
Abstract
Objective
This study aimed to describe the clinical and genetic characteristics of Chinese patients with congenital fibrosis of the extraocular muscles (CFEOM), and to evaluate the phenotype–genotype correlations in these patients.
Methods
This was a retrospective study. Patients with CFEOM underwent detailed ophthalmic examinations and magnetic resonance imaging (MRI). Panel-based next-generation sequencing was performed to identify pathogenic variants of disease-causing genes.
Results
Sixty-two patients with CFEOM were recruited into this study. Thirty-nine patients were diagnosed with CFEOM1 and 23 with CFEOM3. Forty-nine of the 62 patients with CFEOM carried either KIF21A (41/49) or TUBB3 variants (8/49). Six known missense variants in the KIF21A and TUBB3 genes, and a novel variant (c.3906T > A, p.D1302E) in the KIF21A gene were detected. Most patients with CFEOM1 carrying the KIF21A mutation displayed isolated CFEOM, whereas patients with CFEOM3 carrying the TUBB3 mutation had a wide range of clinical manifestations, either CFEOM alone or syndromes. Nystagmus was also present in 12 patients with CFEOM. Furthermore, the MRI findings varied, ranging from attenuation of the extraocular muscles to dysgenesis of the cranial nerves and brain structure.
Conclusions
The novel variants identified in this study will further expand the spectrum of pathogenic variants in CFEOM-related genes. However, no phenotype–genotype correlations were established because of the diversity of the clinical characteristics of these patients.
Similar content being viewed by others


Introduction
Congenital fibrosis of the extraocular muscles (CFEOM) is a group of rare ocular motor disorders with an estimated prevalence of 1:230,000 [1]. This condition develops after birth and is characterized by nonprogressive ophthalmoplegia and ptosis in one or both eyes. Some patients with CFEOM may have an abnormal head posture, usually with a chin-up position caused by hypotropia and ptosis. Recent advances in genetics and neuroanatomy have proven that CFEOM is caused by maldevelopment of the cranial nerves and their motor neurons [2]. To date, mutations in seven genes, i.e., KIF21A (OMIM *608,283), PHOX2A (OMIM *602,753), TUBB3 (OMIM*602,661), TUBB2B (OMIM *612,850), TUBA1A (OMIM *602,529), ECEL1 (OMIM *605,896), and COL25A1 (OMIM *610,004), have been associated with CFEOM [3, 4]. However, with the exception of TUBB3, for which a phenotype–genotype correlation has been suggested in some pathogenic variants, no correction between phenotype and genotype has been established for these genes [5,6,7].
KIF21A is located on chromosome 12q12 and is expressed in the brain, spinal cord, and other structures of the nervous system. It encodes kinesin-like motor proteins that interact directly with microtubules in the nervous system and regulate microtubule dynamics. Microtubules are composed of alpha and beta tubulin and play an important role in transporting essential cellular components along axons and dendrites [8]. The TUBB3 gene is located on chromosome 16q24.3 and is widely expressed in neurons, the brain, the spinal cord, sensory organs, and, to a lower extent, in the testes. This gene encodes a Class III member of beta tubulin that heterodimerizes with alpha tubulin and assembles to form the microtubules that are involved in neurogenesis and axonal guidance and maintenance [7]. Mutations in KIF21A and TUBB3 are suggested to alter microtubule dynamics and the tubulin heterodimer angle, resulting in CFEOM [9]. However, it is not known whether the TUBB3-associated CFEOM and KIF21A-associated CFEOM variants share a common pathogenic mechanism.
Based on the clinical features and inheritance pattern, CFEOM is subdivided into five subtypes (1 to 5) [3]. CFEOM1 is the most typical and common type of all forms of CFEOM; it is inherited in an autosomal dominant manner with complete penetrance, and is caused by mutations in KIF21A, resulting in dysgenesis of the superior division of the third cranial nerve and the motor neurons, leading to atrophy of its innervated superior rectus muscles and the levator palpebrae superioris. Patients with CFEOM1 are unable to raise their eyes above the midline, presenting as hypotropia and ptosis, with their eyes fixed in an infra-ducted position [8, 10].
CFEOM2 is inherited in an autosomal recessive manner and is caused by mutations in the PHOX2A gene, which is prominently expressed in developing oculomotor and trochlear motor neurons and is essential for their survival. Mutations in the PHOX2A gene would result in dysgenesis of the third and fourth cranial nerves, leading to symptoms similar to those of complete third nerve palsy, presenting with severe ptosis and large exodeviation. In addition, these patients may have small pupils with a poor response to light [11].
