
Abstract
Background
Limb-girdle muscular dystrophy recessive 1 calpain3-related (LGMDR1), previously known as LGMD2A, is a disease caused by mutations in the CAPN3 gene. It is characterized by progressive weakness and muscle degeneration. Frizzled related protein (FRZB), upregulated in LGMDR1, was identified as a key regulator of the crosstalk between Wnt and integrin signalling pathways. FRZB gene silencing showed a recovery in the expression of some of the costamere protein levels in myotubes.
Results
Here, we performed a comprehensive characterization of Frzb−/− mice muscles to study the absence of Frzb in skeletal muscle and eventual links with the molecular characteristics of LGMDR1 patient muscles. Frzb−/− mice showed reduced muscle size and strength. Gait analysis showed that Frzb−/− mice moved more slowly but no impaired regeneration capacity was observed after muscle injury. Additionally, Frzb−/− mice muscle showed an increased number of mesoangioblasts. Lack of Frzb gene in Frzb−/− mice and its increased expression in LGMDR1 patients, showed contrary regulation of Rora, Slc16a1, Tfrc and Capn3 genes. The reciprocal regulation of Frzb and Capn3 genes further supports this axis as a potential target for LGMDR1 patients.
Conclusions
Our data confirm a role for Frzb in the regulation of Rora, Slc16a1, Tfrc, and Capn3 genes in muscle cells. In vivo, reduced muscle strength and gait in the Frzb−/− mice are intriguing features. The reciprocal relationship between FRZB and CAPN3 further supports a key role for this axis in patients with LGMDR1.
Similar content being viewed by others


Muscular dystrophies are a heterogeneous group of genetic disorders characterized by progressive weakness and muscle degeneration. Among these, Limb-girdle muscular dystrophy recessive 1 calpain3-related (LGMDR1), previously known as LGMD2A, is a disease caused by mutations in the CAPN3 gene [1]. LGMDR1 was considered the most frequent type of LGMD worldwide [2,3,4,5] up till now, although new data suggest that this may not apply to some regions of Latin America [6]. In LGMDR1 patients, disease symptoms caused by muscle wasting and gradual degeneration of the proximal muscle groups, usually first present during the second decade of life and progressively worsen with patients becoming wheelchair-dependent after less than 25 years of evolution of the disease [7, 8]. The pathophysiological mechanisms underlying the process of muscle degeneration in the absence of functional CAPN3 protein are still largely unknown.
Loss of CAPN3 leads to abnormal sarcomere formation [9] as well as changes in the expression of several genes in the muscles of LGMDR1 patients [10]. Sarcomere assembly and stabilisation are dependent on a protein complex called the costamere [11,12,13]. Its function is to enable the adhesion between the sarcomere in the muscle and the extracellular matrix [14]. This linkage is partially mediated by integrins [15]. In LGMDR1 myotubes, the physiologically required replacement of the integrin β1 isoforms (β1A substitution by β1D) is disturbed and may be the cause of incorrect costamere assembly. Our studies on integrin interacting proteins as well as proteins implicated in costamere regulation, identified frizzled related protein (FRZB) as a key regulator of the crosstalk between integrin and Wnt signalling pathways [16].
FRZB was originally identified as a secreted antagonist of the Wnt signalling pathway, blocking the effects of Wnt-1, Wnt-8, Wnt-5a and Wnt-9a [17,18,19,20]. In muscle tissue of LGMDR1 patients FRZB gene expression is strongly upregulated [10] and FRZB is a negative regulator of myogenesis [21]. Upon silencing of FRZB gene expression in LGMDR1 patients’ myotubes, we earlier showed a recovery in the expression of some of the costamere protein levels such as integrin β1D, melusin and anosmin-1 [16].
Frzb-deficient mice were shown to be highly susceptible to the development of osteoarthritis characterized by damage to the articular cartilage, and also have increased cortical bone density [22]. Of note, Frzb−/− mice showed reduced voluntary exercise performance in running wheels [23]. Here, we aimed to further study the effects of absence of Frzb in skeletal muscle of Frzb−/− mice, and eventual links with the molecular characteristics of LGMDR1 patient muscles.
Results
Skeletal muscle analysis
Routine histological analysis throughout our experiments did not reveal any striking differences in muscle structure between wild type and Frzb−/− mice. At the age of 5 weeks, male and female Frzb−/− mice were on average 4.92 g [95%CI: 3.09–6.75; p < 0.0001] and 3.95 g [95%CI: 2.04–5.86; p < 0.0001] smaller in weight compared to wild-type mice [F(1,25) = 64.2 (p < 0.001) - 17.6 (p = 0.003) for genotype and sex by two-way ANOVA]. At the age of 8 weeks, these differences persisted only in female mice [difference between means 1.35 g (95%CI: 0.29–2.40; p = 0.010) with F (1,27) = 9.4 (p = 0.005) for interaction between sex and genotype by two-way ANOVA] (Fig. 1a-b). This weight difference was not observed earlier in Frzb−/− mice on an outbred Swiss/CD1 background [23].

Frzb−/− mice are smaller than wild-types (WT) with reduced muscle size and strength. a-b Body weight (g) of 5- and 8-week-old WT and Frzb−/− mice [n = 8 WT-females, 7 WT-males, 6 Frzb−/−-females and 8 Frzb−/−-males; *** p < 0.0001, p < 0.0001 and **p = 0.01, Bonferroni-corrected for two tests in two-way ANOVA]. c Muscle weights (mg) of 4–5-week-old WT and Frzb−/− mice [n = 15 WT and 14 Frzb−/− mice, *** p < 0.0001, **p = 0.009, Bonferroni-corrected for four tests in two-way ANOVA]. d Real-time PCR analysis of Myh1, Myh2 and Myh4 in Tibialis anterior and Soleus of WT and Frzb−/− mice (n = 3 WT and 3 Frzb−/− mice for Tibialis anterior and 2 WT and 4 Frzb−/− for Soleus samples). e Muscle strength reflected by hanging time in male WT and Frzb−/− mice [n = 9 WT and 7 Frzb−/− mice, * p = 0.048 for effect of genotype, p = 0.8 for effect of weight in two-way ANOVA]. Error bars indicate mean ± SEM
To determine whether the absence of Frzb in mice affects baseline muscle mass and characteristics, we isolated different muscle groups (Gastrocnemius, Tibialis anterior, Quadriceps and Biceps brachii) from male and female wild-type and Frzb−/− mice aged 4 to 5 weeks. Muscle weight for the different muscle groups was significantly lower in Frzb−/− mice compared to wild-type controls. There was a statistically significant interaction between muscle type and genotype on weight [F(1,25)=5.455 (p = 0.019) by two-way ANOVA]. The Gastrocnemius, Quadriceps and Biceps muscle were on average 28.3, 28.02 and 14.98 mg heavier in wild-types compared to Frzb−/− mice [(95%CI: 18.22–38.41; p < 0.001), (95%CI: 17.92–38.11, p < 0.0001) and (95%CI: 4.875–25.066, p = 0.009), Bonferroni-corrected for four tests in two-way ANOVA] (Fig. 1c). When muscle weight was normalized to body weight, no differences were observed (data not shown) suggesting that the differences may be related to growth retardation.
Earlier analysis in Frzb−/− mice on the Swiss/CD1 background did not reveal differences in type I vs type IIa fibres in the Soleus or in type IIa vs type IIb fibres in the Extensor digitorum longus [23]. Here, we further analysed fibre composition at the gene expression level: different myosin heavy chain isoforms (Myh1, Myh2 and Myh4) were analysed in 10-week-old male and female Frzb−/− and wild-type mice. The distinct genotypes did not show differences in the expression of the respective myosins (Fig. 1d). As expected Myh4 (myosin present in 2B fibre type) followed by Myh1 (myosin present in 2X fibre type) were highly expressed in the Tibialis anterior while Myh2 showed only minimal expression (myosin present in 2A fibre type). In the Soleus expression of Myh1 and Myh2 was balanced while Myh4 was minimally expressed.
To screen for a potential functional impact of these observations, we assessed mouse muscle strength and endurance by the four-limb hanging test. Male Frzb−/− mice were performing inferior to wild-type mice in this set-up, independent of weight [difference between means 895.52 s (95%CI: 10.58–1780.47) in 2-way ANOVA with F(1,12) = 4.861; p = 0.048 for genotype] (Fig. 1e). Taken together, these analyses suggest that Frzb−/− mice have reduced muscle size and strength that is not explained by obvious changes in fibre types.
