
Abstract
Background
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS), characterized by neuroinflammation, demyelination, and neurodegeneration. Considering the increasing prevalence among young adults worldwide and the disabling phenotype of the disease, a deeper understanding of the complexity of the disease pathogenesis is needed to ultimately improve diagnosis and personalize treatment opportunities. Recent findings suggest that bioactive lipid mediators (LM) derived from ω-3/-6 polyunsaturated fatty acids (PUFA), also termed eicosanoids, may contribute to MS pathogenesis. For example, disturbances in LM profiles and especially those derived from the ω-6 PUFA arachidonic acid (AA) have been reported in people with MS (PwMS), where they may contribute to the chronicity of neuroinflammatory processes. Moreover, we have previously shown that certain AA-derived LMs also associated with neurodegenerative processes in PwMS, suggesting that AA-derived LMs are involved in more pathological events than solely neuroinflammation. Yet, to date, a comprehensive overview of the contribution of these LMs to MS-associated pathological processes remains elusive.
Main body
This review summarizes and critically evaluates the current body of literature on the eicosanoid biosynthetic pathway and its contribution to key pathological hallmarks of MS during different disease stages. Various parts of the eicosanoid pathway are highlighted, namely, the prostanoid, leukotriene, and hydroxyeicosatetraenoic acids (HETEs) biochemical routes that include specific enzymes of the cyclooxygenases (COXs) and lipoxygenases (LOX) families. In addition, cellular sources of LMs and their potential target cells based on receptor expression profiles will be discussed in the context of MS. Finally, we propose novel therapeutic approaches based on eicosanoid pathway and/or receptor modulation to ultimately target chronic neuroinflammation, demyelination and neurodegeneration in MS.
Short conclusion
The eicosanoid pathway is intrinsically linked to specific aspects of MS pathogenesis. Therefore, we propose that novel intervention strategies, with the aim of accurately modulating the eicosanoid pathway towards the biosynthesis of beneficial LMs, can potentially contribute to more patient- and MS subtype-specific treatment opportunities to combat MS.
Graphical Abstract

Similar content being viewed by others


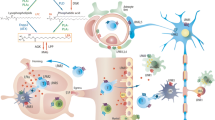
Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS) with an increasing global incidence among young adults (between the age of 20–40 years). In 2020, 35.9 out of 100,000 people were estimated to have MS, which corresponds to 2.8 million people with MS (PwMS) worldwide [1]. Central to the disease is the targeting of the lipid-rich myelin sheath by the immune system, giving rise to its breakdown, a process known as demyelination. As the primary functions of the myelin sheath imply providing nutrients and protection to neurons as well as acting as an electrical insulator for proper neuronal signalling, demyelination often leads to axonal damage and neurodegeneration [2]. In MS, this neurodegeneration can be translated into clinical symptoms, such as vision and cognitive impairments, or physical disabilities (e.g., balance or movement), depending on the location and size of these insults within the CNS [3].
Traditionally, MS is believed to start with a primary neuroinflammatory phase characterized by the infiltration of T- and B lymphocytes into the CNS, which, subsequently, attracts peripheral monocytes [3]. Both infiltrating leukocytes as well as locally activated glial cells create a pro-inflammatory environment within the CNS, through the secretion of pro-inflammatory cytokines (e.g., interferon γ (IFNγ), interleukin-1 (IL-1) and tumour necrosis factor (TNF)), neurotoxic factors (reactive oxygen species (ROS)) and matrix metalloproteinases (MMPs) [4]. In turn, this pro-inflammatory environment further triggers glial activation, demyelination and axonal damage [5] (Fig. 1). Moreover, leukocyte infiltration into the CNS is accompanied by a transient disruption of the blood–brain barrier (BBB), a selective barrier comprised of brain endothelial cells, pericytes, and astrocytes that, under healthy conditions, restricts the passage of pathogens, large hydrophilic molecules, and peripheral immune cells into the CNS [6]. BBB disruption and the associated neuroinflammation, besides demyelination and neurodegeneration, therefore, form critical hallmarks of MS pathogenesis, leading to MS lesion formation and disease progression [7, 8]. Nonetheless, large individual differences in the progression of MS among PwMS exist, which can be attributed to numerous genetic and environmental factors [9,10,11,12,13]. For instance, individuals carrying the HLA–DRB1*15:01 allele, individuals who have had a previous infection with the Epstein–Barr virus, and smokers display a higher risk of developing MS. As a result, MS is an extremely heterogeneous and complex disease with an unknown aetiology.

Simplified overview of the traditional perspective on multiple sclerosis pathogenesis. Pathogenesis is mediated by an accumulation and activation of T/B lymphocytes and monocyte-derived macrophages within the CNS (1–2a) and activated microglia and astrocytes (2b, 3b). This leads to the release of a plethora of inflammatory mediators (3a, 3c), targeting myelin sheats and oligodendrocytes surrounding axons (4–5)
Clinical diagnosis of MS has been roughly divided into three different subtypes: (I) relapsing–remitting MS (RRMS), (II) primary progressive MS (PPMS), and (III) secondary progressive MS (SPMS) [14]. Overall, 85% of PwMS display the RRMS subtype that generally depicts the first phase, defined by recurrent relapses lasting at least a day, followed by partial or full recovery (remission). Here, neuroinflammation, mainly driven by the CNS-infiltrating T- and B-lymphocytes, and demyelination driven by monocyte-derived macrophages are the most common pathological hallmarks. The majority of people with RRMS (PwRRMS), however, gradually develop a more progressive variant of MS, termed SPMS. In this subtype, neurodegeneration becomes more prominent and the innate immune system is suggested to be the main driver of progression (e.g., infiltrating peripheral monocyte-derived macrophages and CNS-resident microglia). Around 15% of PwMS display this progressive course from disease onset and are classified as people with PPMS (PwPPMS). Diagnosis of these subtypes is based on a combination of clinical, biochemical and radiological features, including biomarkers, such as neurofilament light (Nfl), symptom evaluation, and location of demyelinating lesions, as measured by magnetic resonance imaging (MRI). Nonetheless, relapses may hide disease progression during early MS stages and specific disease outcomes, such as progression independent of relapse activity (PIRA), might be more clinically useful instead [15]. Considering the great heterogeneity in disease onset, course, progression and dependence on lesion location, no definitive test for subtype-specific MS diagnosis is currently available.
A better understanding of MS pathogenesis is, therefore, crucial, and current research focuses on biomarker discovery that may enable a more accurate disease course prediction as well as a better distinction between the different MS subtypes to optimize and personalize treatments. Part of this research is focussed on the neuroinflammatory and neurodegenerative components of MS and includes biomarkers, such as Nfl, glial fibrillary acidic protein (GFAP), and IL-1β [16,17,18]. Recently, bioactive lipid mediators (LMs) derived from ω-3/-6 poly-unsaturated fatty acids (PUFA) have gained interest due to their potential role in MS progression, as they are effective regulators of inflammation, both during onset as well as during inflammation–resolution [19,20,21]. In addition, derivatives of the ω-6 PUFA arachidonic acid (AA) or eicosanoids [e.g., prostaglandin E2 (PGE2) and 15-hydroxyeicosatetraenoic acid (15-HETE)], are found to be elevated in PwMS and correlate with clinical parameters, such as the expanded disability status scale (EDSS), Nfl and MRI parameters [19, 20, 22, 23]. These observations suggest that the eicosanoid pathway may fulfil a broader role in MS than solely driving neuroinflammation. This review, therefore, summarizes and discusses the current knowledge on the eicosanoid biosynthetic pathway and its contribution to key pathological hallmarks of MS during different disease stages with a specific focus on AA derivatives in MS pathogenesis.
The group IVA cytosolic phospholipase A2 (cPLA2-α)-dependent lipid mediator pathway with arachidonic acid (AA) as its substrate
AA is an ω-6 PUFA abundantly present in the CNS, liver and muscles, where it is stored in glycerophospholipids within cellular membranes. Upon cellular stimulation, calcium-dependent cPLA2-α is phosphorylated and activated by members of the mitogen-activated protein (MAP) kinase pathway, which promotes its translocation to the Golgi, endoplasmic reticulum (ER) and/or nuclear envelope [24,25,26,27,28]. Here, it catalyses the hydrolysis of AA on the sn-2 position of glycerophospholipids, which triggers the release of AA from the cellular membranes to make it accessible for cytochrome P450 (CYP), cyclooxygenase (COX) and lipoxygenase (LOX) enzymes that can reside at these membranes (Fig. 2) [28, 29]. These enzymes can convert AA further into a plethora of downstream LMs, all having an unique set of biological actions, often defined by interactions with LM-specific receptors (Tables 1, 2).

Schematic overview of the molecular signalling that leads to AA hydrolysis from glycerophospholipids in cellular membranes of the Golgi system and the nuclear envelope. Environmental stimuli (e.g., pro-inflammatory cytokines) that either activate the MAPK-signalling pathway (1) or raise intracellular Ca2+ levels (2) result in the phosphorylation of group IVA cytosolic phospholipase A2 (cPLA2-α) (3). This results in the translocation of cPLA2-α towards the cellular membranes of the Golgi system and the nuclear envelope, where it interacts with esterified AA incorporated in glycerophospholipids, which will make AA accessible for further metabolism (4). Enzymes with oxidative properties, such as cyclooxygenases (COXs) and lipoxygenases (LOXs), that reside in these cellular membranes can interact with this hydrolyzed AA and convert it into a variety of bioactive LMs (5). *for simplicity, only MAPK without further upstream signalling is shown
Cyclooxygenase (COX)-derived prostanoids in MS
The most extensively investigated enzymes of the eicosanoid pathway are COX-1 and COX-2, responsible for the biosynthesis of prostanoids (e.g., thromboxanes, prostaglandins and prostacyclin) through the formation of meta-stable prostaglandin G2 (PGG2). Where COX-1 is constitutively expressed and thought to have cytoprotective and homeostatic functions, COX-2 expression is tissue-specific, with relatively high expression levels in tissue, such as the kidney, heart and brain, which can be increased in response to growth factors and pro-inflammatory stimuli (e.g., TNF) [30]. Importantly, several studies have shown that COX-2 expression is significantly elevated in PwMS and experimental murine models of MS (i.e., Theiler’s murine encephalomyelitis virus-induced demyelinating disease), specifically in microglia and macrophages [31,32,33,34]. These findings have made COX-2 a prominent target in the context of MS pathology, where it is currently considered to mediate both beneficial as well as detrimental processes depending on the biosynthesis of its downstream LMs.
COX-2 contains two catalytic properties through which it oxidizes the liberated AA, generating the short-living intermediate PGG2 (Fig. 3). PGG2 is rapidly converted by peroxidase activity into prostaglandin H2 (PGH2), which forms the central precursor for the synthesis of all other downstream prostanoids, including prostaglandin D2 and E2 (PGD2 and PGE2, respectively), prostacyclin (PGI2) and thromboxane A2 (TxA2). Interestingly, inhibition of COX-2 in the experimental autoimmune encephalitis (EAE) murine model for MS was found to reduce clinical signs by preventing the proliferation of autoreactive T lymphocytes and the production of pro-inflammatory cytokines [35]. However, this approach should be treated with caution, as long-term COX-2 inhibition may translate into severe vascular side effects, such as non-fatal myocardial infarction, non-fatal stroke, or vascular death [36]. In addition, as the synthesis of anti-inflammatory prostanoids, such as 15-deoxy-(12,14)-PGJ2 (15d-PGJ2), might be affected upon COX-2 inhibition, selective blocking of enzymatic targets further downstream in the prostanoid pathway might be more valuable to combat the neuroinflammatory component of MS to avoid severe side-effects.