CFEOM3 is inherited in an autosomal dominant manner with incomplete penetrance. Its clinical characteristics vary and are less typical than those of CFEOM1 and CFEOM2. Patients with CFEOM3 may have unilateral or bilateral ophthalmoplegia and ptosis, with a variable degree of restriction in the different gaze directions. CFEOM3 is subdivided into three subtypes, i.e., the CFEOM3A–3 C forms, with CFEOM3A being caused by mutations in the TUBB3 gene, CFEOM3B being caused by mutations in the KIF21A gene, and no responsible gene having been identified for CFEOM3C [7, 12, 13]. CFEOM4, also known as Tukel syndrome, has a similar phenotype to that of CFEOM3 and presents with postaxial oligodactyly/oligosyndactyly of the hands [14]. Finally, CFEOM5 is inherited in an autosomal recessive manner and is caused by mutations in the COL25A1 gene. Similar to CFEOM3, CFEOM5 has variable clinical characteristics. Patients with CFEOM5 may even present with bilateral Duane retraction syndrome [15]. In this study, we aimed to investigate the clinical and genetic characteristics of 62 Han Chinese patients with CFEOM.
Methods
Participants
This is a retrospective study. All patients underwent molecular and clinical diagnostics prior to enrollment. Patient clinical data and genetic test results were used as part of a retrospective study, subject to informed consent from the patient and approval by the ethics committee of Beijing Children’s Hospital.
Sixty-two Han Chinese patients with CFEOM were enrolled in this study and underwent careful examinations, including slit lamp for the anterior segment, retinoscopy for fundus, refractive status, ocular alignment and motility, and levator palpebrae superior function assessments. The binocular sensory status was evaluated using the Worth 4 dot test or the Bagolini striated glasses at near and distance. The diagnosis of CFEOM is based on the following criteria: (1) onset of nonprogressive restrictive ophthalmoplegia in one eye or both eyes within 6 months of birth and (2) involvement of more than two extraocular muscles in one eye. Magnetic resonance imaging (MRI) was performed using a 3.0-T GE DISCOVERY MR750 scanner with an 8-channel head coil. Genetic testing was performed on all patients and their affected family members under their informed consent, which was obtained from the parents/legal guardians of participants who were younger than 18 years. The study was approved by the ethics committee of Beijing Children’s Hospital and was in accordance with the tenets of the Declaration of Helsinki.
Genetic testing
Three microliters of peripheral blood samples were collected from all participants and available nuclear family members, and genomic DNA was extracted using a Blood DNA Kit (Roche Biochemical, Inc). Panel-based next-generation sequencing (NGS) was commercially performed by the Mygenomic Biochemical Company (Beijing, China). A DNA library was constructed and sequenced on an Illumina HiSeq X Ten platform (Illumina, San Diego, USA). The amplified DNA was captured using the self-designed GenCap Capture Kit (MyGenostics Inc, Beijing, China), which consists of 235 eye-disease-causing genes selected from the Orphanet (https://www.orpha.net) and OMIM databases (https://www.omim.org/), in which the probe is designed to capture the whole coding and non-coding regions known to be involved in pathogenic variants associated with eye diseases, including CFEOM. The 235 genes included in the GenCap Capture Kit are listed in Supplementary Table 2. Based on methods reported previously, the quality of the reads was controlled by a serious of procedures [16,17,18,19]. First, the low-quality (< 80 bp) and duplicated reads were removed from the raw data using the Cutadapt (https://cutadapt.readthedocs.io/en/stable/) and Sentieon (https://www.sentieon.com/) software, respectively. The clean reads (< 150 bp) were aligned to the UCSC hg19 human reference genome and then analyzed using the Genome Analysis Toolkit (GATK) program, to identify single-nucleotide variants (SNVs) and small insertions or deletions (indels). Only variants with a GATK-assigned quality criterion score > 50.0 and a minor allele frequence < 0.01 were considered for downstream analysis. Variants were further annotated using the ANNOVAR software (http://annovar.openbioinformatics.org/en/latest/), which combines multiple public databases, including 1000 Genomes, ESP6500, dbSNP, genomAD, ClinVar, HGMD, and the commercial MyGenostics database. Copy number variants were detected using the CNV kit based on the read-depth algorithm [20]. The pathogenicity of missense mutations was predicted using PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant from Tolerant (SIFT, https://sift.bii.a-star.edu.sg/), and Combined Annotation Dependent Depletion (CADD) (https://cadd.gs.washington.edu/). The splicing effects of the variants were assessed using the SpliceAI program [21] and CADD. All potential pathogenic variants identified by NGS were further validated using the Sanger sequencing method. A co-segregation analysis of variants in the family was performed using Sanger sequencing. The primer pairs used in the Sanger method are listed in Supplementary Table 3. The pathogenicity of the variant was interpreted according to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [22]. A 3-dimension (3D) model of the non-synonymous mutant protein was generated and visualized using the PyMOL program (https://pymol.org/) [23].