Gait analysis
We earlier demonstrated that deletion of Frzb reduces voluntary running exercise performance in mice using running wheels [23]. We therefore set-out to analyse the gait of the Frzb−/− mice compared to wild-type controls to evaluate the global mobility of these mice by Catwalk gait analysis [24]. Frzb−/− mice paws spent significantly more time in contact with the glass plate and in the air as shown by stand and swing phase analysis respectively (Fig. 2a-b). There was a statistically significant interaction between the effects of genotype, sex and front- or hind-paw on stand [F(1,34)=4.84, p = 0.04] and swing phase [F(1,34)=12.53, p = 0.01] by two-way ANOVA, subsequently Bonferroni corrected post-hoc tests were carried out. For male mice, stand was different between wildtype and Frzb−/− mice in both front- and hind paws [p = 0.002 and p < 0.001 respectively], but there was no evidence that the stand differed in females. Male Frzb−/− mice had on average 0.053 [95%CI: 0.028–0.077] and 0.066 [95%CI: 0.041–0.091] seconds longer stand time in front and hind-paws compared to wildtypes. Similarly, swing was different between male wildtype and Frzb−/− mice in both front- and hind-paws [p < 0.001 and p = 0.005 respectively], but there is no evidence that the stand differed in females. Male Frzb−/− mice had on average 0.045 [95%CI: 0.029–0.060] and 0.029 [95%CI: 0.014–0.044] seconds longer swing time in front and hind-paws compared to wildtypes. As a consequence, step cycle was on average 0.097 s longer in male Frzb−/− mice [95%CI: 0.060–0.134, p < 0.001] (Fig. 2c). Although Frzb−/− mice moved more slowly (increased stand and swing phase), the distance covered by their paws, stride length, did not differ between WT and Frzb−/− mice (Fig. 2d).

Frzb−/− mice moved more slowly with no differences in stride length. a-d Gait analysis of 8-week-old female and male wild-type and Frzb−/− mice. Average stand, swing phase, step cycle and stride length analysed with ANOVA accounting for genotype (WT, KO), gender (yes, no), paw [front paw (FP), hind paw (HP)] and all interactions. Error bars indicate mean ± SEM. *** p < 0.0001, **p < 0.01 by Bonferroni-corrected test for four tests in two-way ANOVA
Treadmill exercise of mice
To document whether Frzb−/− and wild-type mice react differently to exercise, animals were subjected to treadmill running. All animals in the experiment were able to perform the selected chronic running exercise protocol and no abnormal behaviour or signs of exhaustion were observed. First, mice body weights were measured, differences between WT and Frzb−/− body weight were gradually diminishing over time, and no differences between trained and not-trained groups were seen [2-way ANOVA (F (11,41) = 5.850 (p < 0.001) for interaction age and genotype, F([11,41) = 0.972 (p = 0.97) for interaction age, genotype and running] (Fig. 3a). Fibre cross-sectional area of Soleus was measured. Although no overall differences between cross-sectional areas were observed between trained and not-trained groups, Frzb−/− mice fibres were smaller than WT fibres, most notable in the not-trained group [2-way ANOVA (F(1,11) = 11.13 (p = 0.0066), 0.51 (p = 0.4865), 0.22 (p = 0.64) for genotype, running and interaction respectively, difference between means 2.7*10− 4 mm2(95%CI: 2.05*10− 5 – 5.2*10− 4; p = 0.034) and 1.7*10− 4 mm2 (95%CI: − 6.46*10− 5 – 4.1*10− 4; p = 0.17) Bonferroni-corrected for 2 tests in non-trained and trained mice] (Fig. 3c). No exercise-induced damage was observed in any of transverse sections of Soleus or Tibialis anterior (Fig. 3d). Muscle fibre type composition was analysed in both study groups in Tibialis anterior muscles and no differences were observed after exercise or between genotypes (Fig. 3d). Gene expression markers for muscle damage were not changed by exercise or genotype, with Fbx2 as a positive control for the effect of training (difference between the means 44.67, 95%CI: 23.49–65.85) (Fig. 3e).

Frzb−/− and wild-type (WT) mice do not respond differently to exercise. a Differences between WT and Frzb−/− body weight during treadmill exercise [n = 4 (WT-not-trained male), 5 (WT-trained male), 2 (Frzb−/−-not-trained male and female) and 3 (Frzb−/−-Trained male), p < 0.001 for Frzb−/− vs WT over time by two-way ANOVA]. b Hematoxylin-eosin staining of Soleus from WT and Frzb−/− mice at the end of the treadmill experiment (scale bar 250 μm) and (c) muscle fibre cross-sectional area measurement [n = 4 (WT-not-trained male), 5 (WT-trained male), 3 (Frzb−/−-not-trained 1 male and 3 female) and 3 (Frzb−/−-Trained male)] * p = 0.0066 for genotype by two-way ANOVA]. d SDH staining of Tibialis anterior from WT and Frzb−/− at the end of the experiment and fibre type quantification. e Real-time PCR analysis of Fbx32, Murf1, Myh3, Myh2, Pax7, Myod and Myog in Gastrocnemius of WT and Frzb−/− mice (n = 3 male for each group). Error bars indicate mean ± SEM
Muscle regeneration capacity after intramuscular cardiotoxin-induced muscle injury
To test whether muscle regenerative capacities of Frzb−/− mice were different from wild-type mice, acute skeletal muscle injury was triggered by cardiotoxin injection in the Soleus and Tibialis anterior muscles. No differences in pathology characteristics were noted between the two mouse strains. Non-injured skeletal muscle shows polygonal myofibres with peripheral nuclei (Fig. 4a-b). At day 3 post injection, muscles showed degenerative myofibres and inflammatory cellular infiltration. In the Soleus, most of the fibres were damaged, while in the case of Tibialis anterior, just the cardiotoxin injection area appeared affected (Fig. 4a-b). One week after injury, small regenerating myotubes with centrally located nuclei were observed with some remnants of inflammation. Regenerating fibers with centrally located nuclei increased their diameter by two weeks and the pattern became homogenous by four weeks (Fig. 4a-b).

Cardiotoxin (CTX) injection trigger no differences in damage between Frzb−/− and wild-type (WT) mice. Hematoxylin-eosin staining of (a) Tibialis anterior and (b) Soleus sections of 10-week-old female and male WT and Frzb−/− mice after 3, 7, 14 and 28 days after cardiotoxin injection. Scale bar (a) 250 μm and (b) 50 μm
In vitro muscle cell differentiation
Taking into account that Frzb affects myogenesis and that myotube differentiation and costamere assembly appear disturbed in LGMDR1 patients and models, we studied muscle cell differentiation in the different genetic backgrounds, using two different cell populations with myogenic potential. First, we isolated satellite cells from four-weeks-old wild-type and Frzb−/− mice using the Biceps brachii, Gastrocnemius, Tibialis anterior and Quadriceps. In proliferation assays, immunofluorescence analysis showed more MyoD positive cells in Frzb−/− mice muscles after enzymatic digestion and increased levels of the proliferation marker protein Ki67, represented as a percentage [P = 0.0046 and P < 0.0001 respectively, t-test] (Fig. 5a). However, after differentiation was induced, at the myotube stage fusion index and numbers of MyoD and myogenin positive nuclei were not different between the wild-type and Frzb−/− mice (Fig. 5b).

Frzb−/− mice have increased MyoD and Ki67 positive satellite cells but no detectable differences in differentiation capacity. a Immunofluorescence analysis of satellite cells extracted from 4-week-old WT and Frzb−/−. Left images stained for MyoD (green) likewise right images for Ki67 (red). Percentage of MyoD and Ki67 positive nuclei [n = 7 (WT 3 male and 4 female) and 6 (Frzb−/− 3 male and 3 female), P = 0.0046 and P < 0.0001 respectively, t-test]. b Immunofluorescence analysis of WT and Frzb−/− myotubes at day 3 of differentiation. Left images stained for nuclear myogenin (red) and cytoplasmatic sarcomeric α-actinin (green). Right images stained for nuclear MyoD (green) and cytoplasmatic MyHC (red). Myotubes fusion index, calculated as the percentage of nuclei inside myotubes on the total amount of nuclei. In all cases nuclei were visualized with Hoechst (blue). Scale bar 250 μm. Error bars indicate mean ± SEM
In addition, we isolated mesoangioblasts (MABs) from five-week-old wild-type and Frzb−/− mice using the Biceps brachii, Gastrocnemius, Tibialis anterior and Quadriceps as source. Freshly isolated cells were analysed by FACs analysis using alkaline phosphatase (ALP) as a cell surface marker. The obtained ALP+ cell distribution was different between wild-type and Frzb−/− mice. Percentage of ALP+ cells from Frzb−/− mice was on average 39.59 higher than from wild-type animals [95%CI: 26.18–53.00; p < 0.0001), Bonferroni corrected for 2 tests in 2-way ANOVA with F(1,22) = 46.73 (p < 0.001) for interaction between genotype and cell type] (Fig. 6a). Further characterization of the ALP+ cells showed that all of them were indeed negative for endothelial cell marker CD31 and hematopoietic cell marker CD45. Nevertheless, while wild-type ALP+ cells were around 81% positive for platelet derived growth factor receptor alpha (CD140A or PDGFRα), Frzb−/− ALP+ cells were only around 43% positive for this marker [p = 0.043 for CD140A, t-test] (Fig. 6b).