Schematic overview of the variety of COX/LOX associated LMs biosynthesized from the hydrolyzed AA
Potential role of PGE 2 –EP2/EP4 signalling and their dual role during neuroinflammation
Examples of such downstream enzymatic targets are microsomal prostaglandin E synthase-1 (mPGES-1) and membrane-bound prostaglandin E synthase-2 (mPGES-2), responsible for the biosynthesis of PGE2 from PGH2 (Fig. 3; Table 1). In MS, PGE2 has been linked to the chronicity of neuroinflammation, where several studies have found increased PGE2 levels in both serum and CSF of both PwRRMS and people with progressive MS (PwPMS) [20, 23, 37]. Moreover, mPGES-1 was found to be expressed by macrophages in demyelinating lesions, yet a direct link between the increased mPGES-1 expression and PGE2 levels has only been shown in EAE mice [38, 39]. Here, the role of mPGES-1-mediated PGE2 in disease development seems to be substantial, as mPGES-1 knock-out (KO) mice showed decreased neuroinflammation and demyelination during EAE, that corresponded with lower PGE2 levels in their spinal cords [38]. Taken together, these findings suggest that PGE2 synthesis is a pivotal contributor to chronic neuroinflammation in MS and that therapeutically targeting of mPGES-1, instead of the more upstream COX-2, may help to attenuate this pathogenic event [40].
Despite these pro-inflammatory characteristics, PGE2 should be considered as a versatile LM, depending both on the timing, its concentration and the receptor it binds to. Four receptors [PGE2 receptor 1–4 (EP1–EP4)] have been identified to date, through which PGE2 can mediate a variety of cellular processes [41] (Table 1). In general, EP1/EP3 receptors promote vasoconstriction and hypertension, whereas EP2/EP4 receptors, in contrast, promote vasodilation and hypotension [42,43,44,45]. EP1 and EP3 have been studied in MS or experimental animal models, but are considered to be of little functional relevance and have not been examined in great detail in this context. Nevertheless, EP1 potentially contributes to the disruption of the BBB, as blocking or genetically deleting EP1 in an ischemic murine model led to reduced BBB permeability, presumably through the downregulation of MMP-9, which in MS is found to be elevated in serum of PwRRMS [46,47,48]. MMPs are enzymes that are involved in BBB breakdown, potentially due to the downregulation of endothelial tight junctions [49]. This suggests that an increase in MMP-9 serum levels facilitates immune cell extravasation into the CNS, potentially in an EP1-dependent manner. Indeed, EP1 may play a larger role in MS development than initially considered, as EP1 gene expression correlates with clinical scores of EAE mice [39]. Notably, PGE2–EP3 signalling does not seem to contribute to MS pathology, as MS-related murine models have shown that EP3 is not present in MS lesions, no correlations are found between EP3 mRNA expression and EAE severity and EAE clinical signs are unaffected in EP3 KO EAE mice in vivo [39, 50].
In contrast, EP2 and EP4 have been associated with MS pathology as both receptors are involved in the regulation of the adaptive and innate immune system (Fig. 4; Table 1) [41]. Both receptors are, for example, expressed on T-helper lymphocytes as well as on microglia and macrophages, whereas EP2 is also expressed on oligodendrocytes (OLs) [51,52,53]. Of these immune cells, T-helper lymphocyte type 1 (Th1) and 17 (Th17) are suggested to be the main drivers of MS pathogenesis, as they accumulate in the CNS and actively reinforce a pro-inflammatory environment [54, 55]. Especially EP2 may promote neuroinflammation as its expression is significantly induced on Th17 lymphocytes of untreated PwRRMS as compared to healthy subjects [51]. In turn, treatment of patient-derived Th17 lymphocytes with the EP2-specific agonist butaprost resulted in increased transcription of IFNγ and granulocyte–macrophage colony-stimulating factor (GM-CSF), thus amplifying the inflammatory response, while EP2 overexpression on Th17 lymphocytes of healthy subjects led to similar results [51].

PGE2–EP2 and PGE2–EP4 signalling in Th17 lymphocytes (left) and microglia/macrophages (right). PGE2 can promote the accumulation of Th17 lymphocytes in the CNS by signalling through its EP2 receptor and promotes the secretion of pro-inflammatory factors such as IFN-y and GM-CSF by signalling through its EP4 receptor. However, in monocyte-derived macrophages and microglia EP2 signalling leads to their polarization towards a pro-inflammatory phenotype, while, in contrast, EP4 signalling in these cells results in the suppression of this pro-inflammatory phenotype
Signalling of PGE2 through EP4, on the other hand, is thought to associate with the accumulation of T-helper lymphocytes through increased proliferation in the CNS of PwMS [56]. This is substantiated by decreased numbers of infiltrated CD4+ T lymphocytes, monocytes and macrophages in the spinal cord of EAE mice with an EP4 deficiency, where it normally may promote Th1 lymphocyte differentiation and Th17 lymphocyte expansion in an IL-23 and IL-1ß-dependent manner [57, 58]. Furthermore, EP4 signalling may also contribute to BBB disruption, as T lymphocytes of EP4-deficient EAE mice show decreased levels of MMP-9 [59]. Taken together, both EP2 and EP4 likely contribute to T lymphocyte-associated detrimental events during early MS pathogenesis, thereby representing promising therapeutic targets for intervention.
During later stages of MS, the innate immune system and especially CNS-infiltrating monocyte-derived macrophages and CNS-resident microglia may become the main drivers of pathology by creating a chronically inflamed environment in the vicinity of MS lesions. PGE2 is also proposed to be involved in this process through EP2/EP4 receptor interactions as it can induce a pro-inflammatory phenotype in both macrophages and microglia through EP2 and, additionally, may promote OLs apoptosis via this signalling pathway [52, 60, 61]. Furthermore, conditional knock-out of EP2 in myeloid cells of lipopolysaccharide (LPS)-challenged mice resulted in reduced hippocampal and cortical IL-6, TNF, IL-1β and inducible nitric oxide synthase (iNOS, a macrophage activation marker) mRNA levels, further demonstrating the pro-inflammatory properties of EP2 signalling [61]. PGE2–EP2 signalling led to increased COX-2 expression and induction of apoptosis in primary rat microglia, which could be prevented with an EP2 antagonist [62]. Intriguingly, EP2-deficient EAE mice did not show an attenuation of EAE development, suggesting that the neuroinflammatory role of EP2 signalling in MS is not essential for disease onset or severity [50].
While both EP2 and EP4 signalling leads to an elevation of intracellular cyclic adenosine monophosphate (cAMP) levels, each receptor-dependent signalling cascade showed differential cAMP production profiles, which were also described to be dose-dependent [63]. In addition, the research on the effects of EP4 signalling in macrophages and microglia highlights a more nuanced role compared to the inflammatory role proposed for EP2 (Fig. 4; Table 1). The usage of a selective EP4 agonist on murine microglial cells in vitro attenuated an LPS-mediated pro-inflammatory response and induced transcription of the anti-inflammatory cytokine IL-10 [53]. On the other hand, conditional deletion of EP4 in myeloid cells of mice challenged with LPS led to increased neural COX-2, TNF, IL-6, and IL-1ß expression and elevated F2-isoprostanes levels, a lipid peroxidation marker [64]. This supports the idea that EP4 may have pro-resolving effects in macrophages and microglia, that could occur either by preventing their polarization towards a pro-inflammatory phenotype or skewing it towards a more pro-resolving phenotype, necessary for tissue recovery. A time-dependent factor might be involved, as EP4 expression was found to decrease over time in mouse microglia treated with LPS for 24 h, whereas an inverse effect was seen for EP2 [53]. To this end, cell-type-specific effects of PGE2 may take place during the different stages of MS pathology, which may explain why solely silencing EP2 may not yield significant beneficial effects during EAE onset, as EP4 signalling on Th17 lymphocytes can still contribute to the pro-inflammatory CNS environment at this stage. Furthermore, antagonizing the EP4 receptor at different timepoints during EAE development resulted in varying degrees of disease severity [50], further substantiating the complexity and temporal impact of PGE2 signalling during the different MS disease phases. Additional insights may be obtained by investigating the effects of an EP4 KO in microglia and macrophages during disease onset as this may hamper EAE development.
The (anti-)inflammatory or demyelinating potential of PGD 2 and its metabolite 15d-PGJ 2
Besides PGE2, other inflammation-mediating prostanoids are identified in PwMS, such as PGD2 and its non-enzymatically formed metabolite 15d-PGJ2 (Fig. 3). PGD2 itself is biosynthesized from PGH2 by two distinct synthases; cytosolic hematopoietic PGD synthase (H-PGDS) and the lipocalin-type PGD synthase (L-PGDS) located on the rough ER and nuclear membrane [65]. In plasma of both PwRRMS and people with SPMS (PwSPMS), PGD2 levels are found to be elevated [20], where it is proposed to have both anti- and pro-inflammatory properties depending on the G protein-coupled receptor (GPCR) it interacts with: the D prostanoid receptor (DP1) or the chemoattractant receptor–homologous molecule on Th2 cells (CRTH2, also known as DP2) (Table 1). PGD2–DP1 signalling is considered to be anti-inflammatory as it inhibits T lymphocyte and basophil migration/activation, whereas PGD2–DP2 signalling can promote T lymphocyte migration and thus can be considered pro-inflammatory [66,67,68]. Intriguingly, PGD2 may even play an indirect role in demyelination through the G protein-coupled F prostanoid receptor FP, which will be addressed more extensively in the PGF2α section [69, 70]. Yet, evidence for a direct contribution of PGD2 to MS pathogenesis is limited.
The non-enzymatically formed PGD2 metabolite 15d-PGJ2, however, is known to suppress astrocytic and microglial-mediated production of pro-inflammatory cytokines, such as TNF and IL-1β [71]. It can exert these effects by binding to the nuclear receptor peroxisome proliferator-activated receptor γ (PPAR-γ), which inhibits the inflammation-promoting transcription factors nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 1 (STAT-1) [72,73,74]. Next to suppressing pro-inflammatory cytokine production, 15d-PGJ2 may regulate macrophage migration, proliferation, and activation in vitro and repress overall EAE development by decreasing toll-like receptor 4 and 9 expression on T lymphocytes in vivo, thereby limiting antigen presentation [75, 76]. However, 15d-PGJ2 treatment of undifferentiated mouse oligodendrocyte precursor cells was found to induce apoptosis, suggesting that it may also contain neurotoxic properties [77]. Overall, additional studies are necessary to determine whether PGD2 plays a role in MS-associated neuroinflammation and/or demyelination. Nevertheless, its derivative 15d-PGJ2 shows several PPAR-γ-mediated anti-inflammatory properties that can be exploited to combat chronic neuroinflammation, although caution is required regarding its potential neurotoxic effects.
The PGF 2α receptor, FP, mediating demyelination
Another prostanoid is PGF2α., that can be biosynthesized either from PGH2 or PGD2 by the aldo–keto reductase family 1 member C3 (AKR1C3) or from PGE2 by the 9-ketoreductase (AKR1C1 and AKR1C2), and exerts its effect through the receptors FPA or FPB (Fig. 3; Table 1) [78]. In PwSPMS, peripheral PGF2α levels have been found to be increased, yet, little is known about the specific function of PGF2α in MS [20]. One study showed that a FP antagonist was able to attenuate demyelination of the corpus callosum in the demyelination-inducing cuprizone murine model for MS [70]. Here, a decrease in TNF expression in the corpus callosum was accompanied by a reduction of glial activation and an increase in motor function, suggesting that PGF2α–FP signalling enhances glial-mediated demyelination. However, such a direct effect by PGF2α still needs to be addressed and, as briefly mentioned before, this effect of FP signalling could also be mediated by PGD2 and PGE2, as these prostanoids are also elevated in PwMS and can bind to the FP receptor, albeit with a lower affinity than PGF2α (Ki = 3.2 nM for PGF2α, 6.7 nM for PGD2 and 116 nM for PGE2 in recombinant HEK293 cells) [69, 79].
