
Abstract
Goatpoxvirus (GTPV), sheeppoxvius (SPPV), and the Lumpy skin disease virus (LSDV) is a Capripoxvirus belonging to the family poxviridae. They can cause significant economic losses in countries where this disease are endemic. However, effective and convenient diagnostic tools against sera antibody are not readily available until now. Toward this goal, a polyclonal antibody competitive enzyme-linked immunosorbent assay (c-ELISA) of detecting serogroup-specific antibody is established based on major LSDV antigen A33. Serum samples (n = 605) were collected to optimize the c-ELISA from different areas. The cut-off value for the c-ELISA was estimate using percent inhibition (PI) values. The diagnostic performance of test including sensitivity (sn) and specificity (sp) were obtained by receiver operator characteristic (ROC) analysis. Among these analysis, > 57.61% PI value was accepted as cut-off of the c-ELISA, the diagnostic sn an diagnostic sp were reached to 96.4% and 98.5%, at > 95% confidence interval. These results show that the developed competitive ELISA is sensitive, specific, and reliable, which make it appropriate for serological investigation.
Similar content being viewed by others


Introduction
Lumpy skin disease (LSD) is a transboundary disease caused by the Lumpy skin disease virus (LSDV), belonging to Capripoxvirus genes [1]. It leads to huge economic losses within the cattle population, such as a drop in milk production, weight loss, skin damage, and temporary or permanent sterility in both bulls and cows [2]. The disease was first reported in northern Rhodesia in 1929, remained endemic to Africa until 1990, but then spread into the Middle East [3, 4]. Up to 2016, the disease was reported in many parts of Europe and Asia [5]. However, the disease was first reported in India, China, and Bangladesh in 2019 [6]. The evidence indicates a high transmission rate of LSD worldwide. Moreover, LSD can also cause acute or subacute disease in cattle and buffalo, thus affecting cattle breeds of all ages [7].
Although experienced veterinarians can readily diagnose these diseases in their acute forms, low virulence strains and other exanthemas in castles, e.g., Pseudocowpox, can present different diagnoses. Laboratory confirmation relies on classical virological techniques PCR to identify the virus in clinical material and virus isolation in cell culture [8]. Serodiagnosis tests available have been based upon immunofluorescence assay (IPMA) [9] and virus neutralization test (VNT) or indirect fluorescent antibody test (IFAT) [10]. Virus neutralization assays are the gold standard for detecting antibodies because they are both specific and sensitive. However, conventional virus neutralization assay requires handling infectious viruses under biosafety level (BSL)-3 condition. Furthermore, it takes several day to obtain results. Diagnostic and screening tests are the primary tools for a successful epidemiological study [11].
Serological diagnosis assays such as enzyme-linked immunosorbent assay (ELISA) are easy to perform at a veterinary clinic and do not require very complicated equipment. Moreover, ELISA requires less operation time, thus enabling many samples to be tested quickly with high sensitivity and specificity. ELISA methods are also important for assessing immune responses in vaccinated animals. Several ELISAs have been developed based on recombinant viral proteins [12]. However, at present, there are no reports about nonstructural protein-specific antibody assay to diagnose LSDV infection.
ORF122, an analog of the A33 protein of “vaccinia virus” (VACV). A33 is a highly conserved specific type II membrane glycoprotein protein mainly present in the extracellular enveloped virus (EEV), essential for efficient EEV formation and long-range viral spread within the host. It also plays an important role in effective cell-to-cell spread within the host [13, 14]. CaPV-A33R is predicted to be highly hydrophilic, with a predicted C-terminal transmembrane domain 183–204 aa with a high antigenic index, as predicted from its amino acid sequence. The poxvirus protein, namely A27L, A33R, B5R, and L1R, has been shown to induce neutralizing antibodies and immunodominant antigens [15].
The competitive ELISA (cELISA) was developed to detect serum antibody responses against many viruses [16, 17]. It is based on a labeled monoclonal antibody or polyclonal antibody specific to a target antigen. This antibody competes with serum antibodies for binding to the antigen, thereby enabling detection and measurement of pathogen-specific antibodies. Furthermore, since cELISA does not require a species-specific secondary antibody, it has an advantage over conventional ELISA in detecting serum antibodies in any animal species.
This study developed a competitive ELISA against LSDV based on LSDV A33 antigen and anti-A33 Polyclonal antibody, which offers the prospect of reliable, high-throughput sero-surveillance on a flock or herd basis.