Frzb−/− mice have an increased percentage of ALP-positive mesangioblasts compared to wild-type (WT) animals. a Percentage of alkaline phosphatase positive and negative cells [n = 7 (WT 4 male and 3 female) and 6 (Frzb−/− 4 male and 2 female), *** p < 0.001 Bonferroni-corrected for two tests in two-way ANOVA]. Error bars indicate mean ± SEM. b FACS data showing CD31, CD45 and CD140A expression with mean ± standard deviation [n = 3 (WT male) and 7 (Frzb−/− 4 male and 2 female), p = 0.043 for CD140A, t-test]
Molecular analysis of the muscles
To screen for molecular differences between the muscles of Frzb−/− and wild-type mouse muscles and find eventual links with LGMDR1, we performed gene expression analysis on isolated muscles. First, we focused on muscle-specific genes. On one hand, no differences were found for Pax7. However the expression of Myod was on average 52.73% higher in Frzb−/− mouse muscles compared to wild types [(95%CI: 18.18–87.33) by Mixed Effect analysis with F(1, 17)= 10.34 (p = 0.005) for genotype]. A similar trend was observed for myogenin [(95%CI: − 1715–76.59] by Mixed Effect analysis with F(1, 17)= 4.07 (p = 0.06) for genotype]. (Fig. 7a). Muscle atrophy-related ubiquitin ligases Fbx32 and Murf1 did not show differences between wild-type and Frzb−/− mice (Fig. 7b). The analysis of adipocyte genes Pparg, Adipo and Fasn, showed no differences between wild-type and Frzb−/− mice (Fig. 7c).

Real-time PCR analysis of Tibialis anterior and Soleus of 10-week-old wild-type (WT) and Frzb−/− mice. a muscle specific genes Pax7, Myod and Myog (> 0.05, p = 0.005 and 0.06 respectively), (b) muscle atrophy-related ubiquitin ligases Fbx32 and Murf1 (all p > 0.05), (c) adipose tissue related genes, Pparg, Adipoq and Fasn (all p > 0.05), (d) genes deregulated in Capn3−/− mice, Park2 and Ky (p = 0.05 and 0.0024), (e) upregulated genes in Frzb−/− mice, Capn3 and Rora (p = 0.014, 0.0025) and (f) genes differentially expressed in muscles from LGMDR1 patients and in the articular cartilage of Frzb−/− mice, Tfrc and Slc16a1 (p = 0.0005, 0.007). Error bars indicate mean ± SEM
We then focused on genes that are differentially expressed in Capn3−/− mice compared to wild-type mice [25]. Park2 expression was on average 17.93% higher in Frzb−/− mice compared to wild-types [(95%CI: − 8 – 43.68) by Mixed Effect analysis with F(1, 7)= 5.29 (p = 0.05) for genotype]. Ky expression was on average 35.21% lower in Frzb−/− mice [(95%CI: 15.15–55.77) by Mixed Effect analysis with F(1, 12)= 4.26 (p = 0.0024) for genotype] (Fig. 7d). These differences were most pronounced in the Soleus muscles.
Other genes of interest were identified from the analysis of LGMDR1 patient muscles with CAPN3 mutations. Interestingly, Capn3 expression was on average 63.76% increased in the Soleus muscle of Frzb−/− mice compared to wildtypes [(95%CI: 21.15–106.4) by t-test with p = 0.0142] but not different in Tibialis anterior or Quadricipes muscles. This was also the case for the Rora gene [79.17% increased (95%CI: 46.76–111.6) by t-test with p = 0.0025] in the Soleus but not in the Tibialis anterior (Fig. 7e). On the other hand, Tfrc and Slc16a1 gene expression were 74.26% [95%CI: 54.34–94.17] and 73.67% [95%CI: 33.63–113.7] downregulated in the Soleus of Frzb−/− mice compared to wildtypes [p = 0.0005 and p = 0.0069 by t-test] (Fig. 7f). Again, no differences were found in the Tibialis anterior muscles. Of note, Tfrc, Rora and Slc16a1 were not only differentially expressed in muscles from LGMDR1 patients but also in the articular cartilage of Frzb−/− mice [10, 26].
FRZB silencing in human muscle cells
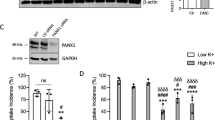
The effect of specific genes of interest were translationally validated in human myotubes by silencing the expression of the human FRZB gene. As shown in Fig. 8a, silencing of FRZB was successful in both control and LGMDR1 patient myotubes. On average FRZB expression was 84.33% reduced [(95%CI: 44.99–123.7) by two-way ANOVA with F(1,8)=24.4 for silencing (p = 0.0011)]. Myogenic markers MYOD and MYOG were not different between FRZB silencing and control conditions (Fig. 8b-c). CAPN3 gene was on average 28.22% upregulated after FRZB silencing [(95%CI: 6.61–49.82) by two-way ANOVA with F(1,8)=9.07 (p = 0.0168) for silencing] but the effect was not different between LGMDR1 patients and the healthy donors (Fig. 8d).

Real-time PCR analysis of healthy individuals and LGMDR1 patients’ myotubes at day 10 of differentiation in FRZB silencing experiments. aFRZB gene expression. bMYOG and (c) MYOD gene expression. dCAPN3 gene expression. Expression levels are relative to housekeeping gene GAPDH, * p = 0.0168 ** p = 0.0011 by two-way ANOVA
Discussion
The potential role of FRZB in LGMDR1 is intriguing as patients have high levels of FRZB expression in their muscles [10]. In myotubes of LGMDR1 patients, correct costamere assembly and cell fusion appears to be disturbed and this has been linked with the absence of the required integrin isoform replacement from β1A to β1D. Remarkably FRZB silencing in myotubes leads to costamere protein rescue. We therefore suggested that FRZB may be a potential therapeutic target for LGMDR1 patients [16]. To better understand the function of endogenous FRZB in muscles, different aspects of muscle biology in the Frzb−/− mouse model were studied at the functional, cellular and molecular level in an exploratory analysis.
Muscle weakness is the hallmark of muscular dystrophies. Our analysis suggests that Frzb−/− mice have a lower muscle mass that may result in reduced performance in the four limb hanging test. These findings seem to correspond to a stunted growth since no differences in body weight were previously observed in 6-month and 1-year old mice [23]. We have no evidence that weakness would evolve over time as this parameter was only evaluated at one time-point.
We also performed gait analysis of Frzb−/− mice to gain insights into the effect of Frzb deficiency on muscle function. Frzb−/− mice showed a longer step cycle, spending more time with the paw in contact with the walkway (stand time) as well as airborne (swing time). Thus, their limb movement was slower, however with no effect on the covered distance. In a previous report, we showed that Frzb−/− mice ran daily significantly lower distances in a voluntary running wheel setup [23]. This reduced voluntary running exercise performance could be attributed to the lower speed rather than to the fact that they spend less time running. Thus, Frzb−/− mice appear to have detectable issues in speed and mobility compared to wild-type controls. Although interesting from a scientific point of view, these observations cannot be translated into clinically meaningful data that could be applied to dystrophy patients. Anomalies that can be noted in muscular dystrophy patients are linked with weakness of the hip abductor muscles, producing a Trendelenburg gait characterized by “waddling” [27] and differ from what was observed in Frzb−/− mice. In addition, the instability and altered step patterns observed among LGMD patients that have been already characterized in a mouse model [27] are different to what was observed in this study.
To better understand the reasons why gait and muscle strength abnormalities were observed, several other factors were analysed. Jackson and collaborators (2015) showed that depletion of Pax7 expressing satellite cells in muscles resulted in reduced voluntary wheel running performance. Hence, Pax7 expression was analysed. No differences with wildtype animals were shown. Increased adipose tissue infiltration of muscles is a common hallmark in neuromuscular disorders [28,29,30]. Frzb−/− mice have lower body and muscle mass. However, Frzb−/− mice did not show fatty infiltration, centrally located nuclei, fibrosis or other dystrophic features in the analysed muscle sections. Regulators of adipocyte differentiation, peroxisome proliferator activated receptor gamma or adiponectin coded by Pparg and Adipoq respectively were not different between the studied mouse strains. In addition to fat infiltration, atrophy can also cause muscle weakness, by upregulation of atrogenes (MAFbx/Atrogin-1 and MuRF1) that lead to loss of muscle mass [31,32,33]. None of the atrogenes were up-regulated in Frzb−/− muscles. Therefore, we have no evidence that fat or atrophy could be the reason why mice spent more time to complete a step cycle.
Gait and other phenotypes reported here might not only be altered by impairment in muscles; but also by changes in the nervous system or bones. So far, there is no clear evidence of alterations in the nervous system of Frzb−/− mice. Some reports confirm dynamic expression of Frzb in neural cells but we are not aware of specific data on motor neurons, motor cortex or cerebellum that would explain our observations [34,35,36,37,38]. Nevertheless, the transgenic mice are known to have thicker cortical bone, with increased stiffness and higher cortical appositional bone formation after loading of the long bones [22]. These differences with wild-type animals cannot be excluded as factors contributing to the changes observed in Frzb−/− mouse gait and other phenotypes.