Prostacyclin (PGI 2 ) synthesis and its potential role in neuroinflammation and demyelination
While it has not been studied extensively in the context of MS, the highly unstable prostacyclin (PGI2) has some beneficial, potentially disease-altering properties worth exploring. PGI2 is biosynthesized from PGH2 by the constitutively expressed enzyme prostaglandin I2 synthase (PTGIS, Fig. 3), present in the cytosol of neurons, microglia, and OLs [80]. Once formed, PGI2 may exert contrasting, cell-type-specific effects on neuroinflammation or demyelination through the prostacyclin (IP) receptor. For example, stimulating CD4+ T lymphocytes with iloprost, a stable PGI2-analog, was found to induce an IP-dependent Th17 lymphocyte differentiation and IL-17 production in vitro [81]. In contrast, iloprost treatment was found to prevent pericyte loss induced by lysophosphatidylcholine (LPC) treatment in an in vitro BBB model and diminished LPC-induced demyelination in vivo [82]. In addition, IP-deficiency in EAE mice was found to reduce the infiltration of mononuclear cells into the spinal cord and delayed EAE development, while it did not affect disease severity, suggesting that PGI2–IP signalling might be involved in the timing of disease onset but not in overall disease development [83] (Table 1). Finally, PGI2 is mostly known to have antithrombotic properties, by counteracting the vasoconstrictor and platelet aggregation-promoting role of thromboxane A2 (TxA2). This interplay between PGI2 and TxA2 is essential for a proper cardiovascular homeostasis and should, therefore, be taken into account when considering PGI2-associated therapies [84].
Thromboxane A 2 , platelet activation and aggregation
As mentioned above, TxA2 is a vasoconstrictor that can promote platelet aggregation [84]. It is biosynthesized from PGH2 by the thromboxane-A synthase (TxAS), in a wide variety of cells but especially in platelets, and interacts mainly with the thromboxane prostanoid (TP) receptor (Fig. 3; Table 1). Similar to other prostanoids, TxA2 is chemically unstable and degrades quickly through hydrolysis into its inactive, but stable metabolite thromboxane B2 (TxB2), which is increased in PwMS [20]. Although no conclusive role for TxA2 has been defined in MS yet, high platelet activation is seen in PwMS and a direct interaction between platelet aggregation and immunity has been observed consistently [85,86,87,88]. In EAE mice, a time-dependent depletion of platelets during disease onset was found to prevent T lymphocyte accumulation in the spinal cord and led to diminished disease and lesion development [87]. More specifically, platelet-activating factors reinforced Th1/Th17 lymphocyte differentiation in early MS and EAE pathogenesis, whereas at later stages of MS, the formation of platelet aggregates and T lymphocytes were associated with diminished T lymphocyte activation [88]. In addition, a low-dose administration of acetylsalicylic acid (ASA, i.e., aspirin), to inhibit platelet activation and aggregation, decreased TxA2 and alleviated clinical symptoms of EAE [89]. Still, a direct role of TxA2 in these processes in MS remains uncertain, as its instability limits the timeframe for proper detection and the ability to investigate whether TxA2 can exert the aforementioned effects in MS pathogenesis before being degraded into TxB2.Furthermore, ASA irreversibly acetylates COX enzymes, leading to the complete inactivation of the downstream prostanoid biosynthesis and not solely to that of TxA2. Instead, the TP receptor might represent a more interesting target, as isoprostanes, which are free radical-catalysed peroxidation products of AA (e.g., 8-iso-PGF2α), are known to promote platelet activation via the TP receptor and have been found to be elevated in the CSF of PwMS as compared to healthy controls [90]. Moreover, TP positively regulates COX-2 expression in endothelial cells and results in increased levels of PGH2, thus potentially fuelling the biosynthesis of other prostanoids [91]. Taken together, platelet activation and aggregation may contribute to early MS by reinforcing Th1/Th17 differentiation, although other factors than TxA2 might be responsible for this effect via the TP receptor.
To summarize, AA-derived prostanoids encompass several LMs with potent inflammatory or demyelinating properties, which seem to be MS-stage-specific and depend not only on the associated receptor but also on the corresponding cell type. This makes the role of this LM family in MS highly complex, but also provides interesting therapeutic targets for personalized and MS-stage-specific treatment. For example, specific targeting of downstream synthases or receptors, such as mPGES-1 or EP2, might provide more optimal disease-stage-specific therapeutic treatments with high efficacy. However, as most LMs are extremely unstable and versatile in a cell-, receptor- and perhaps even time- and concentration-dependent manner, extensive research is warranted to further understand their exact role in the context of MS pathology.
Lipoxygenase (LOX)-associated AA-derivatives in MS
Besides the COX-mediated biosynthesis of eicosanoids, an increasing amount of research is focusing on other enzymes with oxygenation properties, such as the lipoxygenases (LOXs), which are thought to be critically involved in microglia-mediated neuroinflammation [92]. A potential reason for this association may involve 5-LOX, which, together with 5-LOX activating protein (FLAP), forms the foundation for the biosynthesis of pro-inflammatory leukotrienes (LTs) (Fig. 3) and is consistently overexpressed in gene expression profiles of peripheral blood mononuclear cells (PBMC)s in PwRRMS [93, 94]. 5-LOX resides in the cytoplasm or nucleoplasm and translocates to the nuclear envelope following stimuli such as stress signals that either increase intracellular calcium levels or promote 5-LOX phosphorylation [95]. At the nuclear envelope, 5-LOX forms an enzymatic complex with FLAP that facilitates the transfer of free AA to 5-LOX. 5-LOX then catalyses the oxygenation of AA, forming 5(S)-HpETE, which, in turn, is rapidly converted into either 5-HETE or into the unstable intermediate leukotriene A4 (LTA4) by an additional enzymatic cycle. Of these products LTA4 is the most interesting in the context of MS as it forms the central precursor for the biosynthesis of other LTs.
Controversy exists regarding the role of 5-LOX in the context of MS. A protective role, for instance, was attributed to 5-LOX during MS pathogenesis based on the observation that EAE progression was exacerbated in 5-LOX-deficient EAE mice [96]. However, in cuprizone mice 5-LOX inhibition with MK-886 attenuated neuroinflammation, motor dysfunction and axonal damage, while it did not reduce the cuprizone-associated demyelination [97]. In addition, administration of flavocoxid, a dual COX-2/5-LOX inhibitor, attenuated EAE pathogenesis presumably by promoting the transition of inflamed microglia towards an anti-inflammatory phenotype [98]. Moreover, in both PwMS and EAE mice, 5-LOX gene expression was upregulated in MS lesions, which was found to be mainly expressed by macrophages in these areas as shown with immunohistochemical analysis [99]. Studies with human monocyte-derived macrophages showed that inhibition of FLAP reduced the biosynthesis of pro-inflammatory LTs, such as leukotriene B4 (LTB4) [100, 101]. In addition, FLAP inhibition has been applied in in vivo models for several inflammatory diseases, including asthma and atherosclerosis, with some inhibitors successfully being applied in clinical trials, showing the potential of this targeting strategy in the context of MS [102, 103].
A possible explanation for these contrasting effects on both inflammation and EAE disease progression may be attributed to the wide range of metabolites 5-LOX can synthesize, similar to the COX enzymes, as 5-LOX is also involved in the biosynthesis of the pro-resolving lipoxins and resolvins [21]. Nonetheless, in human leukocytes these pro-resolving LMs are present in low quantities and often cannot be detected, in contrast to the abundant release of LTs under inflammatory conditions [104]. Of interest, in contrast to blocking 5-LOX, targeting FLAP in macrophages can efficiently suppress LT formation without reducing resolvin levels [100, 101]. Therapeutic targeting of downstream LT-associated synthases might be a more direct approach to steer the direction of LM biosynthesis towards more beneficial LMs during specific MS disease stages and targeting of the LTB4 receptor 1 (BLT1) may provide such a tool.
LTB 4 : a potent chemoattractant for migrating leukocytes towards the CNS
LTB4 is the most common leukotriene implicated in MS pathogenesis and is biosynthesized from LTA4 by the LTA4 hydrolase (LTA4H) (Fig. 3). It exerts its effect mainly via two GPCRs called BLT1 and BLT2, and through PPAR-α, through which it promotes chemotaxis of lymphocytes, T lymphocyte activation, and ROS production (Table 2) [105,106,107,108]. In MS, these chemoattractant properties can mediate the migration of Th17 lymphocytes into the CNS, as BLT1 is not only highly expressed on Th17 lymphocytes, but these cells also migrate along an LTB4-dependent gradient in vitro (Fig. 5) [108]. In addition, LTB4 levels are found to be almost twice as high in the CSF of people with clinically active MS when compared to healthy controls [109]. This suggests that the CNS infiltration of lymphocytes during MS depends to a certain extent on signalling through the LTB4–BLT1 axis. In line with this, BLT1 deficient mice show reduced CNS infiltration of T lymphocytes, neutrophils and peripheral monocyte-derived macrophages during EAE [110]. This coincided with a delay in EAE onset in combination with reduced disease severity and diminished production of pro-inflammatory cytokines, such as IFNγ, TNF, IL-6 and IL-17, stressing the importance of the LTB4–BLT1 axis in EAE pathogenesis. Intriguingly, migration of Th17 lymphocytes towards a high LTB4 concentration also diminished after treatment of EAE mice with montelukast, a type 1 cysteinyl leukotriene receptor (CysLTR1)-specific antagonist, indicating that the LTB4-associated chemotaxis of Th17 lymphocytes may not solely depend on the BLT1 receptor [111].

Hypothetically, LTB4/LTD4 can act as chemo-attractants through either the BLT-1 or CysLTR1 receptor on Th-17 lymphocytes thereby mediating their influx across the disrupted BBB towards high LTB4/LTD4 levels in the CNS of MS patients
LTB4–BLT2 signaling, on the other hand, might be less involved in mediating neuroinflammation, as LTB4 is known to have a much lower affinity for BLT2 than for BLT1 [112]. In addition, other AA derivatives, such as 12-HHTrE, a byproduct of TxA2, and 15-HETE, which will be addressed later in detail, display a greater affinity for BLT2 and compete with LTB4 for BLT2 binding, but not BLT1 [112, 113]. As a result, BLT2 may mediate distinct biological as well as pathophysiological processes compared to BLT1, such as epidermal wound healing [114]. Finally, LTB4 signaling via PPAR-α could exert anti-inflammatory effects, as signaling via this route was found to play a role in macrophage apoptosis in vitro [115, 116]. However, no clear link with MS pathology can be drawn for LTB4–PPAR-α signaling considering that EAE progression and severity in PPAR-α KO EAE mice was similar to that of WT EAE mice, and PPAR-α protein levels, unlike PPAR-γ, are unaltered in the CSF of PwMS [117, 118]. Together, LTB4 potentially influences MS pathogenesis via the BLT1 receptor, through which it can promote the chemotaxis of Th17 lymphocytes into the CNS, where they fuel a pro-inflammatory environment. Specific BLT1 blocking or preventing LTB4 biosynthesis may, therefore, be considered as potent therapeutic strategies, especially during the early stages of MS pathology, which are dominated by profound lymphocyte CNS infiltration. Finally, while most studies focus on the role of LTB4 on lymphocyte CNS infiltration which indirectly leads to demyelination and neurodegeneration, a local, more direct contribution of LTB4 to these pathological events is also plausible given its pro-inflammatory nature, as suggested by the autocrine effects of LTB4 on microglial activation through the BLT1 receptor [119].
