression of SIX2 was reversed by PFN2 knockdown in MKN-45 cells (Fig. 4B and D and Figure S2C). Additionally, si-PFN2 was found to decrease the tumor-initiating ability of MKN-45 cells and weakened SIX2 overexpression-mediated promotion on tumor-initiating ability (Fig. 4E and H). The knockdown of SIX2 also extremely repressed the tumor-initiating potential of MKN-45 cells, which was recovered by PFN2 overexpression (Fig. 4E and H). Thus, these findings indicated that SIX2 assist GC cell stemness relying on PFN2.

PFN2 is accountable for the stemness regulation of GC cells mediated by six2. (A and B) The percentage of Aldefluor-positive cells were detected via Flow cytometry analysis in MKN-45cells with different treatment. (C and D) Spheroid formation (10x) in MKN-45 cells. (E and F) Tumors size and number of MKN-45 cells with different treatment (Vector, Si-NC, PFN2, Si-SIX2), respectively (E) and calculate the tumor formation rate (F). (G and H) ELDA analysis was used to calculate the stem cell frequency and pairwise tests for differences in these groups. Data are presented as mean ± SD, n = 3, *P < 0.05 vs. vector, or si-NC
PFN2 elevates the stemness of GC cells
PFN2 was then stably overexpressed in cells and knocked down in MKN-45 cell lines by lentivirus infection. The expression of stemness markers (ALDH1A1, SOX2, OCT3/4) was elevated via over-expressing PFN2, while knocking down PFN2 resulted in the contrary outcomes (Fig. 5A and C). Moreover, PFN2 overexpression promoted cell spheroid formation, as manifested by an increase in spheroid size and number, while PFN2 knockdown displayed an opposite effects (Fig. 5B). Flow cytometry analysis showed an increase in the percentage of Aldefluor-positive cells in PFN2 overexpression cells and a decrease in cells with PFN2 knockdown (Fig. 5D). Moreover, we observed that PFN2 overexpression increased the tumorigenic ability of GC cells, which was clearly indicated by a raised rate of tumor formation (Fig. 5E). Thus, these findings suggested that PFN2 could enhance the stemness of GC cells. Next, RNA fluorescence in situ hybridization analysis on a tissue microarray containing 75 cases of GC and adjacent tissues revealed that PFN2 expression was generally higher in adjacent tissues than in cancer tissues, except for 12 cases of the opposite (Figure S3). As shown in (Figure S4A), We analyzed the expression level of PFN2 in gastric cancer tissue using GEPIA (Gene Expression Profiling Interactive Analysis) [35], and found that the expression of PFN2 protein in GC tissue was noticeably higher than that in normal tissue. We also evaluated the survival rate of GC patients with PFN2 expression through Kaplan Meier plotter database (http://kmplot.com) and detected that the higher the level of PFN2 expressed in GC patients, the worse of their OS, PPS, and FP probability (Figure S4B). These results are consistent with the survival rate of SIX2 in GC patients. Meanwhile, we assessed the relevance between PFN2 and SIX2 using R2, and obtained that the SIX2 expression was positively associated with PFN2 expression in GC (Figure S4C). We also estimated the expression correlation between PFN2 and stemness markers (SOX2, Nanog and ALDH1A1), and found that PFN2 expression is positively associated with the expression of stemness markers in gastric tumor tissues (Figure S4D-S4F). Above, we confirm that PFN2 promotes the stemness of GC cells and plays an important role in GC.