Materials and methods
Sera
Cattles were experimentally infected with different capripoxvirus isolates, kindly provided by the Lanzhou veterinary research Institute, and sera were collected at various time intervals following vaccination. Cattles were infected with LSDV with Gansu in China isolates. Orf virus-positive sera were collected from sheep experimentally infected with the orf virus [18]. All animal were handled in accordance with the Good Animal Practice Requirements of the Animal Ethics Procedures and.
Guidelines of the People’s Republic of China, and the protocol was reviewed and approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, Chinese Academy of gricultural Science. Cattle sera from native animals or animals infected with LSDV, either experimentally or in field outbreaks, were obtained from Lanzhou.
Construction of A33 expression vector
A primer set (A33R-F/A33R-R) targeting A33R gene sequence (201–591), encoding for a truncated length A33R without the trans-membrane region as well as C-terminal region (aa) was designed based on a reference gene sequence available in GenBank (Acc. # DQ184476), forward primer A33R-F 5’- ccgGAATTCATGTTAGCATTTTTTAATAATAC-3’, and reverse primer A33R-R 5’- ccgAAGCTTTTAAAAAAAAGATCTTACACAG-3’. These oligonucleotides had added restriction enzyme sites (underlined) for EcoRI and Hind III, respectively, at the 5′ ends and primer tags (small letters).
The DNA extracted from LSDV/Xinjiang/China/2019 using a viral DNA extraction kit (TaKaRa) as the manufacturer’s protocol was used as a template in a PCR reaction mixture. A partial A33R gene was amplified using gene-specific primers (A33R-F and A33R-R). The PCR reaction mixture (50 µl) was comprised of primers (10 pmol of each) along with 5×Taq PCR Master mix (MBI Fermentas, USA) and a final volume made up by nuclease-free-water. The cycling condition was: initial denaturation at 95 °C for 5 min, followed by 34 cycles of denaturation at 94 °C for the 40 s, annealing at 55 °C for 40 s, extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. The amplified PCR product and pET30a vector were digested with EcoRI and Hind III enzymes and gel purified. In a standard reaction, the insert was ligated into the pET30a vector, and the recombinant plasmid (pET30a-A33R) was transferred into E. coli JM109 strain initially and subsequently into expression host cells E. coli BL21 (DE3) cells. The recombinant strain with pET30a-A33R plasmid named RpET30a-A33R and sequenced by Shanghai Biological Co., Ltd.
Expression, purification of the recombinant A33 fusion protein
RpET30a-A33R was cultured at 37 °C for 3 h with 200 rpm agitation after the OD600 value was reached 0.6, 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) was added and kept at 37℃ for 6 h. The sediments were collected and washed three times using PBS. The protein of LSDV A33 was purified by Ni-bind@ Resin (GE Healthcare bio-science, Sweden). The purified protein was dialyzed against phosphate-buffered saline (PBS), diluted 4:1 in glycerol after SDS-PAGE analysis, and stored at -70 °C before use in ELISA and immunization.
For confirmation, the proteins were resolved by electrophoresis on 12% Bis-Tris polyacrylamide gels (Shanghai Sangon Biotech, China) and transferred to polyvinylidene fluoride membranes (Immobilon-P Transfer membranes, Millipore). Membranes were blocked for 12 h at 4 °C in 5% (wt/vol) Tris-buffered saline supplemented with 0.1% Tween 20 (TBST)-diluted milk (BSA, Amresco) buffer. Membranes were incubated with primary antibody diluted in 5% (wt/vol) BSA and 1 × TBST at room temperature. The detection was carried out with 1:400 diluted LSDV positive serum as primary antibody and 1:15000 diluted rabbit anti-bovine IgG horseradish peroxidase (HRP) conjugate (Stan cru, USA) as a secondary antibody. The blots were detected by the enhanced chemiluminescence detection kit (#1705062, Bio-Rad) after incubation with an appropriate secondary antibody conjugated to horseradish peroxidase. All the membranes were imaged with the ChemiDoc XRS + system (Bio-Rad).
The polyclonal antibody preparation
Three experimental rabbits of three-month-olds were immunized subcutaneously with A33 protein antigen diluted in PBS and emulsified with an equal volume of complete Freud’s adjuvant (Sigma, USA). The rabbits were injected two times at an interval of 14 d with the same concentration using incomplete Freud’s adjuvant as a boost. Test bleeding was done after 14 d of final immunization, and an indirect ELISA confirmed the titer of the specific antibody.