Linking cysteinyl-leukotrienes to lymphocyte infiltration
Besides LTB4, LTA4 can also be converted into cysteinyl leukotrienes (CysLTs), which comprises LTC4, LTD4 and LTE4 (Fig. 3) [120]. This LT cascade starts with the conversion of LTA4 into LTC4 a process that is catalysed by the LTC4 synthase in conjugation with glutathione. LTC4 is subsequently secreted into the extracellular space via the multi-drug resistance protein 1 (MRP-1), where it can be further converted into LTD4 and LTE4 by extracellular synthases, such as γ-glutamyl transferase and LTD4 dipeptidase-1 and -2 (Fig. 3). All CysLTs exert their actions via one of the two GPCRs CysLTR1 or CysLTR2, where LTD4 has a high affinity for CysLTR1 and both LTC4 and LTE4 for CysLTR2 (Table 2) [120]. Of these LMs, LTD4 is considered to be the most relevant for MS due to its high affinity for CysLTR1, the receptor associated with Th17 lymphocyte migration and EAE disease severity as previously described [111]. Selective blocking of CysLTR1 with Montelukast prevented this migration towards high LTB4 concentrations, but also towards high LTD4 concentrations in vitro [121, 122].
The importance of CysLTR1-signaling in MS is gaining more interest as CysLTR1 is found to be elevated in the blood of PwMS, with an increase on CD4+ T lymphocytes, but also on astrocytes and microglia in MS lesions compared to normal appearing white matter in post-mortem brain tissue [123]. In addition, more CysLTR1-positive Th-lymphocytes were found inside MS lesions of these PwMS as compared to normal appearing white matter. As elevated levels of both LTB4 and LTD4 have been observed in the CSF of clinically active PwRRMS, it is tempting to speculate that these LTs might be related to the increased number of Th-17 lymphocytes via CysLTR1 [109, 123]. Nevertheless, whether these findings are directly linked to one another remains to be addressed.
Hydroxyeicosatetraenoic acids (HETEs) in MS
A generally understudied LM subclass of AA-derivatives in the context of MS are the hydroxyeicosatetraenoic acids (HETEs), of which 5-, 11-, 12- and 15-HETE will be discussed (Fig. 3). In our previous work, we have shown that relative plasma levels of 5-HETE were increased in PwPMS as compared to PwRRMS and healthy controls and correlated positively with EDSS and serum Nfl levels, suggesting a link with disease progression [19]. However, no evidence for local 5-HETE levels in the CNS is available to further substantiate these initial findings. In addition, its oxidised metabolite 5-OxoETE, which is formed under oxidative stress by the microsomal enzyme 5-hydroxyeicosanoid dehydrogenase (5-HEDH), may be of importance in neuroinflammation [124]. 5-OxoETE can function as a chemoattractant for monocytes synergistically with chemokine (C–C motif) ligand (CCL) 2 and 7. It also acts as a potent activator of GM-CSF secretion by monocytes via the oxoeicosanoid receptor 1 (OXER1), also known as the GPR170 in humans (Table 2) [124,125,126]. Based on these properties, 5-OxoETE may promote monocyte migration towards lesions, where they can induce a pro-inflammatory environment by GM-CSF secretion. However, whether 5-HETE, 5-OxoETE or GPR170 signalling actually contribute to MS disease progression warrants further investigation.
11-HETE, lipid peroxidation and other pathological hallmarks of MS
One of the AA-metabolites of this subclass is 11-HETE, which can be biosynthesized either by COX-1/2, CYP, or non-enzymatically as byproduct of AA auto-oxidation [127, 128]. Mainly due to this auto-oxidative biosynthesis, 11-HETE is described as a marker for lipid peroxidation, a process known to occur in MS and thought to be related to inflammation, demyelination and neurodegeneration [129, 130]. However, no receptors for 11-HETE have been identified to date and 11-HETE itself has never been linked to MS before. Nevertheless, other lipids associated with lipid peroxidation have been studied in the context of MS, for example, increased levels of the classical oxidative low-density lipoproteins (ox-LDL) and high-density lipoproteins (ox-HDL), for instance, have been found in both the brain, plasma and CSF of PwMS, where their neurotoxic properties are considered to promote oxidative damage [131, 132]. Thus, it remains to be determined whether the AA-metabolite 11-HETE displays these neurotoxic properties as well.
12-HETE: potential promotor of neuroinflammation and ROS-mediated demyelination
The platelet-type 12-lipoxygenase (12-LOX or ALOX12) is the predominant producer of 12-HETE, and is found to be increased in the plasma of both PwPMS and PwRRMS in remission [20, 133]. In contrast, 12-LOX expression in PBMCs of PwRRMS during a relapse was found to be significantly lower than that of healthy subjects [20]. Despite the lack of a clear contribution of this LM to MS pathology, one can speculate that 12-HETE can have significant pathological implications in inflammatory diseases by promoting the chemotaxis of leukocytes and induction of oxidative stress through receptor interactions (i.e., GPR31 and BLT2) [114, 134,135,136]. 12-LOX-associated ROS production was, for example, found to induce apoptosis of mature OLs both in vitro and in vivo through an ERK1/2–12-LOX–ROS pathway, suggesting that 12-LOX, and presumably 12-HETE, may contribute to demyelination in MS [137]. Furthermore, 12-HETE can both stimulate and inhibit platelet aggregation thereby affecting T lymphocyte accumulation and differentiation, critical processes during early MS and EAE pathogenesis [87, 138]. Additional studies are, therefore, required to assess the contribution of 12-LOX and 12-HETE to MS pathogenesis with a specific focus on demyelination, mediated by the loss of OLs, and neuroinflammation.
15-HETE: a link between lipids and MS lesions?
The last LM of this subclass of monohydroxylated AA metabolites addressed here is called 15-HETE and can be biosynthesized from AA by several lipoxygenases, including 15-lipoxygenase-1 (15-LOX-1 or ALOX15) and 15-LOX-2 (or ALOX15B) (Fig. 3). In our recent work, relative plasma levels of 15-HETE, together with disease duration, Nfl and GFAP, were revealed as possible predictors of MS disability (as measured by EDSS) in PwPMS [19]. In addition, negative correlations were observed for 15-HETE with MRI parameters such as total brain and deep grey matter volumes in PwPMS and indicate a potential link between this AA-metabolite and neurodegenerative processes. These findings are in line with other studies, where increased levels of 15-HETE were observed in CSF and plasma of PwMS [20, 23, 37]. Aside from peripheral production, we hypothesize that local 15-HETE is biosynthesized primarily by 15-LOX-2 in demyelinated areas, potentially as a result of hypoxia or oxidative stress, as these stimuli have been linked to induction of 15-LOX-2 gene expression [131, 139,140,141,142]. 15-LOX-1, which also generates 15-HETE, may contribute to these elevated 15-HETE levels as well, since efferocytosis (referred to as the effective clearance of apoptotic cells) initiates ALOX15-1 expression in macrophages in vitro and has been observed in demyelinating areas [143, 144]. However, ALOX15-1 moderately converts AA into 15-HETE and may prefer the ω-6 PUFA linoleic acid (LA) as its substrate [145].
The potential function of 15-HETE in the CNS of PwMS is still relatively unknown, although 15-HETE can regulate several cellular processes via its two receptors: BLT2 and PPAR-γ (Table 2) [112, 146, 147]. Detrimental effects might be ascribed to 15-HETE as it is able to induce ROS production, apoptosis and macrophage foam cell formation, processes that are all observed in PwMS [147,148,149,150]. The latter process is, in particular, interesting in MS, as CNS-infiltrating macrophages are known to become oversaturated with oxidized lipids derived from the deteriorating myelin sheath, thereby turning into foam cells [151]. 15-HETE may promote this lipid uptake as it can induce membrane glycoprotein CD36 expression in these cells, a scavenger receptor that recognizes oxidized phospholipids and lipoproteins and mediates their internalization [152, 153]. Increased 15-LOX-2 expression is also found in atherosclerotic plaques known to contain foamy macrophages and silencing of ALOX15-B in an atherosclerotic mouse model resulted in decreased lipid accumulation and inflammatory markers in macrophages [150]. On the other hand, 15-HETE was also found to inhibit LTB4-induced chemotaxis of polymorphonuclear (PMNs) leukocytes in vitro and may promote a pro-resolving phenotype of macrophages/microglia via binding to the nuclear PPAR-γ receptor, thereby potentially promoting tissue recovery in the MS lesion vicinity [154, 155]. Additional studies, including in vivo studies, are, therefore, crucial to unravel the relevance between the elevated 15-HETE levels in PwPMS and MS-associated neuropathological events.
Summary and future perspectives
MS is a heterogeneous disease of the CNS, where current therapeutic strategies are mainly focussed on symptom management, predominantly by targeting specific parts of the immune system. Disease-modifying therapies including interferon beta, leukocyte migration inhibitors and monoclonal antibodies that result in lymphocyte depletion are currently on the market, which generally reduce relapse rate, but, unfortunately, have a limited effect on disease progression and are accompanied by unwanted side effects. Therefore, a high and unmet need remains to design therapeutic strategies that incorporate anti-inflammatory, remyelination-promoting and/or neuroprotective effects to slow down disease progression with as little side-effects as possible.
The involvement of AA-derived LMs in various pathogenic processes such as the MS-associated neuroinflammation, demyelination and neurodegeneration suggest that they may have versatile and disease-altering properties. By delving deeper into the role of LMs in MS, one may gain new insights into MS subtype-specific occurrences, ultimately leading to the development of subtype-specific intervention strategies and accompanying biomarkers. In this review, we substantiated the importance of targeting receptors associated with LMs or the downstream biosynthetic enzymes to dampen pathogenic processes and fuel the protective characteristics of LM biosynthesis in MS while minimizing the risk of side effects. This necessity is demonstrated by the example of COX-1/2 inhibitors such as nonsteroidal anti-inflammatory drugs that are used to alleviate flu-related symptoms, highlighting their beneficial and anti-inflammatory properties. However, these medications (i.e., COX-2-selective coxibs) have also been associated with severe cardiovascular-associated side effects, presumably due to imbalances in the prostanoid pathway, for example, between PGI2 and TXA2 (Fig. 3). COX-2 inhibition affects the metabolism of a broad range of LMs, and each of these could be detrimental or beneficial in the context of MS, such as PGE2, PGI2, PGD2 and its derivative 15d-PGJ2. Instead, targeting downstream biosynthetic enzymes in this pathway, such as mPGES-1 and 2, should be considered as improved intervention strategies as these enzymes comprise the final step for PGE2 biosynthesis and their inhibition may, therefore, have no negative impact on beneficial prostanoids. Similarly, targeting LTB4/LTD4 metabolism by modulation of enzymes involved in the leukotriene biosynthetic pathway, such as FLAP, LTA4H or LTC4H, may also be beneficial in MS as this may potentially disrupt the (LTB4-related) chemotaxis signal that drives T lymphocytes infiltration into the CNS of PwMS during early disease stages.
Moreover, cell-type-specific targeting of LM-associated receptors bears the potential to affect disease pathogenesis. For example, blocking the PGE2 receptors EP2 and EP4 and the LTB4/LTD4 receptors BLT-1 and CysLTR1 on Th-lymphocytes during early MS stages (e.g., relapse phase) and EP2 on microglia/macrophages at later stages (e.g., progressive phase) may provide useful tools to influence disease-specific pathological events. Other parts of the AA pathway, such as monohydroxylated HETEs, have not been investigated thoroughly, yet may provide additional targets for intervention. Overall, in this review we provide evidence that the AA metabolome is strongly intertwined with pathological processes in MS and indicate the need for strategies targeting this molecular pathway, to create novel patient- and MS subtype-specific therapeutic options against MS.
Availability of data and materials
Not applicable.