Exercise has direct effects on muscles, triggering changes in CSA of the fibres, fibre type distribution, weight change and potentially muscle injury [39]. Although physical training induces beneficial adaptive changes in skeletal muscle of healthy individuals, its effects in patients with muscular dystrophy remain controversial. While some studies attributed a beneficial effect without reporting muscle injury, other studies reported training-induced muscle damage and creatine kinase elevations in high-intensity training programs in patients, or even an earlier onset of symptoms associated with exercise in LGMD2B patients [40,41,42,43,44]. Since distinct muscular dystrophies show different progression of muscle degeneration and strength loss, leading to diverse exercise tolerance, endurance treadmill training tolerance and muscle changes were studied in Frzb−/− model. The lack of structural changes in Frzb−/− mice after exercise suggests the absence of a severe phenotype.
The CSA of muscle fibres in Frzb−/− mice was smaller. Consequently, we studied muscle fibre type composition. Tibialis anterior and Soleus fibre type distribution were within normal range values as has been described for C57Bl/6 mice [45, 46]. Previous studies in Soleus and Extensor digitorum longus analysed by immunofluorescence showed, similar fibre composition in WT and Frzb−/− mice [23]. Additionally, myosin gene expression as well as SDH activity were analysed obtaining the same result: fibre composition in Soleus and Tibialis anterior Frzb−/− did not vary from wild-type mice.
Several muscular dystrophy models, such as syntrophin α1 null mice and murine models for LGMDR12 (LGMD2L) and LGMDR1, showed aberrant muscle regeneration with longstanding necrosis and impaired exercise and contractile properties with aberrant neuromuscular junctions [47,48,49]. However, in Frzb−/− mice, after cardiotoxin injection, no aberrant or impaired regeneration capacity or fibrosis was noticed. In summary, Frzb−/− mice muscles showed normal fibre composition and they do not display altered regeneration capacity.
Murine primary cell cultures have been widely studied for myogenesis and muscular dystrophies in vitro analysis such as LGMDR1, LGMD2I or DMD [9, 16, 50,51,52,53,54,55]. We isolated satellite cells and we found that Frzb−/− mice cells showed enrichment for MyoD and Ki67 nuclear proteins. FRZB inhibits MyoD expression at RNA and protein level [21, 56]. We here show that in the absence of FRZB, MyoD was upregulated in our cells. As myotube formation was not altered, myogenesis may not be strongly impaired, but further studies will be required to identify the consequences of MyoD during myogenesis in the absence of FRZB.
On the other hand, the increased Ki67 expression, which is a proliferation marker, suggested an increased proliferative capacity in Frzb−/− muscles. However, there is controversy about the way in which the presence or absence of FRZB could affect proliferation. Some studies described that FZRB inhibits the growth of mesoangioblasts and suppressed cell proliferation in gastric cancer [57, 58], but other authors suggested that Frzb suppression reduced proliferation in alveolar rhabdomyosarcoma [59]. Moreover, tissue dependent differences have been observed in the same model, since Frzb−/− mice chondrocytes proliferated less than those obtained from WT mice, contrary to the observation in satellite cells in the same mice.
Considering that some muscle resident cell population are able to generate muscle, both in vitro and in vivo [60,61,62] pericyte-derived adult MABs were also studied. Pericyte–derived adult MABs are isolated from adult muscles and they retain similar characteristics of embryonic MABs [63,64,65,66]. Detailed analysis of the ALP+ cells showed significantly less PDGFRα expression. So far, two types of pericytes have been described: type-1 and type-2. Both of them differ in their cell surface markers as well as in their differentiation capacity. Type-1 are Nestin−/PDGFRα+ and are characterized by their ability to differentiate into adipocytes while type-2 are Nestin+/PDGFRα- and do not differentiate into adipocytes but form myotubes in culture [67, 68]. The lower PDGFRα expression could indicate that Frzb−/− mice have more type-2 pericytes.
In previous studies, differentially expressed genes have been analysed in the articular cartilage-subchondral bone biomechanical unit of Frzb−/− mice [22, 26]. However, gene expression analyses in muscle have not been carried out. Thus, one aim of this study was to establish whether Frzb deficiency impairs muscle gene expression in Frzb−/− mice. The analysis was focused mainly on the Soleus, as Soleus showed the greatest molecular similarities to human skeletal muscles [69] and since together with diaphragm these are the most affected muscles in Capn3−/− mice [9].
Myogenesis, a process that takes place during growth and regeneration in adult, depends on satellite cell activation of Pax7 cells and it is regulated by muscle-specific transcription factors such as MyoD and Myogenin [70, 71]. In the studied samples Pax7 was not upregulated, MyoD was upregulated in Frzb−/− and there was some increase in Myog. The same was observed in the FRZB silenced human samples; MYOD expression was upregulated. In Xenopus, Frzb inhibits axis duplication induced by Xwnt8 and also muscle development by blocking MyoD induction [18, 56, 72]. In mammals, myogenesis inhibition by Frzb accompanied by reduction in Myf5 and MyoD expression was reported [21]. Although most of the works were carried out during embryonic development, the possibility that Frzb has a role in adult myogenesis or muscle maintenance, modualting MyoD levels, should be considered. Although Myod was upregulated in Frzb−/− mouse muscles, increased myogenesis was not observed (centrally located nuclei were absent and different size fibres were not observed). MyoD increase after siFRZB in LGMDR1 patients could be considered as a beneficial consequence given that muscle degeneration stimulus is occurring and consequently new myofibres formation would be necessary. Nevertheless, further studies will be required to analyse if this increase improves cell physiology.
Among the selected genes deregulated in C3KO mice [25] the Ky gene showed expression changes in Frzb−/− mice. Its protease activity targets different proteins and its absence could disrupt muscle cytoskeleton homeostasis [73]. Natural ky mutant mice has smaller muscles with slower contraction time and are weaker than controls [74, 75]. When focusing on deregulated genes in LGMDR1 patients, it is noteworthy to mention that Rora, Slc16a and Tfrc genes showed the same direction of differential expression in cartilage and muscle of Frzb−/− mice. These differences were opposite to what was observed in LGMDR1 patients where FRZB is upregulated.
Tfrc is implicated in muscle biology. On one hand, it participates in iron acquisition in skeletal muscle [76]. On the other hand, Tfrc has been already described as a Wnt target gene [77]. Tfrc is elevated in regenerating fibres in patients with Duchenne muscular dystrophy as well as in facioscapulohumeral muscular dystrophy (FSHD) [78, 79]. However, the effects of TFRC upregulation in LGMDR1 patients have so far not been studied.
In skeletal muscle, retinoic acid receptor-related orphan receptor-α (Rorα) has been described as positive regulator of myogenesis by its interaction with MyoD and p300 cofactor which lead to activate muscle-specific genes transcription [80]. Furthermore Rorα is involved in the regulation of glucose and lipid metabolism in skeletal muscle [81, 82]. On the other hand, a role in Wnt signalling has been described since Wnt5a/PKCα-dependent as well as PGE2/PKCα-dependent Rorα phosphorylation exerts inhibitory function of the expression of Wnt / β-catenin target genes [83, 84]. Altogether, rescue of Rora expression by Wnt signalling pathway activation in the absence of FRZB could be beneficial for LGMDR1 patients due to its importance in muscle homeostasis.
Slc16a1, coding for a proton-linked monocarboxylate transporter, is highly expressed in oxidative fibres (type I fibres) consistent with the role of SLC16A1 in mediating lactate uptake for oxidative metabolism [85]. Its deregulation may be responsible for some of the metabolic impairment in LGMDR1 patients. No previous relation between Slc16a1 and the Wnt pathway was reported so far, but its downregulation in mouse muscle and in cartilage as well as its upregulation in LGMDR1 patients, where FRZB is overexpressed, suggests a direct interaction in its regulation.
One of the most striking findings was the upregulation of the Capn3 gene in Frzb−/− mice Soleus and its upregulation after FRZB silencing in human myotubes since no genetic regulatory mechanism of Capn3 expression has been described so far. FRZB upregulation in CAPN3 deficient LGMDR1 patients was already described [10], but the reciprocal regulation has not been reported. However, the increase in Frzb expression has been discarded as a beneficial compensatory mechanism since silencing of the gene increased several proteins that were upregulated in LGMDR1 patients [16]. These findings could be interesting not only for LGMDR1 patients, but for dysferlinopathy and titinopathy patients in whom a secondary reduction of CANP3 has been described [86, 87].
Conclusion and limitations
In summary, the result presented here confirm a role for Frzb in the regulation of Rora, Slc16a1, Tfrc, and Capn3 genes, which is of interest in understanding molecular changes observed in LGMDR1 patients. However, the studies and results failed to demonstrate a clinical correlate or clinically meaning effect that can be applied to our understanding of LGMDR1. Our specific in vivo and ex vivo analyses may not have captured all differences between the loss of function and wildtype mice, in particular due to the limitations in experimental design in particular when selecting the age of the mice used in the experiments. Even if not directly clinically revelevant for patients with LGMDR1, the involvement of FRZB in myogenesis was confirmed, since this gene regulates MyoD gene expression and the Frzb−/− mice show reduced muscle strength and gait abnormalities. However, lack of Frzb did not alter skeletal muscle regeneration capacity and neither induced modifications after exercise, with the caveat that our observations can be dependent on the age of the mice studied. Our data thus uncover new roles for FRZB in muscle and support a specific role for FRZB in patients with LGMDR1.