Development of the competitive ELISA
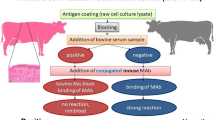
The competitive ELISA conditions were optimized by checkerboard titration using serial dilutions of A33 protein tested against serial dilutions of known positive and negative bovine sera.
A checkerboard titration was performed to determine the optimal working dilution of the coating antigen, serum, competitive antibody, and horseradish peroxidase-labeled mouse anti-rabbit IgG (HRP-IgG) (Santa Cruz, sc-2357) using a 96-well ELISA plate. Antigen coating concentrations were 16 µg/ml, 8 µg/ml, 4 µg/ml, 2 µg/ml, 1 µg/ml and 0.5 µg/ml per well, and serum dilutions were 1:2, 1:4, 1:8, 1:16, 1:32 and 1:64. The dilutions that gave the smallest difference between positive and negative serum (P/N) by absorbance at 450 nm were selected. Test sera included positive, negative, and blank sample controls, controls without serum.
After optimization, competitive ELISA was performed using the following procedure. Ninety-six well ELISA plates (Costar) were diluted in carbonate buffer with 1000 folds (protein concentration 2 mg/ml) coated with 100 µl and incubated overnight at 4°C. Then plates were sealed with 5 g/LCasein (Sigma) and incubated for 2 h at 37°C. After three washes with phosphate-buffered saline (PBS) containing 0.05% Tween 20 (PBST), serum samples were diluted 1:2 in dilution buffer (PBS), in a 50 µl volume well, the competitive antibody in a 50 µl volume well mixing and incubated for 30 min at 37°C, internal controls including positive, negative controls serum and 100% control wells without serum. After four washes, horseradish peroxidase-conjugated mouse anti-rabbit serum was added in the same dilution buffer at an appropriate working concentration, 100 µl per well, and incubated at 37°C for 30 min. After three washes, color was developed with 3,3’,5,5’-tetramethylbenzidine (TMB, Sigma), and the reaction was stopped after 15 min with 2 M H2SO4. The optical density (OD) of the samples were read at 450 nm with a microplate reader (Model 680. Bio-Rad).
The Percent inhibition (PI) value = (1- mean OD value at 450 nm of sample/ mean OD of 100% control well without serum at 450 nm)×100. Those normally applied in studies conducted in our laboratory were needed for considering a run valid: mean OD of 100% control wells = 1.5 ± 0.5, the PI of positive control ≥ 60%.
Panel sera employed for the validation of the method
A panel sample was employed, including 605 negative sera and 110 LSDV positive sera, divided into detailed categories.
350 field samples without LSDV exposure, collection of serum samples was performed in 3 Gansu province farms during 2017 (no LSD in china).
100 field samples of Yak without LSDV exposure, collection of serum samples was performed in Gansu province farms during 2018(there has no LSD in china).
155 field samples without LSDV exposure, collection of serum samples was performed in 2 Gansu province farms during 2020, and these were confirmed based on Capripox double antigen multi-species ELISA (ID.Vet).
40 positive sera came from the experimental infection (Lanzhou Veterinary Research Institute P3) and conformed based on PCR detection and Capripox double antigen multi-species ELISA kit (ID.Vet).
37 positive sera came from the immune bovine with inactivated Goatpox vaccine (Lanzhou veterinary research institute) and conformed based on Virus Neutralization Test (According to the OIE operating procedure).
33 positive sera with LSDV exposure were collected in Guangdong province, Xinjiang, and Hebei provinces and confirmed based on Capripox Double Antigen multi-species ELISA (ID.Vet).
1 positive serum of FMDV, 1 positive serum of BVDV, 1 positive serum of Brucella, 1 positive serum of bluetongue, and 1 Orf virus-positive sera were stocked in Lanzhou Veterinary Research Institute.
Cut-off estimation, statistical validation, and determination of the C-ELISA
A receiver operating characteristic (ROC) curve is a graphical representation of the relative effects of the false negative and false positive rates for every possible cut-off, and it is an effective method of evaluating the quality or performance of diagnostic tests. Therefore, the discriminating power of the competitive ELISA for anti-A33R antibody detection was evaluated by a ROC curves analysis [19] using Medcalc statistical software version 20.008 (https://www.medcalc.org/), trial version. The accuracy of a diagnostic test is displayed in an interactive dot diagram, where data of the negative and positive groups are displayed as dots on two vertical axes.