PFN2 elevates the stemness of GC cells. (A) Western blot detection of protein expression levels (ALDH1A1, SOX2, and OCT4) in MKN-45 cells with or without PFN2 overexpression or knockdown. (B) Spheroid formation (10x) of MKN-45cells depicted in (A). (C) qPCR detection of mRNA levels (ALDH1A1, SOX2, and OCT4) in MKN-45cells depicted in (A). (D) The percentage of Aldefluor-positive cells were detected via Flow cytometry analysis in MKN-45 cells depicted in (A). (E) Tumors size and tumor number of MKN-45 cells with different treatment, respectively. The tumor formation rate and the ELDA analysis was used to analyze the frequency of stem cells. Data are presented as mean ± SD, n = 3, *P < 0.05 vs. vector or si-NC
PFN2 regulates the transcriptional activity and mRNA stability of SIX2 by recruiting NONO and YBX-1, and the SIX2/PFN2 loop activates the MAPK/JNK pathway to promote GC stemness
We accidentally noticed that overexpression of PFN2 significantly upregulated the mRNA level of SIX2, while PFN2 knockdown decreased the mRNA level of SIX2 (Fig. 6A and Figure S5A). Consistent results were obtained when testing the effect of PFN2 on the protein level of SIX2 (Fig. 6B). Considering the above results, we speculate that PFN2 may promote the transcription of SIX2. This hypothesis was supported by luciferase reporter analysis, which indicated that the luciferase activity of SIX2 promoter was upregulated with PFN2 overexpression (Fig. 6C). Furthermore, mRNA stability experiments showed that PFN2 overexpression enhanced the mRNA stability of SIX2 (Fig. 6D). Although PFN2 did not possess the function of a transcription factor or a RNA-binding protein [36], we hypothesized that PFN2 may promote SIX2 transcription and increase the mRNA stability of SIX2 by recruiting transcription factors or RNA-binding proteins. To verify this hypothesis, we used IP-MS technology to identify potential binding proteins of PFN2. The protein sequence information of proteins with higher scores after IP-MS screening is shown in Fig. 6E. Meanwhile, we predicted transcription factors that may bind to the SIX2 promoter. Intersecting the predicted results with the protein information after IP-MS, we found that NONO and KDM1A were duplicated (Figure S5B). On the basis of the above-mentioned results, we focused on non-POU domain-containing octamer-binding protein (NONO) and YBX-1, which are predicted transcription factors that may bind to the SIX2 promoter. Next, we verified the interaction of endogenous PFN2 with NONO and YBX-1 by CO-IP in MKN-45 cells (Fig. 6F), as well as the interaction of exogenous PFN2 with NONO and YBX-1 in 293T cells using CO-IP (Fig. 6G). Moreover, we found that overexpression of YBX-1 enhanced the stability of SIX2 mRNA (Fig. 6H), while the luciferase activities of SIX2 promoter had no changes with NONO or YBX-1 overexpression (Fig. 6I). These results illustrate that PFN2 can recruit YBX-1, which can bind to SIX2 mRNA and enhance its stability, forming a SIX2/PFN2 feedback loop.

PFN2 regulates the transcription activity and mRNA stability of SIX2 by recruiting NONO and YBX-1. (A) qRT-PCR detected SIX2 mRNA levels in MKN-45 cells with or without PFN2 overexpression. (B) Western blot detection of SIX2 protein expression levels in MKN-45 cells with or without PFN2 overexpression or knockdown. (C) Relative luciferase activity measured by Luciferase report experiment. (D) The mRNA stability of SIX2 was detected in MKN-45 cells depicted in (B). (E) The protein sequence information of proteins with higher scores after IP-MS screening. (F) CO-IP method for detecting the endogenous PFN2 with NONO and YBX-1 in MKN-45 cells. (G) The interaction of exogenous PFN2 with NONO and YBX-1 were detected in 293T cells using CO-IP. (H) The mRNA stability of SIX2 was measured in MKN-45 cells with or without YBX-1 overexpression or knockdown. (I) Luciferase report experiment for detecting relative luciferase activity in MKN-45 cells with different treatment. Data are presented as mean ± SD, n = 3, *P < 0.05 vs. vector, si-NC or Control
We conducted further investigation into the downstream signaling pathways affected by SIX2/PFN2 regulatory loop. Transcriptome sequencing results revealed a significant enrichment of the MAPK signaling pathway in GC cells overexpressing SIX2 (Figure S6A). Then MKN-45 cells with or without SIX2 and PFN2 overexpression were treated with MEK signaling pathway inhibitor (Mirdametinib), as shown in Figure S6B, treatment with Mirdametinib obviously attenuated the SIX2/PFN2 regulatory loop-induced promotion on the expression of stemness markers. To explore the specific MAPK signaling pathways involved, we checked the expression levels of the marker proteins for the P38, ERK1/2, and JNK pathways, which play well-characterized roles in regulating cellular activities [37]. Our results presented that overexpression of SIX2 or PFN2 promoted the expression of phosphorylated JNK (Figure S6C), indicating that the SIX2/PFN2 regulatory loop activates the JNK signaling pathway. To confirm this, we treated MKN-45 cells with or without SIX2 and PFN2 overexpression with JNK inhibitor (SP600125). This treatment also alleviated the SIX2/PFN2 regulatory loop-induced enhancing effects on the expression of stemness markers (Figure S6D), and cell spheroid formation capacity (Figure S6E). Additionally, MKN-45 cells, either with or without overexpression of SIX2 and PFN2, were treated with Mirdametinib or SP600125, respectively. As shown in Figure S6F, the treatment with Mirdametinib or SP600125 significantly decreased the levels of p-JNK and attenuated the promoting effects of the SIX2/PFN2 regulatory loop on the JNK signaling pathway. Hence, our findings indicate that the SIX2/PFN2 regulatory loop elevates the stemness of GC cells through the MAPK/JNK pathways.