Abbreviations
- AA:
-
Arachidonic acid
- AKR1C:
-
Aldo-keto reductase family 1 member C
- ASA:
-
Acetylsalicylic acid
- BBB:
-
Blood brain barrier
- BLT:
-
Leukotriene B4 receptor
- COX:
-
Cyclooxygenases
- cPLA2-α:
-
Group IVA cytosolic phospholipase A2
- CRHTH2:
-
Chemoattractant receptor–homologous molecule on Th2 cells
- cAMP:
-
Cyclic adenosine monophosphate
- CYP:
-
Cytochrome P450
- CysLTR:
-
Cysteinyl leukotriene receptor
- DP:
-
D prostanoid receptor
- EAE:
-
Experimental autoimmune encephalitis
- EDSS:
-
Expanded disability status scale
- EP:
-
Prostaglandin E2 receptor
- ER:
-
Endoplasmic reticulum
- FLAP:
-
5-Lipoxygenase-activating protein
- FP:
-
F prostanoid receptors
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- GPCR:
-
G protein-coupled receptor
- GSH:
-
Glutathion
- HETE:
-
Hydroxyeicosatetraenoic acid
- H-PGDS:
-
Hematopoietic PGD synthase
- HIF-1α:
-
Hypoxia-inducible factor 1 alpha
- IFNγ:
-
Interferon gamma
- IL:
-
Interleukin
- IP:
-
Prostacyclin receptor
- LM:
-
Lipid mediator
- iNOS:
-
Inducible nitric oxide synthase
- LOX:
-
Lipoxygenase
- LPC:
-
Lysophosphatidylcholine
- L-PGDS:
-
Lipocalin-type PGD synthase
- LPS:
-
Lipopolysaccharide
- LT:
-
Leukotriene
- LTA4H:
-
Leukotriene A4 hydrolase
- MAP:
-
Mitogen-activated protein
- MMP:
-
Matrix metalloproteinase
- mPGES-1:
-
Microsomal prostaglandin E synthase-1
- mPGES-2:
-
Membrane-bound prostaglandin E synthase-2
- MRP-1:
-
Multi-drug resistance protein 1
- MS:
-
Multiple sclerosis
- NF-κB:
-
Nuclear factor kappa B
- Nfl:
-
Neurofilament light
- NO:
-
Nitric oxide
- OLs:
-
Oligodendrocytes
- OXER1:
-
Oxoeicosanoid 1
- Ox-HDL:
-
Oxidized high-density lipoprotein
- Ox-LDL:
-
Oxidized low-density lipoprotein
- PBMC:
-
Peripheral blood mononuclear cells
- PG:
-
Prostaglandin
- PGHS:
-
Prostaglandin endoperoxide synthase-1
- PGI2 :
-
Prostacyclin
- PGIS:
-
Prostaglandin I2 synthase
- PPAR-γ:
-
Peroxisome proliferator-activated receptor γ
- PMN:
-
Polymorphonuclear
- PMS:
-
Progressive multiple sclerosis
- PPMS:
-
Primary progressive multiple sclerosis
- PUFA:
-
Polyunsaturated fatty acid
- PwMS:
-
People with multiple sclerosis
- PwPMS:
-
People with progressive multiple sclerosis
- PwPPMS:
-
People with primary progressive multiple sclerosis
- PwRRMS:
-
People with relapsing-remitting multiple sclerosis
- PwSPMS:
-
People with secondary progressive multiple sclerosis
- ROS:
-
Reactive oxygen species
- RRMS:
-
Relapsing–remitting multiple sclerosis
- SPMS:
-
Secondary progressive multiple sclerosis
- STAT-1:
-
Signal transducer and activator of transcription 1
- Th17:
-
T-helper lymphocyte type 17
- TNF:
-
Tumor necrosis factor
- TP:
-
Thromboxane receptor
- Tx:
-
Thromboxane
- TxAS:
-
Thromboxane-A synthase
- 5-HEDH:
-
5-Hydroxyeicosanoid dehydrogenase
- 15d-PGJ2 :
-
15-Deoxy-(12,14)-PGJ2
References
Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, Robertson N, la Rocca N, Uitdehaag B, van der Mei I, Wallin M, Helme A, Angood Napier C, Rijke N, Baneke P. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Multiple Scler J. 2020;26(14):1816–21. https://doi.org/10.1177/1352458520970841.
Kuhn S, Gritti L, Crooks D, Dombrowski Y. Oligodendrocytes in development, myelin generation and beyond. Cells. 2019;8(11):1424. https://doi.org/10.3390/cells8111424.
Garg N, Smith TW. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015;5(9):e00362. https://doi.org/10.1002/brb3.362.
Rauf A, Badoni H, Abu-Izneid T, Olatunde A, Rahman MM, Painuli S, Semwal P, Wilairatana P, Mubarak MS. Neuroinflammatory markers: key indicators in the pathology of neurodegenerative diseases. Molecules. 2022;27(10):3194. https://doi.org/10.3390/molecules27103194.
Cencioni MT, Mattoscio M, Magliozzi R, Bar-Or A, Muraro PA. B cells in multiple sclerosis—from targeted depletion to immune reconstitution therapies. Nat Rev Neurol. 2021;17(7):399–414. https://doi.org/10.1038/s41582-021-00498-5.
Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1): a020412. https://doi.org/10.1101/cshperspect.a020412.
Ortiz GG, Pacheco-Moisés FP, Macías-Islas MÁ, Flores-Alvarado LJ, Mireles-Ramírez MA, González-Renovato ED, Hernández-Navarro VE, Sánchez-López AL, Alatorre-Jiménez MA. Role of the blood–brain barrier in multiple sclerosis. Arch Med Res. 2014;45(8):687–97. https://doi.org/10.1016/j.arcmed.2014.11.013.
Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA. Multiple sclerosis. Nat Rev Dis Primers. 2018;4(1):43. https://doi.org/10.1038/s41572-018-0041-4.
Alcina A, Abad-Grau Mdel M, Fedetz M, Izquierdo G, Lucas M, Fernández O, Ndagire D, Catalá-Rabasa A, Ruiz A, Gayán J, Delgado C, Arnal C, Matesanz F. Multiple sclerosis risk variant HLA-DRB1*1501 associates with high expression of DRB1 gene in different human populations. PLoS ONE. 2012;7(1):e29819. https://doi.org/10.1371/journal.pone.0029819.
Bjornevik K, Cortese M, Healy B, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296–301. https://doi.org/10.1126/science.abj8222.
Healy BC, Ali EN, Charles GRG, Chitnis T, Glanz BI, Houtchens M, Stazzone L, Moodie J, Berger AM, Duan Y, Bakshi R, Khoury S, Weiner H, Ascherio A. Smoking and disease progression in multiple sclerosis. Arch Neurol. 2009;66(7):858–64. https://doi.org/10.1001/archneurol.2009.122.
Horjus J, van Mourik-Banda T, Heerings MAP, Hakobjan M, de Witte W, Heersema DJ, Jansen AJ, Strijbis EMM, de Jong BA, Slettenaar AEJ, Zeinstra EMPE, Hoogervorst ELJ, Franke B, Kruijer W, Jongen PJ, Visser LJ, Poelmans G. Whole exome sequencing in multi-incident families identifies novel candidate genes for multiple sclerosis. Int J Mol Sci. 2022;23(19):11461. https://doi.org/10.3390/ijms231911461.
van der Vuurst de Vries RM, Mescheriakova JY, Runia TF, Siepman TAM, Wokke BHA, Samijn JPA, Hintzen RQ. Smoking at time of CIS increases the risk of clinically definite multiple sclerosis. J Neurol. 2018;265(5):1010–5. https://doi.org/10.1007/s00415-018-8780-4.
Klineova S, Lublin FD. Clinical course of multiple sclerosis. Cold Spring Harbor Perspect Med. 2018;8(9):a028928. https://doi.org/10.1101/cshperspect.a028928.
Kappos L, Wolinsky JS, Giovannoni G, Arnold DL, Wang Q, Bernasconi C, Model F, Koendgen H, Manfrini M, Belachew S, Hauser SL. Contribution of relapse-independent progression vs relapse-associated worsening to overall confirmed disability accumulation in typical relapsing multiple sclerosis in a pooled analysis of 2 randomized clinical trials. JAMA Neurol. 2020;77(9):1132–40. https://doi.org/10.1001/jamaneurol.2020.1568.
Biernacki T, Kokas Z, Sandi D, Füvesi J, Fricska-Nagy Z, Faragó P, Kincses TZ, Klivényi P, Bencsik K, Vécsei L. Emerging biomarkers of multiple sclerosis in the blood and the CSF: a focus on neurofilaments and therapeutic considerations. Int J Mol Sci. 2022;23(6):3383. https://doi.org/10.3390/ijms23063383.
Ayrignac X, Le Bars E, Duflos C, et al. Serum GFAP in multiple sclerosis: correlation with disease type and MRI markers of disease severity. Sci Rep. 2020;10:10923. https://doi.org/10.1038/s41598-020-67934-2.
de Jong BA, Huizinga TW, Bollen EL, Uitdehaag BM, Bosma GP, van Buchem MA, Remarque EJ, Burgmans AC, Kalkers NF, Polman CH, Westendorp RG. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126(1–2):172–9. https://doi.org/10.1016/s0165-5728(02)00056-5.
Broos JY, Loonstra FC, De Ruiter LRJ, Gouda MTEE, Fung WH, Schoonheim MM, Heijink M, Strijbis EMM, Teunissen CE, Killestein J, De Vries HE, Giera MA, Uitdehaag B, Kooij G. Association of arachidonic acid-derived lipid mediators with disease severity in patients with relapsing and progressive multiple sclerosis. Neurology. 2023;101:e533.
Kooij G, Troletti CD, Leuti A, Norris PC, Riley I, Albanese M, Ruggieri S, Libreros S, van der Pol SMA, van het Hof B, Schell Y, Guerrera G, Buttari F, Mercuri NB, Centonze D, Gasperini C, Battistini L, de Vries HE, Serhan CN, Chiurchiù V. Specialized differentially pro-resolving lipid mediators are altered in peripheral blood of patients with multiple sclerosis and attenuate monocyte and blood-brain barrier dysfunction. Haematologica. 2020;105(8):2056–70. https://doi.org/10.3324/haematol.2019.219519.
Gilbert NC, Newcomer ME, Werz O. Untangling the web of 5-lipoxygenase-derived products from a molecular and structural perspective: The battle between pro- and anti-inflammatory lipid mediators. Biochem Pharmacol. 2021;193:114759. https://doi.org/10.1016/j.bcp.2021.114759.
Mallinger J, Wildfeuer A, Mehlber L. Leukotrienes in the cerebrospinal fluid of multiple sclerosis patients. Acta Neurol Scand. 1992;86(6):586–7. https://doi.org/10.1111/j.1600-0404.1992.tb05491.x.
Mattsson N, Yaong M, Rosengren L, Blennow K, Månsson JE, Andersen O, Zetterberg H, Haghighi S, Zho I, Pratico D. Elevated cerebrospinal fluid levels of prostaglandin E2 and 15-(S)-hydroxyeicosatetraenoic acid in multiple sclerosis. J Intern Med. 2009;265(4):459–64. https://doi.org/10.1111/j.1365-2796.2008.02035.x.
Miura K, Walter MD, Hubbard C, MacGlashan DW Jr. Phosphorylation of cytosolic phospholipase A2 by IL-3 is associated with increased free arachidonic acid generation and leukotriene C4 release in human basophils. J Allergy Clin Immunol. 1998;102(3):512–20. https://doi.org/10.1016/s0091-6749(98)70142-3.
Hefner Y, Börsch-Haubold AG, Murakami M, Wilde JI, Pasquet S, Schieltz D, Ghomashchi F, Yates JR, Armstrong CG, Paterson A, Cohen P, Fukunaga R, Hunter T, Kudo I, Watson SP, Gelba MH. Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J Biol Chem. 2000;275(48):37542–51. https://doi.org/10.1074/jbc.M003395200.
Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72(2):269–78.
Evans JH, Spencer DM, Zweifach A, Leslie CC. Intracellular calcium signals regulating cytosolic phospholipase A 2 translocation to internal membranes. J Biol Chem. 2001;276(32):30150–60. https://doi.org/10.1074/jbc.M100943200.