Validation, repeatability, and comparison of the c-ELISA
The coefficient of variation (CV) was calculated between plates (inter-assay variation) and within the same plate (intra-assay variation) for 30 sera samples (20 positive, 10 negative) to validate the test and evaluate repeatability. For inter-assay CV, each sample was tested on three different plates on different occasions, and for intra-assay CV, four replicates within each plate were assayed. A total of 30 experimentally generated evaluated by the ELISA and MNT.
Results
Construction of expression plasimids
The A33 gene fragment of 393 bp was amplified from LSDV DNA. The construction of the pET30a-A33R plasmid was confirmed by restriction enzyme digestion and sequencing analysis (Fig. 1).

Construction of pET-30a- A33R expression plasmid. Lane 1: The recombinant plasmid was digested by EcoRI and Hind III (∼393 bp). The length of A33R gene fragments is 393 bp, the length of the vector fragments is about 5422 bp. Lane 2: Agarose gel electrophoresis showing PCR amplified A33R gene, A33R gene fragments is 393 bp. M: DNA2000 Marker
Expression, purification and Western blot analysis of the pET-30a-A33R protein
The pET30a-A33R plasmid transformed BL-21 Escherichia coli was inoculated in LB medium with kan+ (50 µg/ml) using 0.5 mM IPTG to detect the expression of the fusion proteins. Total proteins were extracted and detected by Western blot with LSDV positive, negative serum, and rabbit anti-bovine mAb. The result showed that A33 protein was properly expressed and yielded a band of the molecular weight of 19 kDa, which is consistent with its predicted size,. The result showed that A33 protein reacted well with LSDV serum (Fig. 2a, b).

Analysis expression of the A33 protein. SDS-PAGE analysis of LSDV A33 expressed in E. coli BL21 (DE3). The expression vector pET-30a-A33 was transformed into the E. coli BL21 (DE3), protein expression was induced by adding 0.5 mM IPTG. Lane 1 The expression product in supernatant, Lane 2–3 Expression product in inclusion body, M: Marker of molecular. b. Western blot analysis of recombinant A33 protein with LSDV infected positive serum and anti-bovine HRP monoclonal antibodies. The result showed A33 protein could react with the LSDV infected positive serum. Lane 1 Expression A33 protein reacted with LSDV positive serum, Lane 2 Expression A33 protein reacted with LSDV negative serum, M: Marker of protein molecular
Detection of the polyclonal antibody
The collection of blood was conducted before every immunization and two weeks after the final inoculation, and antibody titer of all serum samples was measured using the enzyme-linked immunosorbent assay (I-ELISA) method in our lab, and the antibody titer was researched about 1:50000 use as the competitive antibody for the c-ELISA.
Optimization of the c-ELISA
The c-ELISA was optimized to make maximum discrimination between positive and negative samples based on the PI values. Different A33 protein concentrations and serum dilutions were used to obtain the maximal differences in the OD values between the known positive and negative control sera. The ELISA’s optimal conditions were 100 µl of the A33 protein at 2 µg/ml in coating buffer overnight, 50 µl serum without dilution and 50 µl competitive antibody, the optimal concentration of competitive antibody was 1:20000 for 30 min, and the concentration of HRP-conjugated mouse anti-rabbit IgG antibody was 1: 8000 for 30 min.
The cutoff values of positive and negative
490 negative samples were tested in duplicate by c-ELISA to set negative/positive cutoff values. The PI value (OD 450 of test serum/negative serum) of the 30 serum samples was compared with the VNT results. The end-point cut-off was determined by analyzing a receiver operating characteristic (ROC) curve based on PI values. Different cut-off values and their respective sensitivity (se) and specificity (sp) were obtained on ROC analysis. Among these, 57.61% PI value was accepted as the cut-off for c-ELISA at which se was 96.4% with 91.0–99.0 (95%) confidence interval, and sp of 98.51% with 97.2–99.3 (95%) confidence interval was achieved (Fig. 3a and b). The area under the curve in the c-ELISA was determined to be 0.986 at 0.974 to 0.993 confidence interval.