Discussion
The dysregulation of developmental genes is a common occurrence in cancer initiation and progression [38]. Our and other previous studies reveal that SIX2 facilitates tumor growth metastasis, and invasion in various cancers [12, 15, 16]. However, SIX2 expression and detailed mechanism in GC remain elusive. Recently, we confirmed that SIX2 elevated the stemness of breast tumor cells by activating the PI3K/Akt and ERK1/2 signaling pathways [12], leading us to hypothesize that SIX2 might also be involved in regulating the stemness of GC cells. In this ongoing study, we clearly elucidated the SIX2/PFN2 regulatory loop and revealed its role in driving the stemness of GC cells. SIX2 enhanced the stemness by directly binding to the promoter of PFN2, while PFN2 can regulate SIX2 promoter activity and mRNA stability by recruiting NONO and YBX-1, respectively. Additionally, the MAPK/JNK signaling pathway was required for the SIX2/PFN2 regulatory loop-mediated regulation of GC cell stemness. Notably, SIX2 and PFN2 expression were negatively related to the OS, PPS, and FP of GC patients, emphasizing the essential roles of this regulatory loop in GC progression. In our understanding, this is the first research to demonstrate the effect of the SIX2/PFN2 regulatory loop on the stemness of GC cells.
SIX2 is a member of SIX gene family, which play a significant role in development as well as tumorigenesis and metastasis by regulating multiple downstream targets [38]. Through the online database KM plotter analyses, we found SIX2 expression was negatively associated with the OS, PPS, and FP of GC patients, highlighting the critical roles of SIX2 in GC progression. Therefore, we speculated that SIX2 might function as an oncogene. However, contrary to our expectations, tissue microarray analysis showed that, the gene expression the expression of SIX2 and PFN2 in GC tissue were lower, comparing with the adjacent tissue, and the SIX2 expression level in GC cells MKN-45 and AGS is significantly lower than that in gastric epithelial cells. This discrepancy puzzled us, and we further explored whether SIX2 plays a promoting or inhibiting role in the stemness of GC cells. Subsequent research revealed that SIX2 indeed increased the stemness of GC cells, as evidenced by the increased cell sub-population with stemness, stemness marker expression, sphere-formation, and tumor-initiating ability. Previous studies have demonstrated high SIX2 expression in cancer tissues, and that SIX2 function as an oncogene in various cancers, including in colorectal cancer [39], breast carcinoma [12], and NSCLC [15]. The function of SIX2 is corresponding to our results that SIX2 enhances the stemness of GC cells. The low expression level of SIX2 puzzled us even more, and we hypothesized that low expression in tumors does not necessarily indicate tumor suppression.