Reddy ST, Herschman HR. Prostaglandin synthase-1 and prostaglandin synthase-2 are coupled to distinct phospholipases for the generation of prostaglandin D2 in activated mast cells. J Biol Chem. 1997;272(6):3231–7.
Bosma KJ, Kaiser CE, Kimple ME, Gannon M. Effects of arachidonic acid and its metabolites on functional beta-cell mass. Metabolites. 2022;12(4):342. https://doi.org/10.3390/metabo12040342.
Herschman HR. Prostaglandin synthase 2. Biochem Biophys Acta. 1996;1299(1):125–40. https://doi.org/10.1016/0005-2760(95)00194-8.
Morel A, Miller E, Bijak M, Saluk J. The increased level of COX-dependent arachidonic acid metabolism in blood platelets from secondary progressive multiple sclerosis patients. Mol Cell Biochem. 2016;420(1–2):85–94. https://doi.org/10.1007/s11010-016-2770-6.
Rose JW, Hill KE, Watt HE, Carlson NG. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J Neuroimmunol. 2004;149(1–2):40–9. https://doi.org/10.1016/j.jneuroim.2003.12.021.
Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006. https://doi.org/10.1186/1471-2377-6-12.
Carlson NG, Rojas MA, Redd JW, Tang P, Wood B, Hill KE, Rose JW. Cyclooxygenase-2 expression in oligodendrocytes increases sensitivity to excitotoxic death. J Neuroinflammation. 2010. https://doi.org/10.1186/1742-2094-7-25.
Ni J, Shu YY, Zhu YN, Fu YF, Tang W, Zhong XG, Wang H, Yang YF, Ren J, Wang MW, Zuo JP. COX-2 inhibitors ameliorate experimental autoimmune encephalomyelitis through modulating IFN-gamma and IL-10 production by inhibiting T-bet expression. J Neuroimmunol. 2007;186(1–2):94–103. https://doi.org/10.1016/j.jneuroim.2007.03.012.
Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332(7553):1302–8. https://doi.org/10.1136/bmj.332.7553.1302.
Prüss H, Rosche B, Sullivan AB, Brommer B, Wengert O, Gronert K, Schwab JM. Proresolution lipid mediators in multiple sclerosis - differential, disease severity-dependent synthesis—a clinical pilot trial. PLoS ONE. 2013;8(2):e55859. https://doi.org/10.1371/journal.pone.0055859.
Takeuchi C, Matsumoto Y, Kohyama K, Uematsu S, Akira S, Yamagata K, Takemiya T. Microsomal prostaglandin e synthase-1 aggravates inflammation and demyelination in a mouse model of multiple sclerosis. Neurochem Int. 2013;62(3):271–80. https://doi.org/10.1016/j.neuint.2012.12.007.
Kihara Y, Matsushita T, Kita Y, Uematsu S, Akira S, Kira J, Ishii S, Shimizu T. Targeted lipidomics reveals mPGES-1-PGE2 as a therapeutic target for multiple sclerosis. Proc Natl Acad Sci USA. 2009;106(51):21807–12. https://doi.org/10.1073/pnas.0906891106.
Wang Q, Li Y, Wu M, Huang S, Zhang A, Zhang Y, Jia Z. Targeting microsomal prostaglandin E synthase 1 to develop drugs treating the inflammatory diseases. Am J Transl Res. 2021;13(1):391–419.
Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: relevance of prostaglandin e receptors. Biochim Biophys Acta Mol Cell Biol Lipids. 2015;1851(4):414–21. https://doi.org/10.1016/j.bbalip.2014.07.008.
Benyahia C, Gomez I, Kanyinda L, Boukais K, Danel C, Leséche G, Longrois D, Norel X. PGE(2) receptor (EP(4)) agonists: potent dilators of human bronchi and future asthma therapy? Pulm Pharmacol Ther. 2012;25(1):115–8. https://doi.org/10.1016/j.pupt.2011.12.012.
Säfholm J, Manson ML, Bood J, Delin I, Orre AC, Bergman P, Al-Ameri M, Dahlén SE, Adner M. Prostaglandin E2 inhibits mast cell-dependent bronchoconstriction in human small airways through the E prostanoid subtype 2 receptor. J Allergy Clin Immunol. 2015;136(5):1232-9.e1. https://doi.org/10.1016/j.jaci.2015.04.002.
Kida T, Sawada K, Kobayashi K, Hori M, Ozaki H, Murata T. Diverse effects of prostaglandin E2 on vascular contractility. Heart Vessels. 2014;29(3):390–5. https://doi.org/10.1007/s00380-013-0374-6.
Norel X, de Montpreville V, Brink C. Vasoconstriction induced by activation of EP1 and EP3 receptors in human lung: effects of ONO-AE-248, ONO-DI-004, ONO-8711 or ONO-8713. Prostaglandins Other Lipid Mediat. 2004;74(1–4):101–12. https://doi.org/10.1016/j.prostaglandins.2004.07.003.
Frankowski JC, DeMars KM, Ahmad AS, Hawkins KE, Yang C, Leclerc JL, Doré S, Candelario-Jalil E. Detrimental role of the EP1 prostanoid receptor in blood–brain barrier damage following experimental ischemic stroke. Sci Reports. 2015. https://doi.org/10.1038/srep17956.
Avolio C, Ruggieri M, Giuliani F, Liuzzi GM, Leante R, Riccio P, Livrea P, Trojano M. Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J Neuroimmunol. 2003;136(1–2):46–53. https://doi.org/10.1016/S0165-5728(03)00006-7.
Lichtinghagen R, Seifert T, Kracke A, Marckmann S, Wurster U, Heidenreich F. Expression of matrix metalloproteinase-9 and its inhibitors in mononuclear blood cells of patients with multiple sclerosis. J Neuroimmunol. 1999;99:19–26.
Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27(4):697–709. https://doi.org/10.1038/sj.jcbfm.9600375.
Esaki Y, Li Y, Sakata D, Yao C, Segi-Nishida E, Matsuoka T, Fukuda K, Narumiya S. Dual roles of PGE2-EP4 signaling in mouse experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2010;107(27):12233–8. https://doi.org/10.1073/pnas.0915112107.
Kofler DM, Marson A, Dominguez-Villar M, Xiao S, Kuchroo VK, Hafler DA. Decreased RORC-dependent silencing of Prostaglandin receptor EP2 induces autoimmune Th17 cells. J Clin Investig. 2014;124(6):2513–22. https://doi.org/10.1172/JCI72973.
Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F. The cyclooxygenase-2 pathway via the PGE2 EP2 receptor contributes to oligodendrocytes apoptosis in cuprizone-induced demyelination. J Neurochem. 2012;121(3):418–27. https://doi.org/10.1111/j.1471-4159.2011.07363.x.
Bonfill-Teixidor E, Otxoa-de-Amezaga A, Font-Nieves M, Sans-Fons MG, Planas AM. Differential expression of E-type prostanoid receptors 2 and 4 in microglia stimulated with lipopolysaccharide. J Neuroinflammation. 2017. https://doi.org/10.1186/s12974-016-0780-7.
Liu R, Du S, Zhao L, Jain S, Sahay K, Rizvanov A, Lezhnyova V, Khaibullin T, Martynova E, Khaiboullina S, Baranwal M. Autoreactive lymphocytes in multiple sclerosis: pathogenesis and treatment target. Front Immunol. 2022;23(13): 996469. https://doi.org/10.3389/fimmu.2022.996469.
van Langelaar J, van der Vuurst De Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, Dankers W, Verjans GMGM, de Vries HE, Lubberts E, Hintzen RQ, van Luijn MM. T helper 171 cells associate with multiple sclerosis disease activity: perspectives for early intervention. Brain. 2018;141(5):1334–49. https://doi.org/10.1093/brain/awy069.
Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through TH1 cell differentiation and TH17 cell expansion. Nat Med. 2009;15(6):633–40. https://doi.org/10.1038/nm.1968.
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910–4. https://doi.org/10.1074/jbc.M207577200.
Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203(7):1685–91. https://doi.org/10.1084/jem.20060285.
Schiffmann S, Weigert A, Männich J, Eberle M, Birod K, Häussler A, Ferreiros N, Schreiber Y, Kunkel H, Grez M, Weichand B, Brüne B, Pfeilschifter W, Nüsing R, Niederberger E, Grösch S, Scholich K, Geisslinger G. PGE2/EP4 signaling in peripheral immune cells promotes development of experimental autoimmune encephalomyelitis. Biochem Pharmacol. 2014;87(4):625–35. https://doi.org/10.1016/j.bcp.2013.12.006.
Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288(13):9293–302. https://doi.org/10.1074/jbc.M113.455816.
Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning-Boǧ AB, Andreasson KI. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J Neurosci. 2013;33(40):16016–32. https://doi.org/10.1523/JNEUROSCI.2203-13.2013.
Fu Y, Yang MS, Jiang J, Ganesh T, Joe E, Dingledine R. EP2 receptor signaling regulates microglia death. Mol Pharmacol. 2015;88(1):161–70. https://doi.org/10.1124/mol.115.098202.
Vleeshouwers W, van den Dries K, de Keijzer S, Joosten B, Lidke DS, Cambi A. Characterization of the signaling modalities of prostaglandin E2 receptors EP2 and EP4 reveals crosstalk and a role for microtubules. Front Immunol. 2021;12(11): 613286. https://doi.org/10.3389/fimmu.2020.613286.
Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E 2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010;184(12):7207–18. https://doi.org/10.4049/jimmunol.0903487.
Beuckmann CT, Lazarus M, Gerashchenko D, Mizoguchi A, Nomura S, Mohri I, Uesugi A, Kaneko T, Mizuno N, Hayaishi O, Urade Y. Cellular localization of lipocalin-type prostaglandin D synthase (-trace) in the central nervous system of the adult rat. J Comparative Neurol. 2000;428:62–78.
Angeli V, Staumont D, Charbonnier AS, Hammad H, Gosset P, Pichavant M, Lambrecht BN, Capron M, Dombrowicz D, Trottein F. Activation of the D prostanoid receptor 1 regulates immune and skin allergic responses. J Immunol. 2004;172(6):3822–9. https://doi.org/10.4049/jimmunol.172.6.3822.
Yoshimura-Uchiyama C, Iikura M, Yamaguchi M, Nagase H, Ishii A, Matsushima K, Yamamoto K, Shichijo M, Bacon KB, Hirai K. Differential modulation of human basophil functions through prostaglandin D2 receptors DP and chemoattractant receptor-homologous molecule expressed on Th2 cells/DP2. Clin Exp Allergy. 2004;34(8):1283–90. https://doi.org/10.1111/j.1365-2222.2004.02027.x.
Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Brief definitive report prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. In J Exp Med. 2001;193(2):255.
Woodward DF, Lawrence RA. Identification of a single (FP) receptor associated with prostanoid-induced Ca2+ signals in Swiss 3T3 cells. Biochem Pharmacol. 1994;47(9):1567–74. https://doi.org/10.1016/0006-2952(94)90533-9.
Iwasa K, Yamamoto S, Takahashi M, Suzuki S, Yagishita S, Awaji T, Maruyama K, Yoshikawa K. Prostaglandin F2α FP receptor inhibitor reduces demyelination and motor dysfunction in a cuprizone-induced multiple sclerosis mouse model. Prostaglandins Leukot Essent Fatty Acids. 2014;91(5):175–82. https://doi.org/10.1016/j.plefa.2014.08.004.
Storer PD, Xu J, Chavis JA, Drew PD. Cyclopentenone prostaglandins PGA2 and 15-deoxy-Δ 12,14 PGJ2 suppress activation of marine microglia and astrocytes: Implications for multiple sclerosis. J Neurosci Res. 2005;80(1):66–74. https://doi.org/10.1002/jnr.20413.