(a). Receiver operating characteristic (ROC) for estimation of cut-off values of c-ELISA. Sensitivity over (100-specificity) at different cut-off values. (b). Interactive dot diagram (0 = negative; 1 = positive); the data of the negative and positive groups are displayed as dots, corresponding to the vertical axes. Cut-off value decided at 57.61% inhibition level (horizontal line) differentiates the positive and negative sera samples
Analytical specificity of cELISA
No cross-reactivity of cELISA was detected with positive sera against a variety of other viruses such as orf, FMDV, BVDV, and Brucella, as all of them gave values below the defined positivity cut-off values (data not shown). These data indicated that the A33 antigen possessed a good analytical specificity.
Validation of the c-ELISA
The inter-assay CV ranged from 1.5 to 4%, and the intra-assay CV ranged from 4 to 7% for 30 different sera selected for validation testing (Table 1).
When detecting the immunity serum with c-ELISA, four cattle were positive for A33 antibody at 14 d first immunization, all cattle (n = 15) were positive at 14, 21, 28 d post-immunization (Table 2). VNT confirmed the results. When detecting the infection serum (Infection (BSL)-3 conditions) with c-ELISA, 2/6, 4/6 cattle were positive for A33 antibody at 9and 12 d post infection respectively, and 6/6 cattle were positive for A33 antibody at 15, 21, 28, 35 d (data not shown).
Discussion
It is important that diagnostic laboratories can efficiently and quickly detect LSDV infection when disease outbreaks occur. A precise and rapid diagnosis of LSDV infection is important for applying virological and serological diagnostic assays. Capripox double antigen multi-species ELISA has already been developed and used to detect antibodies to LSDV in other countries [20]. Indirect ELISA based on p32 and A27 Capripox virus particles as antibodies have been reported [21, 22]. However, the competitive ELISA used to detect antibodies to LSDV has not been reported. The present study describes a c-ELISA method based on the A33 antigen. A33 is a high conservative extracellular enveloped virus (EEV)-specific type II membrane glycoprotein in all capripoxvirus and contains an antigenic determinant and a target for neutralizing antibody responses against EEV [14]. Thus, A33 is important in the pathogenicity, diagnosis, prevention, and control of capripoxvirus [15]. Therefore, establishing ELISA detection methods with A33 protein as the antigen is essential since in LSDV- infected /immunity animals, middle and late-stage antibodies are produced against A33 protein. In this study, LSDV A33 protein was expressed well in E.coli system, and expressed product is mainly inclusion body, after purification, dialysis and refolding, was stored at − 40 ℃. Furthermore, the protein had good stability, it was used to coat plates and test the reference serum samples after stored for 24 months at − 40 ℃, the OD450 value didn’t decrease.
The optimization of a c-ELISA between the group-specific polyclonal antibody against A33 and a concentration of recombinant A33 antigen. The competitive ELISA was optimized to make maximum discrimination between positive and negative samples based on the PI values. The test was optimized to obtain the OD 450 of control without serum close to 1.5 while the OD 450 value of the positive control remains below 0.4. The A33 antigen, when adsorbed at a concentration of 200 ng/well, was found to be optimum to appreciate the competition, and the optimization dilution of serum and anti-A33 antibody at the of 1:1 and 1:20000, respectively. The optimum dilution of the secondary antibody conjugate (anti-Rabbit HRP) at this condition was found to be 1:8000.
A panel of serum samples of known antibody status was collected from cattle using ROC analysis. On ROC analysis, different cut-off values and their respective se and sp were obtained. When 57.61% PI value was accepted as the cut-off for develop c-ELISA, the se was 96.4% with 91.0–99.0 (95%) confidence interval and the sp of 98.5% with 97.2–99.3 (95%) confidence interval (Fig. 3a and b). The area under the curve in the c-ELISA was determined to be 0.987 (SE ¼ 0.00445) at 0.978 to 0.993 confidence interval, this showed a high specificity and sensitivity of the develop c-ELISA. The results also indicate the c-ELISA’s base on A33 protein has diagnostic potential for detecting specific antibodies in serum. But it is not enough good, perhaps the intervals of serum sample collected post-infection or immunization can affect the analytical sensitivity and specificity of detection. The coefficient of variation (CV) calculated for the normalized data obtained from replicates of the strong positive, weak positive, and negative serum are shown in Table 1. The result of the %CV of intra-inter batch duplicability tests was less than10%, which showed that the develop c-ELISA had good repeatability and specificity. In particular, the diagnostic se and sp of c-ELISA were evaluated by detection serum come from immune and infection bovine comparting it with VNT and clinical signs. All of 6 cattle were infected in (BSL)-3 laboratory, the serum was collected in different infection d, in which antibody could be detected starting 9 to 12 d, but VNT could be detected starting 12d, the result of c-ELISA was consistent with the VNT from post-infection 15 d (data not shown).VNT was recommended as gold standard, we confirmed that anti-A33 antibody has neutralizing activity, in this study, 6 infection serum in different days was detected, the result was agree with VNT after infection 15 day, the number of the samples was limited, in the follow up work, in order to improve the applicability more accurately, more serum samples that known bankground was detected by developing c-ELISA method. as expect, it is need to test more serum samples to confirm whether it can replace virus neutralization test.