Through RNA-sequencing and bioinformatics analysis, we identified PFN2 as the downstream target of SIX2 and confirmed that SIX2 can regulate the levels of PFN2 by directly bind to PFN2 promoter. PFN2, a member of the actin-binding protein Profilin family, is known to regulate the dynamics of actin polymerization [36]. Emerging evidence has linked PFN2 in various types of cancer. For example, Cui at al. demonstrated that PFN2 promoted esophageal squamous cell carcinoma progression and metastasis, and portending a poor prognosis [23]; Tang et al. showed that PFN2 promoted lung cancer growth and metastasis [40]. Additionally, a mouse xenograft model demonstrated that of PFN2 overexpression dramatically increases small cell lung cancer growth and vasculature formation [22]. Nonetheless, the biological function and mechanism of PFN2 in GCSCs have not been revealed. Recently, Hu et al. found that the expression of PFN2 protein in GC tissue was noticeably higher than that in adjacent tissue, and high expression of PFN2 protein substantially predicted worse prognosis in GC patients [41], which is consistent with our findings. Regarding the discrepancy in PFN2 expression levels in between GC tissues and normal gastric epithelial tissues, we speculate that this may be due to the limited sample size, and that more cases are needed to confirm this observation. In addition, the observed discrepancy between the overexpression of PFN2 leading to significant upregulation of ALDH1A1, SOX2, and OCT in Fig. 5, contrasted with the less striking difference in the protein levels, raises the question of whether there is post-transcriptional control influencing the expression of these genes. Although PFN2 appears to play a potent role in regulating the transcription of these genes, the limited impact on their expression following PFN2 knockdown suggests the involvement of additional regulatory mechanisms beyond transcriptional control. Further investigation into post-transcriptional regulatory pathways may provide insights into the observed discrepancies in gene expression levels.
Latest investigations have revealed that RBPs play a key role in cancer [42], but the clinical significance and underlying mechanisms of the bulk of RBPs in GCSCs remain unknown. Our research has identified a novel interaction between SIX2 and PFN2 in regulating the stemness of GC cells. Interestingly, we found that PFN2 can also affect the expression and the mRNA stability of SIX2 by chance. Through IP-MS and Co-Immunoprecipitation, we discovered that PFN2 can regulate SIX2 promoter activity and mRNA stability by recruiting NONO and YBX-1, respectively. NONO belongs to the Drosophila behavior/human splicing (DBHS) family, which is a typical RBP with two highly conserved RNA-recognition motifs. Seeing that NONO participates in virtually every step of gene regulation, its emerging paradigm is described as a “multipurpose molecular scaffold [43]. The multi-functional characteristics of NONO are related to the protein partners it binds to. [44]. Prior researches have demonstrated that NONO is involved in triple-negative breast cancer growth and EGFR-mediated tumorigenesis of [44]. Kim et al. also found the role of NONO in stabilizing STAT3 RNA, and which directly binding the STAT3 protein and increasing its stability and activity [45]. We found the luciferase activities of SIX2 promoter had no changes with NONO overexpression, indicating that although PFN2 can interact with NONO, this interaction does not regulate the activity of the SIX2 promoter. YBX-1 is a DNA and RNA binding protein that is the main protein associated with mRNA, and is involved in numerous RNA-dependent events, such as mRNA splicing, RNA stabilization, translational repression, and transcriptional regulation [46, 47]. Previous research has found that the GC patients with high YBX-1 expression showed a worse survival [48]. Additionally, YBX-1 has been indicated to regulate cell proliferation and migration in GC cells [49]. Similarly, we found that YBX-1 positively regulates SIX2 mRNA stability, but it does not affect its promoter activity. This suggests that the regulatory role of YBX-1 on SIX2 is not mediated through its DNA-binding protein activity, but rather through its RNA-binding protein activity. These results imply that PFN2 may regulate the promoter activity of SIX2 by recruiting other transcription factors. Additionally, based on our findings, we propose that YBX-1 binds to SIX2 mRNA and enhances its stability. PFN2 appears to play a role in this process by recruiting YBX-1 to the SIX2 mRNA, forming a regulatory complex. However, further experiments are needed to definitively determine the sequence of binding and whether both proteins have distinct binding sites on SIX2 mRNA. And it must be admitted that our data did not specifically address whether PFN2 activates NONO or YBX-1. However, we observed that the overexpression of YBX-1 alone could enhance the stability of SIX2 mRNA, suggesting that YBX-1 can function independently. The role of PFN2 might be more related to facilitating or stabilizing the interaction between YBX-1 and SIX2 mRNA rather than directly activating YBX-1. Similarly, the interaction between PFN2 and NONO requires further investigation to determine any potential activation effects.