Methods
Animals
Frzb knockout (Frzb−/−) mice were previously generated [22]. C57BL/6 mice were at least in the 19th -20th generation of backcrossing. Wild-type C57Bl/6 mice (WT) were purchased from Janvier (Le Genest St Isle, France). Mice were housed in groups of 4–5 mice in Static micro-insulator cage with Macrolon filter with bedding material, under conventional laboratory conditions (14 h light – 10 h dark; 23+/− 2 °C), with standard mouse chow food (Sniff, Soest, Germany) and water provided ad libitum. All studies were performed with the approval from the Ethics Committee for Animal Research (P034/2016; KU Leuven, Belgium) in accordance with relevant guidelines and regulations. Several groups of mice were used for different experiments, their specific characteristics are available in Table 1.
Human samples
All participants gave informed consent, using forms approved by the Ethics Committee on the Use of Human Subjects in Research at Donostia University Hospital (ASP-FRZ-2017-01) and all the experiments were performed in accordance with relevant guidelines and regulations. Muscle biopsy specimens were obtained from adult patients with LGMDR1 (genetically confirmed) and healthy donors (Supplementary Table S1). Primary human skeletal muscle cell culture and FRZB gene silencing experiments were performed in healthy and LGMDR1 patients’ myotubes as previously described [16].
Muscle strength and endurance analysis
The four limb hanging test was used to monitor muscle strength and endurance (Treat-NMD Neuromuscular Network (SOP (ID) Number DMD_M.2.1.005)) [88]. Five to six-week-old mice were placed once on a crosslinked wire grid. The grid was inverted and the ‘time to fall’ was monitored.
Mouse gait analysis
The CatWalk™ XT system (Noldus, CatWalk 7.1, The Netherlands) was used to assess gait and locomotion [24]. Mice were placed on the runway for three consecutive runs. Runs were analysed separately and an average of these three runs was used as an individual value. The following parameters were evaluated: stand (paw contact time with the glass plate during the step cycle in seconds), swing phase (paw time in the air during the step cycle in seconds), step cycle (the sum of stand and swing time in seconds) and stride length (distance covered by a paw in mm).
Treadmill exercise
Six-week-old mice were subjected to a 5-week chronic exercise protocol on a four-lane modular treadmill (Columbus Instruments). The exercised group ran 30 min on a horizontal treadmill at 12 m/min twice a week for 5 weeks [89, 90] after a warm-up exercise consisting in 2 min at 4.2 m/min followed by 8 min at 7.8 m/min. Four WT and 2 Frzb−/− mice were included in a non-training control group. All mice weights were monitored every training day and muscles were dissected.
Cardiotoxin injection
Ten-week-old mice were anesthetized by intraperitoneal injection and 3 μl of 50 μM cardiotoxin (CTX; Latoxan, Portes-lès-Valence, France) was injected into the Tibialis anterior and 3 μl of 16.7 μM of cardiotoxin into the Soleus [91]. Tibialis anterior and Soleus muscles were dissected at three days, one, two and four weeks after cardiotoxin injection.
Histology and immunofluorescence
OCT compound (Tissue-Tek) immersed Tibialis anterior and Soleus were directly frozen into cold 2-methylbutane (Thermo Fisher Scientific). Frozen muscles were sectioned and stained with hematoxylin and eosin. For immunohistochemistry muscle cryosections were fixed (4% paraformaldehyde (PFA) (Electron microscopy sciences; Hatfield, PA, USA)) followed by permeation (0.3% Triton-X (Sigma-Aldrich; San Luis, MO, USA) in PBS) and blocked (PBS containing 3% bovine serum albumin (BSA) (Biowest; Nuaillé, France) solution). For immunostaining, muscles were incubated at 4 °C overnight with the primary antibodies against alkaline phosphatase, ALP (R&D Systems, AF2910; 10 μg/ml), platelet derived growth factor receptor beta, PDGFRβ (CST, #3169; 1:50), neural/glial antigen 2, NG2 (Millipore, AB5320; 1:500) or alpha smooth muscle actin, α SMA (Abcam, ab5694; 1:500) in a PBS containing 3% BSA solution. Isolated cells were fixed (4% PFA in PBS for 10 min), permeabilized (0.2% Triton-X100 in 1% BSA) and blocked (donkey serum 1:10 in PBS; VWR international, Radnor, PA, USA). Primary antibodies were incubated incubated overnight at 4 °C against monoclonal mouse anti-Ki67 (BD Bioscience, San Jose, CA, USA, 556003; 1:300), polyclonal rabbit anti-MyoD (Santa Cruz biotechnology, SC-760; 1:50), mouse anti-MyHC (Developmental Studies Hybridoma Bank -DSHB- Iowa City, IA, USA, 1:20), mouse anti-myogenin (DSHB; 1:10) and polyclonal rabbit anti-sarcomeric α-actinin (Abcam, ab72592; 1:500) in a PBS containing 0.1% Triton-X100 and 0.5% BSA solution. Secondary antibodies were incubated for 1 h at room temperature; goat anti-rabbit conjugated to Alexa-Fluor 555 (A-21428), and 488 (A-11034), donkey anti-mouse conjugated to Alexa-Fluor 594 (A-21203) and donkey anti-goat conjugated to Alexa-Fluor 488 (A-11055, Thermo Fisher Scientific; 1:500). Nuclei were visualized with 10 μg/ml containing Hoechst (Sigma-Aldrich) solution. Fluor Save reagent (Millipore) was used as mounting medium. Muscle structure was analysed using a Nikon 80i microscope and the NIS-Element software. For satellite cell immunofluorescence analysis, the percentage of positive nuclei was counted in randomly selected 6 fields of view. Fusion index was calculated in myotubes as the percentage of nuclei inside myotubes, being MyHC or sarcomeric α-actinin, myotubes markers. Between 5 and 6 field of view were counted for each sample. For fibre type classification SDH enzymatic activity was used [92]. The fibres were assigned to four different groups depending on the intensity (in pixels) value obtained with the ImageJ program (strong< 100, medium-stron 100–150, medium-weak 150–200 and weakly coloured > 200 pixels). All fibres of one section of Tibialis anterior per mouse in to the chronic exercise protocol experiment were measured.
Muscle fibres cross-sectional area
ImageJ software was used to measure fibres cross-sectional area (CSA). One hundred fibres from 4 different fields of view were measured.
Mouse primary cells
Satellite cells
Murine satellite cells from 4-week-old mice were isolated from Gastrocnemius, Tibialis anterior, Quadriceps and Biceps as previously described [93]. Cells were plated in triplicate and at confluency, the differentiation was induced by switching medium to DMEM high glucose supplemented with 2% horse serum (Gibco-Thermo Fisher Scientific) and 1 mM (100 mg/ml) sodium pyruvate, 100 U/ml penicillin and streptomycin, 2 mM L-glutamine. Cells were incubated at 37 °C, 5% CO2, 5% O2.
Mesoangioblasts
Mesoangioblasts (MABs) were isolated from Gastrocnemius, Tibialis anterior, Quadriceps and Biceps explant culture, by Fluorescence Activated Cell Sorter (FACS; BD FACS ARIA III) for alkaline phosphatase (ALP) + cells (R&D Systems-Biotechne, Minneapolis, MN, USA) from 5 week-old mice as previously described [94]. Flow cytometry analysis of the ALP+ fraction was carried out in 3 WT and 7 Frzb−/− samples. Protein tyrosine phosphatase receptor type C (CD45), platelet and endothelial cell adhesion molecule 1 (CD31) and platelet derived growth factor receptor alpha (PDGFRα or CD140α; Thermo Fisher Scientific) cell surface proteins presence were analysed by flow cytometry (FACs; BD Canto AIG) and analysed by BD FACSDiva software.
RNA extraction
Muscle samples were homogenised in a Tissue Lyser mixer-mild disruptor (QIAgen, Hilden, Germay) in Trizol (QIAzol® lysis reagent, QIAgen). Total and small RNAs were purified using miRNeasy mini kit (QIAgen) following the manufacturer’s instructions.