Results
Demographics and clinical features
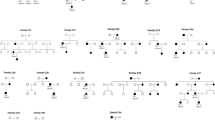
Of the 62 affected individuals, 33 patients were recruited from 12 pedigrees with an autosomal dominant inheritance pattern (Fig. 1; Table 1), whereas 29 patients were sporadic cases (Table 1). Based on their clinical characteristics, 39 patients (62.90%, 39/62) were identified as having CFEOM1 and 23 (37.10%, 23/62) were diagnosed as having CFEOM3. Nine of the 12 pedigrees were identified as being CFEOM1 and three as CFEOM3. Twelve patients with sporadic disease (41.38%, 12/29) were diagnosed with CFEOM1 and 17 (58.62%, 17/29) with CFEOM3. Their anterior and fundus were normal under slit-lamp and retinoscopy examinations. None of the cooperative patients had binocular vision, as evaluated by the Worth 4 dot test or the Bagolini striated glasses. Thirty-two patients (51.61%, 32/62) with CFEOM had a sluggish pupillary response to light, even among patients with CFEOM1. Among the 53 patients who cooperated with undergoing refractive examination, 95 eyes had astigmatism over 0.25D, 50 eyes (52.63%) had rule astigmatism, 29 eyes (30.52%) and against-the-rule astigmatism, and 16 eyes (16.84%) had oblique astigmatism. All patients with CFEOM1 had bilateral blepharoptosis, ophthalmoplegia with both eyes fixed in an infra-ducted position, an inability to move the eyes above the horizontal midline, and a compensatory head position of chin-up. Among the patients with CFEOM1, 30 presented (76.92%, 30/39) with exotropia, four (10.26%, 4/39) with esotropia, and five (12.82%, 4/39) with orthophoria in the horizontal direction. Patients with CFEOM3 had restrictive strabismus in one or both eyes. Six out of 23 patients (26.09%, 6/23) with CFEOM3 presented with monocular ophthalmoplegia, and 17 (73.91%, 17/23) of them exhibited binocular ophthalmoplegia. Their strabismus may appear as a combination of deviations in the horizontal and vertical directions, either exotropia, esotropia, or orthophoria in the horizontal direction, with or without vertical deviations. Seventeen (73.91%, 17/23) of the 23 patients with CFEOM3 had variable degrees of ptosis, and six (26.09%, 6/23) did not exhibit ptosis.

Pedigrees of CFEOM families carrying variants. (A) Pedigrees of CFEOM1 families carrying KIF21A variants. (B) Pedigrees of CFEOM3 families carrying KIF21A and TUBB3 variants
Nystagmus was detected in only 12 patients. Horizontal jerk nystagmus was found in six patients with CFEOM1 and four patients with CFEOM3 (S17, S19, S27, S28). Upbeat and multidirectional nystagmus was respectively identified from each of two patients with CFEOM3 (S21, and S16). In addition, other symptoms were observed in patients with CFEOM3, including facial weakness in three patients (S16, S17, S27), developmental delay in four patients (S16, S17, S26, S27), Kallmann syndrome (hypogonadotropic hypogonadism with anosmia) in four patients (S16, S17, S26, S27), vocal cord paralysis in two patients (S16, S17), and scoliosis in one patient (S22) (Table 1; Fig. 2).

Clinical spectrum and spine X-ray of patient S22 with CFEOM3. (a) Facial photograph showing the normal bilateral palpebral fissure widths and orthophoria on the primary eye position. (b) Body photograph indicating scoliosis. (c) Thoracic X-ray film of scoliosis. (d) Nine gaze photographs showing limited eye movements in both the horizontal and vertical directions, which differentiates the condition of the patient from horizontal gaze palsy with progressive scoliosis
Twenty-nine patients underwent MRI. The MRI findings included attenuation of the extraocular muscles in all patients, hypoplasia of oculomotor nerve in 24 (82.76%, 24/29) patients (15 CFEOM1, 9 CFEOM3), dysgenesis or agenesis of the sixth cranial nerve in five patients (S2, S16, S20, S24, and P12-III1), dysgenesis of the seventh cranial nerve in a patient with CFEOM3 (S16), hypoplasia of the corpus callosum in four patients with CFEOM3 (S16, S17, S21 and S26), aplasia of the olfactory bulb and sulcus in a patient with CFEOM3 (S17), and temporal horn enlargement of the lateral ventricle in a patient with CFEOM3 (S16). The demographics and detailed clinical characteristics of the patients with CFEOM are listed in Table 2 (additional information is provided in Supplementary Table 1).