The c-Jun N-terminal kinase (JNK) pathway is one of the three well-characterized MAPK pathways that regulate cellular activities, including cancer cell proliferation, in which the MAPK cascade is a complex signaling pathway that plays an important role in regulating cell proliferation and apoptosis [37]. Previous studies have shown the activation of JNK pathway promotes cell invasion and autophagy-dependent JNK activity facilitates GC cell survival by maintaining normal mitochondrial function [50, 51]. Furthermore, studies have revealed that the activation of JNK-MAPK/JUN axis by ubiquitous mitochondrial creatine kinase facilitates the migration, invasion, and liver metastasis in GC cells [52]. Our study demonstrated that the SIX2/PFN2 regulatory loop-mediated regulation of GC cell stemness is partially due to the activation of the MAPK/JNK signaling pathway. However, the underlying mechanisms through which the SIX2/PFN2 regulatory loop activates the MAPK/JNK signaling pathway are still not fully understood and require further investigation for clarification in succeeding research.
Ultimately, our work suggests that the SIX2/PFN2 feedback loop provides potential targets for developing novel strategies to target and compromise the stemness of GC, and that overexpression of PFN2 may serve as a molecular biomarker which is clinically useful and unfavorable in the prognosis of GC patients.
Conclusions
Our work reveals the mechanism that SIX2 regulated the stemness of gastric cancer cells, defines a novel SIX2/PFN2 regulatory loop responsible for this regulation, and present the potentiality of targeting the SIX2/PFN2 loop to inhibit the stemness of GC.
Data availability
All relevant data supporting the key findings of this study are available within the article and its Supplementary Information files or from corresponding authors on request.
References
Ferlay JEM, Lam F, Colombet M, Mery L, Piñeros M, Znaor A, Soerjomataram I, Bray F. Global Cancer Observatory: Cancer Today. : Lyon, France: International Agency for Research on Cancer; (2020) [ https://gco.iarc.fr/today
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388(10060):2654–64.
Zhang R, Liu L, Wang F, Zhao W, Liu K, Yu H, et al. AKAP8L enhances the stemness and chemoresistance of gastric cancer cells by stabilizing SCD1 mRNA. Cell Death Dis. 2022;13(12):1041.
Uchihara T, Miyake K, Yonemura A, Komohara Y, Itoyama R, Koiwa M, et al. Extracellular vesicles from Cancer-Associated fibroblasts containing annexin A6 induces FAK-YAP activation by stabilizing β1 integrin, enhancing Drug Resistance. Cancer Res. 2020;80(16):3222–35.
Zhang H, Jiang H, Zhang H, Liu J, Hu X, Chen L. Ribophorin II potentiates P-glycoprotein- and ABCG2-mediated multidrug resistance via activating ERK pathway in gastric cancer. Int J Biol Macromol. 2019;128:574–82.
Zhang F, Li K, Yao X, Wang H, Li W, Wu J, et al. A miR-567-PIK3AP1-PI3K/AKT-c-Myc feedback loop regulates tumour growth and chemoresistance in gastric cancer. EBioMedicine. 2019;44:311–21.
Song Y, Wang Y, Tong C, Xi H, Zhao X, Wang Y, et al. A unified model of the hierarchical and stochastic theories of gastric cancer. Br J Cancer. 2017;116(8):973–89.
Qi Y, Wei J, Zhang X. Requirement of transcription factor NME2 for the maintenance of the stemness of gastric cancer stem-like cells. Cell Death Dis. 2021;12(10):924.
Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18(11):669–80.
Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, et al. Coexpression of Oct4 and nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70(24):10433–44.
Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014;511(7508):246–50.
Zheng L, Guo Q, Xiang C, Liu S, Jiang Y, Gao L, et al. Transcriptional factor six2 promotes the competitive endogenous RNA network between CYP4Z1 and pseudogene CYP4Z2P responsible for maintaining the stemness of breast cancer cells. J Hematol Oncol. 2019;12(1):23.
You L, Guo X, Huang Y. Correlation of Cancer stem-cell markers OCT4, SOX2, and NANOG with Clinicopathological features and prognosis in operative patients with rectal Cancer. Yonsei Med J. 2018;59(1):35–42.
Kobayashi A, Valerius MT, Mugford JW, Carroll TJ, Self M, Oliver G, et al. Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell. 2008;3(2):169–81.
Hou H, Yu X, Cong P, Zhou Y, Xu Y, Jiang Y. Six2 promotes non-small cell lung cancer cell stemness via transcriptionally and epigenetically regulating E-cadherin. Cell Prolif. 2019;52(4):e12617.