In WOAH Terrestrial Manual 2023, ELISA method has been recommended as a diagnostic tool for evaluating the immune response and infection to LSDV because of the technology is simple and rapid. In this study, the obtained cut-off value gave an assay with a high degree of specificity and sensitivity. The assay also had good repeatability and promised to be useful in clinical application.
Conclusion
In this study, we established a c- ELISA using the A33 protein of LSDV as an antigen. The assay provides an alternative, inexpensive, and rapid serological detection method suitable for LSDV antibody detection on a large scale.
Data availability
Data and materials are available upon request from the corresponding author.
References
Hunter P, Wallace D. Lumpy skin disease in southern Africa: a review of the disease and aspects of control. J South Afr Veterinary Association-Tydskrif Van Die Suid-Afrikaanse Veterinere Vereniging. 2001;72:68–71.
Aye G, Haftu R, Jemberie S, Belay A, Gelaye E, Sibhat B, Skjerve E, Asmare K. Lumpy skin disease in cattle in central Ethiopia: outbreak investigation and isolation and molecular detection of the virus. Revue Scientifique Et Technique-Office Int Des Epizooties. 2014;33:877–87.
Alves RC, Carneiro RD, Kommers GD, de Souza AP, de Galiza GJN, Dantas AFM. 2020. Systemic candidosis in Dogs Associated with Canine Distemper Virus. Acta Sci Veterinariae 48.
Fagbo S, Coetzer JAW, Venter EH. 2014. Seroprevalence of Rift Valley fever and lumpy skin disease in African buffalo (Syncerus caffer) in the Kruger National Park and Hluhluwe-Imfolozi Park, South Africa. J S Afr Vet Assoc 85.
Tuppurainen ESM, Antoniou SE, Tsiamadis E, Topkaridou M, Labus T, Debeljak Z, Plavsic B, Miteva A, Alexandrov T, Pite L, Boci J, Marojevic D, Kondratenko V, Atanasov Z, Murati B, Acinger-Rogic Z, Kohnle L, Calistri P, Broglia A. 2020. Field observations and experiences gained from the implementation of control measures against lumpy skin disease in South-East Europe between 2015 and 2017. Preventive Veterinary Medicine 181.
Hamdi J, Boumart Z, Daouam S, El Arkam A, Bamouh Z, Jazouli M, Tadlaoui KO, Fihri OF, Gavrilov B, El Harrak M. 2020. Development and evaluation of an inactivated lumpy skin disease vaccine for cattle. Vet Microbiol 245.
Namazi F, Tafti AK. Lumpy skin disease, an emerging transboundary viral disease: a review. Veterinary Med Sci. 2021;7:888–96.
Sadri R, Khedmati K, Fallahi R, Mirsaidi FM. 2008. Standardization of a New Rapid Test to Detect Specific Antibody or Soluble Antigen in Viral Animal Disease (Pox disease in Sheep and Goats) by In vitro. 15th Congress of the Federation of Asian Veterinary Associations, Fava-Oie Joint Symposium on Emerging Diseases, Proceedings:P277-P278.
Haegeman A, De Leeuw I, Mostin L, Van Campe W, Aerts L, Vastag M, De Clercq K. 2020. An immunoperoxidase monolayer assay (IPMA) for the detection of lumpy skin disease antibodies. J Virol Methods 277.
Gari G, Biteau-Coroller F, LeGoff C, Caufour P, Roger F. Evaluation of indirect fluorescent antibody test (IFAT) for the diagnosis and screening of lumpy skin disease using bayesian method. Vet Microbiol. 2008;129:269–80.
Greiner M, Gardner IA. Application of diagnostic tests in veterinary epidemiologic studies. Prev Vet Med. 2000;45:43–59.