Gene expression analysis
RNA was reverse-transcribed to cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems; Foster City, CA, USA) according to the manufacturer’s instructions. For gene expression analyis Taqman single assays (Thermo Fisher Scientific; Walthman, MA, USA) and custom-designed SYBR green panels (Bio-Rad, Hercules, CA, USA) were used. As TaqMan probes the following genes were studied; myogenic or skeletal muscle specific markers, Pax7 (Mm01354484_m1), Myh3 (Mm01332463_m1), Myod (Mm00440387_m1) and Myog (Mm00446194_m1). Fbx32 (Mm00499523_m1) and Murf1 (Mm01185221_m1) for muscular atrophy. Adipoq (Mm00456425_m1) and Pparg (Mm01184322_m1) for adipose tissue infiltration measurement. Capn3 (Mm00482985_m1), Ky (Mm01224823_m1) and Park2 (Mm00450186_m1) were selected from a list of deregulated genes in Capn3 knockout (C3KO) mice [25]. Gapdh (Mm99999915_g1) and Tpb (Mm00446973_m1) were used as housekeeping. The entire list of the custom-designed SYBR green panels can be found as Supplementary Table S2 online. a) muscle specific genes, b) deregulated genes in LGMDR1 patients’ muscles [10, 95], c) deregulated genes in Frzb−/− mice articular cartilage and LGMDR1 patients’ muscles [23, 26] d) genes coding for proteins participating in Wnt signalling pathway. Gapdh and Tpb were used as housekeeping. In human origin myotubes FRZB (Hs00173503_m1), MYOD (Hs00159598), MYOG (Hs01072232) and CAPN3 (Hs00181057_m1) genes were analysed. GAPDH (Hs99999905_m1) was used as housekeeping. For RT-QPCR analysis the CFX384 Touch PCR System (Bio-Rad) and CFX Manager Software was used. Relative gene expression levels between WT mice and Frzb−/− mice muscles were calculated using the 2-ΔΔCT method.
Statistical analysis
Data are presented as mean and SEM, or as individual data points. Statistical analyses were performed where appropriate with GraphPad Prism software (version 8) or R Studio (Version 1.0.15) for analyses with multiple within-subject variables, using the aov_car function from the afex package. Data distribution was evaluated based on parameter characteristics, QQ plots and graphs of the residuals. T-tests or ANOVA-tests were applied. Data are reported with estimates of differences of means between groups (95% confidence intervals). Datasets with within-subject variables (repeated measurements) were analysed with 2-way ANOVA or a general linear model (GLM) in case of missing data. When different groups were compared by ANOVA or GLM tests, in some set-ups pair-wise t-tests were subsequently performed applying a Bonferroni correction for multiple comparisons to control for Type I errors in rejecting the null hypothesis.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Abbreviations
- Adipoq:
-
Adipose most abundant gene transcript 1 protein
- ALP:
-
alkaline phosphatase
- BSA:
-
Bovine serum albumin
- C3KO:
-
Capn3−/− mouse
- CAPN3:
-
Calpain 3
- CSA:
-
cross-sectional area
- CTX:
-
Cardiotoxin
- DMD:
-
Duchenne Muscular Dystrophy
- Fasn:
-
Fatty acid synthase
- Fbx32:
-
F-Box protein 32
- FP:
-
Front paw
- FRZB:
-
Frizzled Related Protein
- FSH:
-
Facioscapulohumeral
- HP:
-
Hind paw
- Ky:
-
Kyphoscoliosis peptidase
- LGMD2A:
-
Limb girdle muscular dystrophy type 2A, calpainopathy
- LGMD2B:
-
Limb girdle muscular dystrophy type 2B, dysferlinopathy
- LGMD2I:
-
Limb girdle muscular dystrophy type 2I
- LGMD2L or LGMDR12:
-
Limb girdle muscular dystrophy type 2 L, anoctaminopathy
- LGMDR1:
-
Limb girdle muscular dystrophy R1 calpain 3-related
- MABs:
-
Mesoangioblasts
- MuRF1:
-
Muscle-specific RING finger protein 1
- MyHC:
-
Myosin heavy chain
- Myod:
-
Myogenic Differentiation 1
- Myog:
-
Myogenin
- NG2:
-
Neural/glial antigen 2
- Park2:
-
Parkin
- RBR E3:
-
Ubiquitin protein ligase
- Pax7:
-
Paired box 7
- PDGFRβ:
-
Platelet derived growth factor receptor beta
- PFA:
-
Paraformaldehyde
- Pparg:
-
Peroxisome proliferator activated receptor gamma
- Rora:
-
RAR Related Orphan Receptor A
- Slc16a1:
-
Solute Carrier Family 16 Member 1
- Tfrc:
-
Transferrin Receptor
- WT:
-
Wild type
- α SMA:
-
Alpha smooth muscle actin
References
Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81(1):27–40.
Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M. Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G. Am J Med Genet. 1999;82(5):392–8.
Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen JEMA, Anderson LVB, et al. Calpainopathy—a survey of mutations and polymorphisms. Am J Hum Genet. 1999;64(6):1524–40.
Pollitt C, Anderson LV, Pogue R, Davison K, Pyle A, Bushby KM. The phenotype of calpainopathy: diagnosis based on a multidisciplinary approach. Neuromuscul Disord. 2001;11(3):287–96.
Bushby KMD, Beckmann JS. The 105th ENMC sponsored workshop: pathogenesis in the non-sarcoglycan limb-girdle muscular dystrophies, Naarden, April 12-14, 2002. Neuromuscul Disord. 2003;13(1):80–90.
Bevilacqua JA, Ehuletche MDRG, Perna A, Dubrovsky A, Franca MC, Vargas S, et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J Rare Dis. 2020;15(1):11..
Fardeau M, Hillaire D, Mignard C, Feingold N, Feingold J, Mignard D, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain J Neurol. 1996;119(Pt 1):295–308.
Urtasun M, Sáenz A, Roudaut C, Poza JJ, Urtizberea JA, Cobo AM, et al. Limb-girdle muscular dystrophy in Guipúzcoa (Basque Country, Spain). Brain J Neurol. 1998;121(Pt 9):1735–47.
Kramerova I, Kudryashova E, Tidball JG, Spencer MJ. Null mutation of calpain 3 (p94) in mice causes abnormal sarcomere formation in vivo and in vitro. Hum Mol Genet. 2004;13(13):1373–88.
Sáenz A, Azpitarte M, Armañanzas R, Leturcq F, Alzualde A, Inza I, et al. Gene Expression Profiling in Limb-Girdle Muscular Dystrophy 2A. PLoS ONE. 2008;3(11):e3750.
Pardo JV, Siliciano JD, Craig SW. A vinculin-containing cortical lattice in skeletal muscle: transverse lattice elements (‘costameres’) mark sites of attachment between myofibrils and sarcolemma. Proc Natl Acad Sci U S A. 1983;80(4):1008–12.
Danowski BA, Imanaka-Yoshida K, Sanger JM, Sanger JW. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J Cell Biol. 1992;118(6):1411–20.
Trimarchi F, Favaloro A, Fulle S, Magaudda L, Puglielli C, Di Mauro D. Culture of human skeletal muscle myoblasts: timing appearance and localization of dystrophin-glycoprotein complex and vinculin-Talin-integrin complex. Cells Tissues Organs. 2006;183(2):87–98.
Burke G, Hillier C, Cole J, Sampson M, Bridges L, Bushby K, et al. Calpainopathy presenting as foot drop in a 41 year old. Neuromuscul Disord. 2010;20(6):407–10.
Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23(4):397–418.
Jaka O, Casas-Fraile L, Azpitarte M, Aiastui A, López de Munain A, Sáenz A. FRZB and melusin, overexpressed in LGMD2A, regulate integrin β1D isoform replacement altering myoblast fusion and the integrin-signalling pathway. Expert Rev Mol Med. 2017;19:e2.
Wang S, Krinks M, Moos M. Frzb-1, an antagonist of Wnt-1 and Wnt-8, does not block signaling by Wnts -3A, −5A, or −11. Biochem Biophys Res Commun. 1997;236(2):502–4.
Leyns L, Bouwmeester T, Kim SH, Piccolo S, De Robertis EM. Frzb-1 is a secreted antagonist of Wnt signaling expressed in the spemann organizer. Cell. 1997;88(6):747–56.
Person AD, Garriock RJ, Krieg PA, Runyan RB, Klewer SE. Frzb modulates Wnt-9a-mediated β-catenin signaling during avian atrioventricular cardiac cushion development. Dev Biol. 2005;278(1):35–48.
Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel KP, et al. Wnt5a functions in planar cell polarity regulation in mice. Dev Biol. 2007;306(1):121–33.
Borello U, Coletta M, Tajbakhsh S, Leyns L, De Robertis EM, Buckingham M, et al. Transplacental delivery of the Wnt antagonist Frzb1 inhibits development of caudal paraxial mesoderm and skeletal myogenesis in mouse embryos. Dev Camb Engl. 1999;126(19):4247–55.
Lories RJU, Peeters J, Bakker A, Tylzanowski P, Derese I, Schrooten J, et al. Articular cartilage and biomechanical properties of the long bones in Frzb-knockout mice. Arthritis Rheum. 2007;56(12):4095–103.
Lories RJU, Peeters J, Szlufcik K, Hespel P, Luyten FP. Deletion of frizzled-related protein reduces voluntary running exercise performance in mice. Osteoarthr Cartil. 2009;17(3):390–6.