Jiang C, Ting AT, Seed B. PPAR-gagonists inhibit production of monocyteinflammatory cytokines. Nature. 1998;391:82–6. https://doi.org/10.1038/34184.
Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Short communication macrophages in human atheroma contain PPAR differentiation-dependent peroxisomal proliferator-activated receptor (PPAR) expression and reduction of MMP-9 activity through PPAR activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153(1):17–23.
Shu H, Wong B, Zhou G, Li Y, Berger J, Woods JW, Wright SD, Cai TQ. Activation of PPARα or γ reduces secretion of matrix metalloproteinase 9 but not interleukin 8 from human monocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267(1):345–9. https://doi.org/10.1006/bbrc.1999.1968.
Meng Y, Chen C, Tian C, Du J, Li HH. Angiotensin II-induced egr-1 expression is suppressed by peroxisome proliferator-activated receptor-γ ligand 15d-PGJ 2 in macrophages. Cell Physiol Biochem. 2015;35(2):689–98. https://doi.org/10.1159/000369729.
Chearwae W, Bright JJ. 15-Deoxy-Δ12,14-prostaglandin J2 and curcumin modulate the expression of toll-like receptors 4 and 9 in autoimmune T lymphocyte. J Clin Immunol. 2008;28(5):558–70. https://doi.org/10.1007/s10875-008-9202-7.
Xiang Z, Lin T, Reeves SA. 15d-PGJ2 induces apoptosis of mouse oligodendrocyte precursor cells. J Neuroinflammation. 2007;4(1):1–10.
Watanabe K, Yoshida R, Shimizu T, Hayaishi O. Enzymatic formation of prostaglandin F(2α) from prostaglandin H2 and D2. Purification and properties of prostaglandin F synthetase from bovine lung. J Biol Chem. 1985;260(11):7035–41. https://doi.org/10.1016/s0021-9258(18)88884-6.
Abramovitz M, Adam M, Boie Y, Carrière M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta. 2000;1483(2):285–93. https://doi.org/10.1016/s1388-1981(99)00164-x.
Siegle I, Klein T, Zou M-H, Fritz P, Kömhoff M, Fischer-Bosch M. Distribution and cellular localization of prostacyclin synthase in human brain. J Histochem Cytochem. 2000;48(5):631.
Zhou W, Dowell DR, Huckabee MM, Newcomb DC, Boswell MG, Goleniewska K, Lotz MT, Toki S, Yin H, Yao S, Natarajan C, Wu P, Sriram S, Breyer RM, FitzGerald GA, Peebles RS. Prostaglandin I2 signaling drives Th17 differentiation and exacerbates experimental autoimmune encephalomyelitis. PLoS ONE. 2012;7(5):e33518. https://doi.org/10.1371/journal.pone.0033518.
Muramatsu R, Kuroda M, Matoba K, Lin H, Takahashi C, Koyama Y, Yamashita T. Prostacyclin prevents pericyte loss and demyelination induced by lysophosphatidylcholine in the central nervous system. J Biol Chem. 2015;290(18):11515–25. https://doi.org/10.1074/jbc.M114.587253.
Jung S, Donhauser T, Toyka KV, Hartung HP. Propentofylline and iloprost suppress the production of TNF-α by macrophages but fail to ameliorate experimental autoimmune encephalomyelitis in Lewis rats. J Autoimmun. 1997;10:519–29.
Gryglewski RJ, Dembínska-Kieć A, Korbut R. A possible role of thromboxane A2 (TXA2) and prostacyclin (PGI2) in circulation. Acta Biol Med Ger. 1978;37(5–6):715–23.
Morel A, Rywaniak J, Bijak M, Miller E, Niwald M, Saluk J. Flow cytometric analysis reveals the high levels of platelet activation parameters in circulation of multiple sclerosis patients. Mol Cell Biochem. 2017;430(1–2):69–80. https://doi.org/10.1007/s11010-017-2955-7.
Sheremata WA, Jy W, Horstman LL, Ahn YS, Alexander JS, Minagar A. Evidence of platelet activation in multiple sclerosis. J Neuroinflammation. 2008. https://doi.org/10.1186/1742-2094-5-27.
D’Souza CS, Li Z, Maxwell DL, Trusler O, Murphy M, Crewther S, Peter K, Orian JM. Platelets drive inflammation and target gray matter and the retina in autoimmune-mediated encephalomyelitis. J Neuropathol Exp Neurol. 2018;77(7):567–76. https://doi.org/10.1093/jnen/nly032.
Starossom SC, Veremeyko T, Yung AWY, Dukhinova M, Au C, Lau AY, Weiner HL, Ponomarev ED. Platelets play differential role during the initiation and progression of autoimmune neuroinflammation. Circ Res. 2015;117(9):779–92. https://doi.org/10.1161/CIRCRESAHA.115.306847.
Vogelsang A, Eichler S, Huntemann N, Masanneck L, Böhnlein H, Schüngel L, Willison A, Loser K, Nieswandt B, Kehrel BE, Zarbock A, Göbel K, Meuth SG. Platelet inhibition by low-dose acetylsalicylic acid reduces neuroinflammation in an animal model of multiple sclerosis. Int J Mol Sci. 2021;22(18):9915. https://doi.org/10.3390/ijms22189915.
Mir F, Lee D, Ray H, Sadiq SA. CSF isoprostane levels are a biomarker of oxidative stress in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2014;1(2): e21. https://doi.org/10.1212/NXI.0000000000000021.
Eckenstaler R, Ripperger A, Hauke M, Petermann M, Hemkemeyer SA, Schwedhelm E, Ergün S, Frye M, Werz O, Koeberle A, Braun H, Benndorf RA. A thromboxane A2Receptor-driven COX-2-dependent feedback loop that affects endothelial homeostasis and angiogenesis. Arterioscler Thromb Vasc Biol. 2022;42(4):444–61. https://doi.org/10.1161/ATVBAHA.121.317380.
Chen S, Zou H. Lipoxygenase metabolism: critical pathways in microglia-mediated neuroinflammation and neurodevelopmental disorders. Neurochem Res. 2022;47(11):3213–20. https://doi.org/10.1007/s11064-022-03645-6.
Arthur AT, Armati PJ, Bye C, Southern MS Genetics Consortium, Heard RN, Stewart GJ, Pollard JD, Booth DR. Genes implicated in multiple sclerosis pathogenesis from consilience of genotyping and expression profiles in relapse and remission. BMC Med Genet. 2008;9:17. https://doi.org/10.1186/1471-2350-9-17.
Kaufmann M, Schaupp AL, Sun R, et al. Identification of early neurodegenerative pathways in progressive multiple sclerosis. Nat Neurosci. 2022;25:944–55. https://doi.org/10.1038/s41593-022-01097-3.
Rådmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem Sci. 2007;32(7):332–41. https://doi.org/10.1016/j.tibs.2007.06.002.
Emerson MR, Levine SM. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res. 2004;1021(1):140–5. https://doi.org/10.1016/j.brainres.2004.06.045.
Yoshikawa K, Palumbo S, Toscano CD, Bosetti F. Inhibition of 5-lipoxygenase activity in mice during cuprizone-induced demyelination attenuates neuroinflammation, motor dysfunction and axonal damage. Prostaglandins Leukot Essent Fatty Acids. 2011;85(1):43–52. https://doi.org/10.1016/j.plefa.2011.04.022.
Kong W, Hooper KM, Ganea D. The natural dual cyclooxygenase and 5-lipoxygenase inhibitor flavocoxid is protective in EAE through effects on Th1/Th17 differentiation and macrophage/microglia activation. Brain Behav Immun. 2016;53:59–71. https://doi.org/10.1016/j.bbi.2015.11.002.
Whitney LW, Ludwin SK, Mcfarland HF, Biddison WE. Microarray analysis of gene expression in multiple sclerosis and EAE identifies 5-lipoxygenase as a component of inflammatory lesions. J Neuroimmunol. 2001;121:40–8.
Kretzer C, Jordan PM, Bilancia R, Rossi A, Gür Maz T, Banoglu E, Schubert US, Werz O. Shifting the biosynthesis of leukotrienes toward specialized pro-resolving mediators by the 5-lipoxygenase-activating protein (FLAP) antagonist BRP-201. J Inflamm Res. 2022;9(15):911–25. https://doi.org/10.2147/JIR.S345510.
Werner M, Jordan PM, Romp E, Czapka A, Rao Z, Kretzer C, Koeberle A, Garscha U, Pace S, Claesson HE, Serhan CN, Werz O, Gerstmeier J. Targeting biosynthetic networks of the proinflammatory and proresolving lipid metabolome. FASEB J. 2019;33(5):6140–53. https://doi.org/10.1096/fj.201802509R.
Pergola C, Gerstmeier J, Mönch B, Çalışkan B, Luderer S, Weinigel C, Barz D, Maczewsky J, Pace S, Rossi A, Sautebin L, Banoglu E, Werz O. The novel benzimidazole derivative BRP-7 inhibits leukotriene biosynthesis in vitro and in vivo by targeting 5-lipoxygenase-activating protein (FLAP). Br J Pharmacol. 2014;171(12):3051–64. https://doi.org/10.1111/bph.12625.
Evans JF, Ferguson AD, Mosley RT, Hutchinson JH. What’s all the FLAP about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sci. 2008;29(2):72–8. https://doi.org/10.1016/j.tips.2007.11.006.
Kahnt AS, Schebb NH, Steinhilber D. Formation of lipoxins and resolvins in human leukocytes. Prostaglandins Other Lipid Mediat. 2023;166: 106726. https://doi.org/10.1016/j.prostaglandins.2023.106726.
Yokomizo T, Kato K, Terawaki K, Izumi T, Shimizu T. A second leukotriene B 4 receptor, BLT2: a new therapeutic target in inflammation and immunological disorders. Int J Exp Med. 2000;192(3):421.
Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARa-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39.
Toda A, Yokomizo T, Shimizu T. Leukotriene B4 receptors. Prostaglandins Other Lipid Mediators. 2002;68:575.
Lee W, Kim HS, Lee GR. Leukotrienes induce the migration of Th17 cells. Immunol Cell Biol. 2015;93(5):472–9. https://doi.org/10.1038/icb.2014.104.
Neu IS, Metzger G, Zschocke J, Zelezny R, Mayatepek E. Leukotrienes in patients with clinically active multiple sclerosis. Acta Neurol Scand. 2002;105:63–6. https://doi.org/10.1034/j.1600-0404.2002.00070.x.
Kihara Y, Yokomizo T, Kunita A, Morishita Y, Fukayama M, Ishii S, Shimizu T. The leukotriene B4 receptor, BLT1, is required for the induction of experimental autoimmune encephalomyelitis. Biochem Biophys Res Commun. 2010;394(3):673–8. https://doi.org/10.1016/j.bbrc.2010.03.049.
Han B, Zhang YY, Ye ZQ, Xiao Y, Rasouli J, Wu WC, Ye SM, Guo XY, Zhu L, Rostami A, Li-Bin W, Zhang Y, Li X. Montelukast alleviates inflammation in experimental autoimmune encephalomyelitis by altering Th17 differentiation in a mouse model. Immunology. 2021;163(2):185–200. https://doi.org/10.1111/imm.13308.
Yokomizo T, Kato K, Hagiya H, Izumi T, Shimizu T. Hydroxyeiconasoids bind to and activate the low affinity leukotriene B4 receptor, BLT2. J Biol Chem Mech Signal Transduct. 2001;276(15):12454–9.
Okuno T, Iizuka Y, Okazaki H, Yokomizo T, Taguchi R, Shimizu T. 12(S)-hydroxyheptadeca-5Z, 8E, 10E-trienoic acid is a natural ligand for leukotriene B4 receptor 2. J Exp Med. 2008;205(4):759–66. https://doi.org/10.1084/jem.20072329.