Wu DW, Lin PL, Wang L, Huang CC, Lee H. The YAP1/SIX2 axis is required for DDX3-mediated tumor aggressiveness and cetuximab resistance in KRAS-wild-type colorectal cancer. Theranostics. 2017;7(5):1114–32.
Jockusch BM, Murk K, Rothkegel M. The profile of profilins. Rev Physiol Biochem Pharmacol. 2007;159:131–49.
Khadka DK, Liu W, Habas R. Non-redundant roles for Profilin2 and Profilin1 during vertebrate gastrulation. Dev Biol. 2009;332(2):396–406.
Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–65.
Li Z, Huo X, Chen K, Yang F, Tan W, Zhang Q, et al. Profilin 2 and endothelial exosomal profilin 2 promote angiogenesis and myocardial infarction repair in mice. Front Cardiovasc Med. 2022;9:781753.
Jiang M, Qiu N, Xia H, Liang H, Li H, Ao X. Long non–coding RNA FOXD2–AS1/miR–150–5p/PFN2 axis regulates breast cancer malignancy and tumorigenesis. Int J Oncol. 2019;54(3):1043–52.
Cao Q, Liu Y, Wu Y, Hu C, Sun L, Wang J, et al. Profilin 2 promotes growth, metastasis, and angiogenesis of small cell lung cancer through cancer-derived exosomes. Aging. 2020;12(24):25981–99.
Cui XB, Zhang SM, Xu YX, Dang HW, Liu CX, Wang LH, et al. PFN2, a novel marker of unfavorable prognosis, is a potential therapeutic target involved in esophageal squamous cell carcinoma. J Transl Med. 2016;14(1):137.
Kim MJ, Lee YS, Han GY, Lee HN, Ahn C, Kim CW. Profilin 2 promotes migration, invasion, and stemness of HT29 human colorectal cancer stem cells. Biosci Biotechnol Biochem. 2015;79(9):1438–46.
Ma CY, Zhang CP, Zhong LP, Pan HY, Chen WT, Wang LZ, et al. Decreased expression of profilin 2 in oral squamous cell carcinoma and its clinicopathological implications. Oncol Rep. 2011;26(4):813–23.
Guo Z, Zhang X, Zhu H, Zhong N, Luo X, Zhang Y, et al. TELO2 induced progression of colorectal cancer by binding with RICTOR through mTORC2. Oncol Rep. 2021;45(2):523–34.
Ni H, Qin H, Sun C, Liu Y, Ruan G, Guo Q, et al. MiR-375 reduces the stemness of gastric cancer cells through triggering ferroptosis. Stem Cell Res Ther. 2021;12(1):325.
Zheng L, Zhang Z, Zhang S, Guo Q, Zhang F, Gao L, et al. RNA binding protein RNPC1 inhibits breast Cancer Cell Metastasis via activating STARD13-Correlated ceRNA Network. Mol Pharm. 2018;15(6):2123–32.
Gao L, Guo Q, Li X, Yang X, Ni H, Wang T, et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine. 2019;41:395–407.
Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–8.
Lánczky A, Győrffy B. Web-based Survival Analysis Tool tailored for Medical Research (KMplot): development and implementation. J Med Internet Res. 2021;23(7):e27633.
Condello S, Morgan CA, Nagdas S, Cao L, Turek J, Hurley TD, et al. β-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene. 2015;34(18):2297–308.
Gao W, Wu D, Wang Y, Wang Z, Zou C, Dai Y, et al. Development of a novel and economical agar-based non-adherent three-dimensional culture method for enrichment of cancer stem-like cells. Stem Cell Res Ther. 2018;9(1):243.
Castro-Mondragon JA, Riudavets-Puig R, Rauluseviciute I, Lemma RB, Turchi L, Blanc-Mathieu R, et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50(D1):D165–73.
Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102.
Murk K, Ornaghi M, Schiweck J. Profilin isoforms in Health and Disease - all the same but different. Front Cell Dev Biol. 2021;9:681122.
Wu Q, Wu W, Fu B, Shi L, Wang X, Kuca K. JNK signaling in cancer cell survival. Med Res Rev. 2019;39(6):2082–104.
Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H, et al. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer. 2016;138(5):1067–75.