Vandeputte C, Taymans J-M, Casteels C, Coun F, Ni Y, Van Laere K, et al. Automated quantitative gait analysis in animal models of movement disorders. BMC Neurosci. 2010;11(1):92.
Jaka O, Kramerova I, Azpitarte M, López de Munain A, Spencer M, Sáenz A. C3KO mouse expression analysis: downregulation of the muscular dystrophy Ky protein and alterations in muscle aging. Neurogenetics. 2012;13(4):347–57.
Lodewyckx L, Cailotto F, Thysen S, Luyten FP, Lories RJ. Tight regulation of wingless-type signaling in the articular cartilage - subchondral bone biomechanical unit: transcriptomics in Frzb-knockout mice. Arthritis Res Ther. 2012;14(1):R16.
Maricelli JW, Lu QL, Lin DC, Rodgers BD. Trendelenburg-like gait, instability and altered step patterns in a mouse model for limb girdle muscular dystrophy 2i. PLoS ONE. 2016;11(9):e0161984.
Lamminen AE, Tanttu JI, Sepponen RE, Suramo IJI, Pinko H. Magnetic resonance of diseased skeletal muscle: combined T1 measurement and chemical shift imaging. Br J Radiol. 1990;63(752):591–6.
McDaniel JD, Ulmer JL, Prost RW, Franczak MB, Jaradeh S, Hamilton CA, et al. Magnetization transfer imaging of skeletal muscle in autosomal recessive limb girdle muscular dystrophy. J Comput Assist Tomogr. 1999;23(4):609–14.
Marden FA, Connolly AM, Siegel MJ, Rubin DA. Compositional analysis of muscle in boys with Duchenne muscular dystrophy using MR imaging. Skelet Radiol. 2005;34(3):140–8.
Fleckenstein JL, Watumull D, Conner KE, Ezaki M, Greenlee RG, Bryan WW, et al. Denervated human skeletal muscle: MR imaging evaluation. Radiology. 1993;187(1):213–8.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294(5547):1704–8.
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412.
Jang M-H, Bonaguidi MA, Kitabatake Y, Sun J, Song J, Kang E, et al. Secreted frizzled-related protein 3 regulates activity-dependent adult hippocampal neurogenesis. Cell Stem Cell. 2013;12(2):215–23.
Sun J, Bonaguidi MA, Jun H, Guo JU, Sun GJ, Will B, et al. A septo-temporal molecular gradient of sfrp3 in the dentate gyrus differentially regulates quiescent adult hippocampal neural stem cell activation. Mol Brain. 2015;8:52.
PećIna-šLaus N, Kafka A, VarošAnec AM, Marković L, Krsnik Ž, Njirić N, et al. Expression patterns of Wnt signaling component, secreted frizzled-related protein 3 in astrocytoma and glioblastoma. Mol Med Rep. 2016;13(5):4245–51.
Kafka A, Tomas D, Lechpammer M, Gabud T, Pažanin L, Pećina-Šlaus N. Expression levels and localizations of DVL3 and sFRP3 in Glioblastoma. Dis Markers. 2017;2017:1–10.
Rich CA, Perera SN, Andratschke J, Stolt CC, Buehler DP, Southard-Smith EM, et al. Olfactory ensheathing cells abutting the embryonic olfactory bulb express Frzb, whose deletion disrupts olfactory axon targeting. Glia. 2018;66(12):2617–31.
Wernig A, Irintchev A, Weisshaupt P. Muscle injury, cross-sectional area and fibre type distribution in mouse soleus after intermittent wheel-running. J Physiol. 1990;428:639–52.
Vignos PJ, Watkins MP. The effect of exercise in muscular dystrophy. JAMA. 1966;197(11):843–8.
McCartney N, Moroz D, Garner SH, McComas AJ. The effects of strength training in patients with selected neuromuscular disorders. Med Sci Sports Exerc. 1988;20(4):362–8.
Sveen M-L, Andersen SP, Ingelsrud LH, Blichter S, Olsen NE, Jønck S, et al. Resistance training in patients with limb-girdle and Becker muscular dystrophies. Muscle Nerve. 2013;47(2):163–9.
Sczesny-Kaiser M, Kowalewski R, Schildhauer TA, Aach M, Jansen O, Grasmücke D, et al. Treadmill training with HAL exoskeleton—a novel approach for symptomatic therapy in patients with limb-girdle muscular dystrophy—preliminary study. Front Neurosci. 2017;11:449.
Moore UR, Jacobs M, Fernandez-Torron R, Jang J, James MK, Mayhew A, et al. Teenage exercise is associated with earlier symptom onset in dysferlinopathy: a retrospective cohort study. J Neurol Neurosurg Psychiatry. 2018;89(11):1224–26.
Augusto V, Padovani CR, Rocha Campos GE. Skeletal muscle fiber types in C57BL6J mice. J Morphol Sci. 2004;21(2):89–94.
Kammoun M, Cassar-Malek I, Meunier B, Picard B. A simplified immunohistochemical classification of skeletal muscle fibres in mouse. Eur J Histochem. 2014;58(2):2254.
Hosaka Y, Yokota T, Miyagoe-Suzuki Y, Yuasa K, Imamura M, Matsuda R, et al. α1-syntrophin–deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J Cell Biol. 2002;158(6):1097–107.
Griffin DA, Johnson RW, Whitlock JM, Pozsgai ER, Heller KN, Grose WE, et al. Defective membrane fusion and repair in Anoctamin5-deficient muscular dystrophy. Hum Mol Genet. 2016;25(10):1900–11.
Yalvac ME, Amornvit J, Braganza C, Chen L, Hussain S-RA, Shontz KM, et al. Impaired regeneration in calpain-3 null muscle is associated with perturbations in mTORC1 signaling and defective mitochondrial biogenesis. Skelet Muscle. 2017;7(1):27.
Robert V, Massimino ML, Tosello V, Marsault R, Cantini M, Sorrentino V, et al. Alteration in calcium handling at the subcellular level in mdx myotubes. J Biol Chem. 2001;276(7):4647–51.
Grefte S, Vullinghs S, Kuijpers-Jagtman AM, Torensma R, Von den Hoff JW. Matrigel, but not collagen I, maintains the differentiation capacity of muscle derived cells in vitro. Biomed Mater. 2012;7(5):055004.
White J, Barro MV, Makarenkova HP, Sanger JW, Sanger JM. Localization of sarcomeric proteins during myofibril assembly in cultured mouse primary skeletal myotubes. Anat Rec. 2014;297(9):1571–84.
Smolina N, Kostareva A, Bruton J, Karpushev A, Sjoberg G, Sejersen T. Primary murine myotubes as a model for investigating muscular dystrophy. Biomed Res Int. 2015;2015:594751.
Manabe Y, Ogino S, Ito M, Furuichi Y, Takagi M, Yamada M, et al. Evaluation of an in vitro muscle contraction model in mouse primary cultured myotubes. Anal Biochem. 2016;497:36–8.
Vannoy CH, Zhou H, Qiao C, Xiao X, Bang AG, Lu QL. Adeno-associated virus–mediated mini-agrin delivery is unable to tescue fisease phenotype in a mouse model of limb girdle muscular dystrophy type 2I. Am J Pathol. 2017;187(2):431–40.
Wang S, Krinks M, Lin K, Luyten FP, Moos M. Frzb, a secreted protein expressed in the Spemann organizer, binds and inhibits Wnt-8. Cell. 1997;88(6):757–66.
Tagliafico E. TGF /BMP activate the smooth muscle/bone differentiation programs in mesoangioblasts. J Cell Sci. 2004;117(19):4377–88.
Qu Y, Li J, Cai Q, Wang Y, Gu Q, Zhu Z, et al. Over-expression of FRZB in gastric cancer cell suppresses proliferation and induces differentiation. J Cancer Res Clin Oncol. 2008;134(3):353–64.
Kephart JJG, Tiller RGJ, Crose LES, Slemmons KK, Chen P-H, Hinson AR, et al. Secreted frizzled-related protein 3 (SFRP3) is required for tumorigenesis of PAX3-FOXO1-positive alveolar rhabdomyosarcoma. Clin Cancer Res. 2015;21(21):4868–80.
De Angelis L, Berghella L, Coletta M, Lattanzi L, Zanchi M, Cusella-De Angelis MG, et al. Skeletal myogenic progenitors originating from embryonic dorsal aorta coexpress endothelial and myogenic markers and contribute to postnatal muscle growth and regeneration. J Cell Biol. 1999;147(4):869–78.
Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, et al. The meso-angioblast: a multipotent, self-renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Dev Camb Engl. 2002;129(11):2773–83.
Sampaolesi M. Cell therapy of -sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301(5632):487–92.
Morosetti R, Mirabella M, Gliubizzi C, Broccolini A, De Angelis L, Tagliafico E, et al. MyoD expression restores defective myogenic differentiation of human mesoangioblasts from inclusion-body myositis muscle. Proc Natl Acad Sci. 2006;103(45):16995–7000.
Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9(3):255–67.