Okuno T, Yokomizo T. Biological functions of 12(S)-hydroxyheptadecatrienoic acid as a ligand of leukotriene B4 receptor 2. Inflamm Regener. 2018. https://doi.org/10.1186/s41232-018-0087-4.
Gao J, Liu Q, Xu Y, Gong X, Zhang R, Zhou C, Su Z, Jin J, Shi H, Shi J, Hou Y. PPARα induces cell apoptosis by destructing Bcl2. Oncotarget. 2015;6(42):44635–42. https://doi.org/10.18632/oncotarget.5988.
Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273(40):25573–80. https://doi.org/10.1074/jbc.273.40.25573.
Szalardy L, Zadori D, Bencsik K, Vecsei L, Klivenyi P. Unlike PPARgamma, neither other PPARs nor PGC-1alpha is elevated in the cerebrospinal fluid of patients with multiple sclerosis. Neurosci Lett. 2017;651:128–33. https://doi.org/10.1016/j.neulet.2017.05.008.
Dasgupta S, Roy A, Jana M, Hartley DM, Pahan K. Gemfibrozil ameliorates relapsing-remitting experimental autoimmune encephalomyelitis independent of peroxisome proliferator-activated receptor-α. Mol Pharmacol. 2007;72(4):934–46. https://doi.org/10.1124/mol.106.033787.
Hijioka M, Futokoro R, Ohto-Nakanishi T, Nakanishi H, Katsuki H, Kitamura Y. Microglia-released leukotriene B4 promotes neutrophil infiltration and microglial activation following intracerebral hemorrhage. Int Immunopharmacol. 2020;85: 106678. https://doi.org/10.1016/j.intimp.2020.106678.
Laidlaw TM, Boyce JA. Cysteinyl leukotriene receptors, old and new; implications for asthma. Clin Exp Allergy. 2012;42(9):1313–20. https://doi.org/10.1111/j.1365-2222.2012.03982.x.
Spada CS, Krauss AH, Nieves AL, Woodward DF. Effects of leukotrienes B4 (LTB4) and D4 (LTD4) on motility of isolated normodense human eosinophils and neutrophils. Adv Exp Med Biol. 1997;400B:699–706.
De Lepeleire I, Reiss TF, Rochette F, Botto A, Zhang J, Kundu S, Decramer M. Montelukast causes prolonged, potent leukotriene D4-receptor antagonism in the airways of patients with asthma. Clin Pharmacol Ther. 1997;61(1):83–92. https://doi.org/10.1016/S0009-9236(97)90184-3.
Hendrickx DAE, van Scheppingen J, van der Poel M, Bossers K, Schuurman KG, van Eden CG, Hol EM, Hamann J, Huitinga I. Gene expression profiling of multiple sclerosis pathology identifies early patterns of demyelination surrounding chronic active lesions. Front Immunol. 2017;21(8):1810. https://doi.org/10.3389/fimmu.2017.01810.
Powell WS, Rokach J. 5-Oxo-ETE and inflammation. In: Steinhilber D, editor. Lipoxygenases in inflammation. Cham: Springer; 2016. https://doi.org/10.1007/978-3-319-27766-0_9.
Sozzani S, Zhou D, Locati M, Bernasconi S, Luini W, Mantovani A, O’Flaherty JT. Stimulating properties of 5-oxo-eicosanoids for human monocytes: synergism with monocyte chemotactic protein-1 and -3. J Immunol. 1996;157(10):4664–71.
Stamatiou PB, Chan CC, Monneret G, Ethier D, Rokach J, Powell WS. 5-Oxo-6,8,11,14-eicosatetraenoic acid stimulates the release of the eosinophil survival factor granulocyte-macrophage colony stimulating factor from monocytes. J Biol Chem. 2004;279:28159–64.
Capdevila J, Marnett LJ, Chacos N, Prough RA, Estabrook RW. Cytochrome P-450-dependent oxygenation of arachidonic acid to hydroxyicosatetraenoic acids. Proc Natl Acad Sci USA. 1982;79(3):767–70. https://doi.org/10.1073/pnas.79.3.767.
Thuresson ED, Lakkides KM, Smith WL. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J Biol Chem. 2000;275(12):8501–7. https://doi.org/10.1074/jbc.275.12.8501.
Wang P, Xie K, Wang C, Bi J. Oxidative stress induced by lipid peroxidation is related with inflammation of demyelination and neurodegeneration in multiple sclerosis. Eur Neurol. 2014;72(3–4):249–54. https://doi.org/10.1159/000363515.
Guido DM, McKenna R, Mathews WR. Quantitation of hydroperoxy-eicosatetraenoic acids and hydroxy-eicosatetraenoic acids as indicators of lipid peroxidation using gas chromatography–mass spectrometry. Anal Biochem. 1993;209(1):123–9. https://doi.org/10.1006/abio.1993.1091.
van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta. 2011;1812(2):141–50. https://doi.org/10.1016/j.bbadis.2010.06.011.
Ferretti G, Bacchetti T. Peroxidation of lipoproteins in multiple sclerosis. J Neurol Sci. 2011;311(1–2):92–7. https://doi.org/10.1016/j.jns.2011.09.004.
Zheng Z, Li Y, Jin G, Huang T, Zou M, Duan S. The biological role of arachidonic acid 12-lipoxygenase (ALOX12) in various human diseases. Biomed Pharmacother. 2020;129:110354.
Kulkarni A, Pineros AR, Ibrahim S, Hernandez-Perez M, Orr KS, Glenn L, Walsh M, Nadler JL, Morris MA, Tersey SA, et al. 12-Lipoxygenase governs the innate immune pathogenesis of islet inflammation and autoimmune diabetes. bioRxiv. 2021.
Guo Y, Zhang W, Giroux C, Cai Y, Ekambaram P, Dilly A, Hsu A, Zhou S, Maddipati KR, Liu J, et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-Hydroxyeicosatetraenoic acid. J Biol Chem. 2011;2011(286):33832–40.
Porro B, Songia P, Squellerio I, Tremoli E, Cavalca V. Analysis, physiological and clinical significance of 12-HETE: a neglected platelet-derived 12-Lipoxygenase product. J Chromatogr B. 2014;2014(964):26–40.
Li S, Vana AC, Ribeiro R, Zhang Y. Distinct role of nitric oxide and peroxynitrite in mediating oligodendrocyte toxicity in culture and in experimental autoimmune encephalomyelitis. Neuroscience. 2011;184:107–19.
Maskrey BH, Rushworth GF, Law MH, Treweeke AT, Wei J, Leslie SJ, Megson IL, Whitfield PD. 12-hydroxyeicosatetraenoic acid is associated with variability in aspirin-induced platelet inhibition. J Inflamm (Lond). 2014;11(1):33. https://doi.org/10.1186/s12950-014-0033-4.
Rydberg EK, Krettek A, Ullström C, Ekström K, Svensson PA, Carlsson LMS, et al. Hypoxia increases LDL oxidation and expression of 15-lipoxygenase-2 in human macrophages. Arterioscler Thromb Vasc Biol. 2004;24(11):2040–5. https://doi.org/10.1161/01.ATV.0000144951.08072.0b.
Yang R, Dunn JF. Multiple sclerosis disease progression: contributions from a hypoxia–inflammation cycle. Multiple Scler J. 2019;25(13):1715–8. https://doi.org/10.1177/1352458518791683.
Yang R, Dunn JF. Reduced cortical microvascular oxygenation in multiple sclerosis: a blinded, case-controlled study using a novel quantitative near-infrared spectroscopy method. Sci Rep. 2015;2015(5):16477.
Colton CA, Gilbert DL. Microglia, an in vivo source of reactive oxygen species in the brain. Adv Neurol. 1993;1993(59):321–6.
Snodgrass RG, Benatzy Y, Schmid T, Namgaladze D, Mainka M, Schebb NH, Lütjohann D, Brüne B. Efferocytosis potentiates the expression of arachidonate 15-lipoxygenase (ALOX15) in alternatively activated human macrophages through LXR activation. Cell Death Differ. 2021;28(4):1301–16. https://doi.org/10.1038/s41418-020-00652-4.
Zhao J, Zhang W, Wu T, Wang H, Mao J, Liu J, Zhou Z, Lin X, Yan H, Wang Q. Efferocytosis in the central nervous system. Front Cell Dev Biol. 2021;9:773344. https://doi.org/10.3389/fcell.2021.773344.
Wecksler AT, Kenyon V, Deschamps JD, Holman TR. Substrate specificity changes for human reticulocyte and epithelial 15-lipoxygenases reveal allosteric product regulation. Biochemistry. 2008;47(28):7364–75. https://doi.org/10.1021/bi800550n.
Choe J, Kwon B. 15-(S)-HETE plays a regulatory role in the immune inflammatory responses. J Immunol. 2019;202(1 Supplement):1259–1259.
Shankaranarayanan P, Nigam S. IL-4 induces apoptosis in A549 lung adenocarcinoma cells: evidence for the pivotal role of 15-hydroxyeicosatetraenoic acid binding to activated peroxisome proliferator-activated receptor gamma transcription factor. J Immunol. 2003;170(2):887–94. https://doi.org/10.4049/jimmunol.170.2.887.
Li Q, Mao M, Qiu Y, et al. Key role of ROS in the process of 15-lipoxygenase/15-hydroxyeicosatetraenoiccid-induced pulmonary vascular remodeling in hypoxia pulmonary hypertension. PLoS ONE. 2016;11(2):e0149164. https://doi.org/10.1371/journal.pone.0149164.
Xue H, Huo Y, Hu Y, et al. The role of ALOX15B in heat stress-induced apoptosis of porcine sertoli cells. Theriogenology. 2022;185:6–15. https://doi.org/10.1016/j.theriogenology.2022.03.018.
Magnusson LU, Lundqvist A, Karlsson MN, et al. Arachidonate 15-lipoxygenase type B knockdown leads to reduced lipid accumulation and inflammation in atherosclerosis. PLoS ONE. 2012;7(8):e43142. https://doi.org/10.1371/journal.pone.0043142.
Grajchen E, Hendriks JJA, Bogie JFJ. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun. 2018;6(1):124. https://doi.org/10.1186/s40478-018-0628-8.
Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal. 2009;2(72):3. https://doi.org/10.1126/scisignal.272re3.
Kotla S, Singh NK, Traylor JG Jr, Orr AW, Rao GN. ROS-dependent Syk and Pyk2-mediated STAT1 activation is required for 15(S)-hydroxyeicosatetraenoic acid-induced CD36 expression and foam cell formation. Free Radic Biol Med. 2014;76:147–62. https://doi.org/10.1016/j.freeradbiomed.2014.08.007.
Ternowitz T, Andersen PH, Bjerring P, Fogh K, Schröder JM, Kragballe K. 15-hydroxyeicosatetraenoic acid (15-HETE) specifically inhibits the LTB4-induced skin response. Arch Dermatol Res. 1989;281(6):401–5. https://doi.org/10.1007/BF00455325.
Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015;2015(2015):549691. https://doi.org/10.1155/2015/549691.
Acknowledgements
All figures were created with Biorender.com
Funding
This review was supported by a Grant from the Dutch Research Council (NWO Vidi Grant 91719305 to G.K.) and a Grant from the Dutch MS Research Foundation (18-1023MS to G.K.).
Author information
Authors and Affiliations
Contributions
JYB, RTMvdB and GK designed the manuscript and JYB created the manuscript figures and tables. JYB, RTMvdB and JK wrote the manuscript. MR, OW, HEdV, MG and GK edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
There are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Broos, J.Y., van der Burgt, R.T.M., Konings, J. et al. Arachidonic acid-derived lipid mediators in multiple sclerosis pathogenesis: fueling or dampening disease progression?. J Neuroinflammation 21, 21 (2024). https://doi.org/10.1186/s12974-023-02981-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02981-w