Fang ZX, Li CL, Wu Z, Hou YY, Wu HT, Liu J. Comprehensive analysis of the potential role and prognostic value of sine oculis homeobox homolog family in colorectal cancer. World J Gastrointest Oncol. 2022;14(11):2138–56.
Tang YN, Ding WQ, Guo XJ, Yuan XW, Wang DM, Song JG. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat Commun. 2015;6:8230.
Hu S, Shi L. [Profilin 2 is highly expressed in gastric cancer and promotes tumor cell proliferation and migration]. Nan Fang Yi Ke Da Xue Xue Bao. 2022;42(2):215–22.
Pereira B, Billaud M, Almeida R. RNA-Binding proteins in Cancer: Old players and New actors. Trends Cancer. 2017;3(7):506–28.
Knott GJ, Bond CS, Fox AH. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44(9):3989–4004.
Shen M, Zhang R, Jia W, Zhu Z, Zhao L, Huang G, et al. RNA-binding protein p54(nrb)/NONO potentiates nuclear EGFR-mediated tumorigenesis of triple-negative breast cancer. Cell Death Dis. 2022;13(1):42.
Kim SJ, Ju JS, Kang MH, Eun JW, Kim YH, Raninga PV, et al. RNA-binding protein NONO contributes to cancer cell growth and confers drug resistance as a theranostic target in TNBC. Theranostics. 2020;10(18):7974–92.
Lyabin DN, Eliseeva IA, Ovchinnikov LP. YB-1 protein: functions and regulation. Wiley Interdiscip Rev RNA. 2014;5(1):95–110.
Mordovkina D, Lyabin DN, Smolin EA, Sogorina EM, Ovchinnikov LP, Eliseeva I. Y-Box binding proteins in mRNP Assembly, translation, and Stability Control. Biomolecules. 2020;10(4).
Li S, Lu C, Li X, Li F, Zhao Y, Xu M, et al. LncRNA HOXA10-AS functions as an oncogene by binding mir-6509-5p to upregulate Y-box binding protein 1 in gastric cancer. Bioengineered. 2022;13(5):11373–87.
Zhang E, He X, Zhang C, Su J, Lu X, Si X, et al. A novel long noncoding RNA HOXC-AS3 mediates tumorigenesis of gastric cancer by binding to YBX1. Genome Biol. 2018;19(1):154.
Ye XL, Zhao YR, Weng GB, Chen YC, Wei XN, Shao JP, et al. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol Rep. 2015;33(6):2746–52.
Almasi S, Kennedy BE, El-Aghil M, Sterea AM, Gujar S, Partida-Sánchez S, et al. TRPM2 channel-mediated regulation of autophagy maintains mitochondrial function and promotes gastric cancer cell survival via the JNK-signaling pathway. J Biol Chem. 2018;293(10):3637–50.
Mi Y, Li Q, Liu B, Wang D, Liu Z, Wang T, et al. Ubiquitous mitochondrial creatine kinase promotes the progression of gastric cancer through a JNK-MAPK/JUN/HK2 axis regulated glycolysis. Gastric Cancer. 2023;26(1):69–81.
Acknowledgements
The Graphical Abstracts in this article is drawn by Figdraw.
Funding
This study was financially supported by the National Natural Science Foundation of China (No.82173842), the Henan Province Science and Technology Research Project (No. 222102310414) and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
W.Z., L.Z. and Q.G. designed the research. L.Z., Q.G., and Y.Z. analyzed the data. Q.G., Y.Z. and H.N. performed the research. Q.G. and L.Z. wrote the paper. Q.G., M.N., S.X., and W.Z. contributed the new reagents or analytic tools. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The collection of human tissues has been approved by the Medical Institutional Review Boards in Shanghai following the ethical guidelines. Title of the approved project is Development and application of gastric cancer tissue chip products. The corresponding approval certificate number is SHYJS-CP-1807020. All animal experiments were approved by the Ethics Committee for Animal Experimentation of China Pharmaceutical University, Title of the approved project is The SIX2/PFN2 feedback loop promotes the stemness of gastric cancer cells (2022-05-040).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Q., Zhou, Y., Ni, H. et al. The SIX2/PFN2 feedback loop promotes the stemness of gastric cancer cells. J Transl Med 22, 832 (2024). https://doi.org/10.1186/s12967-024-05618-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05618-5