Crisan M, Yap S, Casteilla L, Chen C-W, Corselli M, Park TS, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–13.
Pierantozzi E, Vezzani B, Badin M, Curina C, Severi FM, Petraglia F, et al. Tissue-specific cultured human pericytes: perivascular cells from smooth muscle tissue have restricted mesodermal differentiation ability. Stem Cells Dev. 2016;25(9):674–86.
Birbrair A, Zhang T, Wang Z-M, Messi ML, Enikolopov GN, Mintz A, et al. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem Cells Dev. 2013;22(16):2298–314.
Birbrair A, Zhang T, Wang Z-M, Messi ML, Enikolopov GN, Mintz A, et al. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res. 2013;10(1):67–84.
Kho AT, Kang PB, Kohane IS, Kunkel LM. Transcriptome-scale similarities between mouse and human skeletal muscles with normal and myopathic phenotypes. BMC Musculoskelet Disord. 2006;7:23.
Buckingham M, Rigby PWJ. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev Cell. 2014;28(3):225–38.
Comai G, Tajbakhsh S. Molecular and cellular regulation of skeletal myogenesis. In: current topics in developmental biology. Elsevier. 2014;110:1–73.
Hoppler S, Brown JD, Moon RT. Expression of a dominant-negative Wnt blocks induction of MyoD in Xenopus embryos. Genes Dev. 1996;10(21):2805–17.
Beatham J, Romero R, Townsend SKM, Hacker T, van der Ven PFM, Blanco G. Filamin C interacts with the muscular dystrophy KY protein and is abnormally distributed in mouse KY deficient muscle fibres. Hum Mol Genet. 2004;13(22):2863–74.
Marechal G, Coulton GR, Beckers-Bleukx G. Mechanical power and myosin composition of soleus and extensor digitorum longus muscles of ky mice. Am J Physiol-Cell Physiol. 1995;268(2):C513–9.
Blanco G, Coulton GR, Biggin A, Grainge C, Moss J, Barrett M, et al. The kyphoscoliosis (ky) mouse is deficient in hypertrophic responses and is caused by a mutation in a novel muscle-specific protein. Hum Mol Genet. 2001;10(1):9–16.
Hofer T, Marzetti E, Seo AY, Xu J, Knutson MD. In: Leeuwenburgh C, Gutiérrez-Merino, editors. Free radicals in biology and medicine; 2008.
Prieve MG, Moon RT. Stromelysin-1 and mesothelin are differentially regulated by Wnt-5a and Wnt-1 in C57mg mouse mammary epithelial cells. BMC Dev Biol. 2003;3:2.
Feero W, Li S, Rosenblatt J, Sirianni N, Morgan J, Partridge T, et al. Selection and use of ligands for receptor-mediated gene delivery to myogenic cells. Gene Ther. 1997;4(7):664–74.
Winokur ST. Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet. 2003;12(22):2895–907.
Lau P, Bailey P, Dowhan DH, Muscat GE. Exogenous expression of a dominant negative RORalpha1 vector in muscle cells impairs differentiation: RORalpha1 directly interacts with p300 and myoD. Nucleic Acids Res. 1999;27(2):411–20.
Lau P, Nixon SJ, Parton RG, Muscat GEO. RORalpha regulates the expression of genes involved in lipid homeostasis in skeletal muscle cells: caveolin-3 and CPT-1 are direct targets of ROR. J Biol Chem. 2004;279(35):36828–40.
Lau P, Fitzsimmons RL, Pearen MA, Watt MJ, Muscat GEO. Homozygous staggerer (sg/sg) mice display improved insulin sensitivity and enhanced glucose uptake in skeletal muscle. Diabetologia. 2011;54(5):1169–80.
Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim HY, et al. RORα attenuates Wnt/β-catenin signaling by PKCα-dependent phosphorylation in colon cancer. Mol Cell. 2010;37(2):183–95.
Shin D, Kim IS, Lee JM, Shin S-Y, Lee J-H, Baek SH, et al. The hidden switches underlying RORα-mediated circuits that critically regulate uncontrolled cell proliferation. J Mol Cell Biol. 2014;6(4):338–48.
Hashimoto T, Masuda S, Taguchi S, Brooks GA. Immunohistochemical analysis of MCT1, MCT2 and MCT4 expression in rat plantaris muscle: MCT expression in rat skeletal muscle fibres. J Physiol. 2005;567(1):121–9.
Anderson LVB, Harrison RM, Pogue R, Vafiadaki E, Pollitt C, Davison K, et al. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul Disord. 2000;10(8):553–9.
Charton K, Sarparanta J, Vihola A, Milic A, Jonson PH, Suel L, et al. CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet. 2015;24(13):3718–31.
Carlson CG, Rutter J, Bledsoe C, Singh R, Hoff H, Bruemmer K, et al. A simple protocol for assessing inter-trial and inter-examiner reliability for two noninvasive measures of limb muscle strength. J Neurosci Methods. 2010;186(2):226–30.
Granchelli JA, Pollina C, Hudecki MS. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul Disord. 2000;10(4–5):235–9.
De Luca A. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther. 2003;304(1):453–63.
Duchen LW, Excell BJ, Patel R, Smith B. Proceedings: light and electron microscopic changes in mouse muscle fibres and motor end-plates caused by the depolarizing fraction (cardiotoxin) of the of the venom of Dendroaspis jamesoni. J Physiol. 1973;234(2):1P–2P.
Degenhardt K, Sassoon DA. A role for engrailed-2 in determination of skeletal muscle physiologic properties. Dev Biol. 2001;231(1):175–89.
Biressi S, Bjornson CRR, Carlig PMM, Nishijo K, Keller C, Rando TA. Myf5 expression during fetal myogenesis defines the developmental progenitors of adult satellite cells. Dev Biol. 2013;379(2):195–207.
Quattrocelli M, Palazzolo G, Perini I, Crippa S, Cassano M, Sampaolesi M. Mouse and human mesoangioblasts: isolation and characterization from adult skeletal muscles. Methods Mol Biol. 2012;798:65–76.
De la Torre C, Illa I, Faulkner G, Soria L, Robles-Cedeño R, Dominguez-Perles R, et al. Proteomics identification of differentially expressed proteins in the muscle of dysferlin myopathy patients. Proteomics Clin Appl. 2009;3(4):486–97.
Acknowledgements
“Not applicable”
Funding
This work was financed through the grants received from the Health Research Fund (PI16/01325) of the Spanish Ministry of Economy and Competitiveness and the European Union (ERDF) and it was in part supported by the Center for Networked Biomedical Research on Neurodegenerative Diseases (CIBERNED), Carlos III Health Institute, Spanish Ministry of Economy and Competitiveness and by GENE (Association of Neuromuscular disease of Gipuzkoa). Research on FRZB biology in the Laboratory of Tissue Homeostasis is supported by grants from the Flanders Research Foundation (FWO Vlaanderen) and from KU Leuven.
LCF was supported by predoctoral fellowship given by the department of Education, Universities and Research of the Basque Government (PRE-2015-1-0117) and received an OARSI (Osteoarthritis Research Society International) fellowship. AR is supported by the predoctoral fellowship given by the department of Education, Universities and Research of the Basque Government (PRE-2016-1-0382) and DC by Association française contre les myopathies (AFM)-Téléthon (#20673).
Author information
Authors and Affiliations
Contributions
Leire Casas-Fraile: Conception or design of the work, data collection, data analysis and interpretation, drafting the article and final approval of the version to be published. Frederique M. Cornelis: Conception or design of the work, data collection, data analysis and interpretation, critical revision of the article and final approval of the version to be published. Domiziana Costamagna: Conception or design of the work, data collection, data analysis and interpretation, critical revision of the article and final approval of the version to be published. Anabel Rico: Critical revision of the article and final approval of the version to be published. Robin Duelen: Data collection and approval of the version to be published. Maurilio M. Sampaolesi: Critical revision of the article and final approval of the version to be published. Adolfo Lopez de Munain: Critical revision of the article and final approval of the version to be published. Rik J. Lories: Conception or design of the work, data analysis and interpretation, critical revision of the article and final approval of the version to be published. Amets Saenz: Conception or design of the work, data analysis and interpretation, critical revision of the article and final approval of the version to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Animal samples: All studies were performed with the approval from the Ethics Committee for Animal Research (P034/2016; KU Leuven, Belgium) in accordance with relevant guidelines and regulations.
Human samples: All participants gave informed consent, using forms approved by the Ethics Committee on the Use of Human Subjects in Research at Donostia University Hospital (ASP-FRZ-2017-01) and all the experiments were performed in accordance with relevant guidelines and regulations.
Consent for publication
“Not applicable”.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Human Tissue Samples. Table S2. Custom-designed SYBR green panel’s gene selection (Bio-Rad).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Casas-Fraile, L., Cornelis, F.M., Costamagna, D. et al. Frizzled related protein deficiency impairs muscle strength, gait and calpain 3 levels. Orphanet J Rare Dis 15, 119 (2020). https://doi.org/10.1186/s13023-020-01372-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01372-1