Babiuk S, Wallace DB, Smith SJ, Bowden TR, Dalman B, Parkyn G, Copps J, Boyle DB. Detection of antibodies against Capripoxviruses using an inactivated Sheeppox Virus ELISA. Transbound Emerg Dis. 2009;56:132–41.
Monticelli SR, Earley AK, Stone R, Norbury CC, Ward BM. 2020. Vaccinia Virus Glycoproteins A33, A34, and B5 form a complex for efficient endoplasmic reticulum to trans-golgi network transport. J Virol 94.
Matho MH, Schlossman A, Meng X, Benhnia MR, Kaever T, Buller M, Doronin K, Parker S, Peters B, Crotty S, Xiang Y, Zajonc DM. Structural and functional characterization of Anti-A33 antibodies reveal a potent cross-species Orthopoxviruses Neutralizer. PLoS Pathog. 2015;11:e1005148.
Pacchioni SM, Bissa M, Zanotto C, Morghen Cde G, Illiano E, Radaelli A. L1R, A27L, A33R and B5R vaccinia virus genes expressed by fowlpox recombinants as putative novel orthopoxvirus vaccines. J Transl Med. 2013;11:95.
Fu Y, Ji Y, Liu B, Dafallah RM, Zhu Q. Development of a solid-phase competition ELISA to detect antibodies against newly emerged duck Tembusu virus. J Virol Methods. 2015;224:73–6.
Mackay DK, Bulut AN, Rendle T, Davidson F, Ferris NP. A solid-phase competition ELISA for measuring antibody to foot-and-mouth disease virus. J Virol Methods. 2001;97:33–48.
Jia HJ, Zhan LL, Wang XX, He XB, Chen GH, Zhang Y, Feng Y, Wei YX, Zhang Y, Jing ZZ. 2017. Transcriptome analysis of sheep oral mucosa response to Orf virus infection. PLoS ONE 12.
Zhou X, H O NA, MeClish DK. 2002. Statistical Methods in Diagnostic Medicine.:Interscience, New York.
Kresic N, Simic I, Bedekovic T, Acinger-Rogic Z, Lojkic I. 2020. Evaluation of Serological tests for detection of antibodies against Lumpy skin Disease Virus. J Clin Microbiol 58.
Tian H, Chen Y, Wu JY, Shang YJ, Liu XT. 2010. Serodiagnosis of sheeppox and goatpox using an indirect ELISA based on synthetic peptide targeting for the major antigen P32. Virol J 7.
Dashprakash M, Venkatesan G, Kumar A, Sankar M, Arya S, Ramakrishnan MA, Pandey AB, Mondal B. Prokaryotic expression, purification and evaluation of Goatpox virus ORF117 protein as a diagnostic antigen in indirect ELISA to detect goatpox. Arch Virol. 2019;164:1049–58.
Acknowledgements
This study was made possible by the invaluable assistance provided by the livestock and poultry zoonotic diseases team staff of Lanzhou Veterinary Institute. We also thank Dr. Bin Dong (Department of chemistry and Biochemistry, University of Arkansas) for the article revision.
Funding
This work was supported by The Science and Technology Program of Lanzhou (2023-1-38). This work was supported by the National Special Project for Monitoring and Prevention of Animal Epidemics (102125211610310009012) and the Independent Project of the State Key Laboratory of Pathogenic Biology of Animal Diseases (SKLVEB2021CGQD03) and The Innovation Program of Chinese Academy of Agricultural Sciences, CAAS-ASTIP-2021-LVRI.
Author information
Authors and Affiliations
Contributions
Conceptualization, G.C. 、 X.H.;Z.Jand Hurisa T.T.; methodology, G.C., and Z.G.; software, G.C; validation, Y.F., H.J. and Jl formal analysis, G.C.; investigation, G.C.; writing—original draftpreparation, G.C.; writing—review and editing, B.F, W.L and Z.J.; supervision, Z.J.; funding acquisition, Z.J and G.C. Allauthors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All trials were constructed with the approval of the Ethical Committee of the Lanzhou Veterinary Research Institute of the Chinese Academy of Agricultural Sciences. All participants were provided with written consent for this study. The study objectives were explained to all participants before obtaining informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The result of this study does not reflect the opinion of the funding sources. All authors have read and approved the final version of this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Chen, G., He, X., Gao, Z. et al. Development of a competitive ELISA based on the LSDV A33 antigen. Virol J 21, 203 (2024). https://doi.org/10.1186/s12985-024-02448-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-024-02448-1