Genetic analysis
Forty-nine of the 62 patients carried variants in the KIF21A and TUBB3 genes, with 41 patients carrying a missense variant of the KIF21A gene and eight patients carrying a missense variant of the TUBB3 gene. Only three types of missense variants, i.e., c.3906T > A(p.Asp1302Glu), c.2860 C > T(p.Arg954Trp), and c.2861G > A(p.Arg954Gln), were identified in the KIF21A gene, and four types of variants, i.e., c.784 C > T(p.Arg262Cys), c.904G > A(p.Ala302Thr), c.1249G > A(p.Asp417Asn), and c.1228G > A(p.Glu410Lys), were detected in the TUBB3 gene. With the exception of c.3906T > A(p.Asp1302Glu) in KIF21A, the remaining variants in KIF21A and TUBB3 had been reported previously and were present in more than one affected individual, especially for c.2860 C > T(p.Arg954Trp) and c.2861G > A(p.Arg954Gln) in KIF21A (Fig. 3). The c.3906T > A(p.Asp1302Glu) variant of KIF21A is regarded as a pathogenic variant because the wild-type aspartic acid (Asp or D) amino acid at codon 1302 is highly conserved among Homo sapiens, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Bos taurus, and Gallus gallus (Fig. 4). Substitution of aspartic acid (D) with glutamic acid (Glu or E) would destroy the stability of the protein, as predicted by the online SIFT, Polyphen-2, MutationTaster, and CADD programs (Table 3) and further confirmed by the 3-D model that was constructed using Dynamut2 (http://biosig.unimelb.edu.au/dynamut2/), in which the mutant type gained an extra hydrogen bond with the serine (S) amino acid at codon 1304 and resulted in a change in the free energy of the structure (Fig. 4; Table 4).

Genomic sequence chromatograms of patients with CFEOM carrying KIF21A and TUBB3 variants. (A) Three heterozygous variants of the KIF21A gene (red arrows). (B) Four heterozygous variants of the TUBB3 gene (red arrows)

Genetic feature of variant c.3906T > A(p.Asp1302Glu) of KIF21A. (A) Domain structure of the KIF21A protein. (B) The D1302 residue is located in the low-complexity region of KIF21A. (C) Multiple sequence alignment demonstrates high conservation of the p.Asp1302 residue (marked with black arrows). (D) Genomic sequence chromatograms of the c.3906T > A(p.Asp1302Glu) variant. E, F. Schematic representation of the structure and spatial distribution of the D1302 residue of the KIF21A protein. G. Interaction force of the Asp1302Glu substitution with the surrounding amino acid residues
Genotype–phenotype correlations
Nine pedigrees (P1–9) with CFEOM1 carried either the c.2860 C > T(p.Arg954Trp) or c.2861G > A(p.Arg954Gln) variant of KIF21A, and two pedigrees (P11–12) with CFEOM3 carried the c.2860 C > T(p.Arg954Trp) variant of KIF21A. One pedigree (P10) with CFEOM3 carried the c.784 C > T(p.Arg262Cys) pathogenic variant of TUBB3 (Fig. 1).
Eleven out of the 12 sporadic patients with CFEOM1 carried either the c.2860 C > T(p.Arg954Trp) or c.2861G > A(p.Arg954Gln) variant of KIF21A, with one of them (S4) carrying the novel c.3906T > A(p.Asp1302Glu) missense variant of KIF21A. Six of the 11 sporadic patients with bilateral ophthalmoplegia who were diagnosed as having CFEOM3 carried the TUBB3 variants of c.904G > A(p.Ala302Thr), c.1249G > A(p.Asp417Asn), and c.1228G > A(p.Glu410Lys), whereas five of them did not carry either KIF21A or TUBB3 mutations.(Table 1).
Patients with the c.3906T > A(p.Asp1302Glu) and c.2861G > A(p.Arg954Gln) variants in KIF21A presented with a typical feature of CFEOM1, whereas patients with the c.2860 C > T(p.Arg954Trp) variant presented with either CFEOM1 or CFEOM3. The clinical characteristics varied among patients with TUBB3 mutations. CFEOM without systemic disorders was present in patients with the c.784 C > T(p.Arg262Cys), c.904G > A(p.Ala302Thr), and c.1249G > A(p.Asp417Asn) variants of TUBB3, whereas a complicated systemic disorder was observed in patients with the c.1228G > A(p.Glu410Lys) variant of TUBB3, including developmental delay and Kallmann syndrome in all four patients (S16, S17, S26, and S27), facial weakness in three patients (S16, S17, and S27), and vocal cord paralysis in two patients (S16 and S17) (Table 1).
Horizontal jerk nystagmus was present in six patients with CFEOM1 carrying the c.2860 C > T(p.Arg954Trp) variant of KIF21A, and four patients with CFEOM3 had the c.904G > A(p.Ala302Thr) and c.1228G > A(p.Glu410Lys) variants of TUBB3. Upbeat nystagmus was found in a patient (S21) with the c.904G > A(p.Ala302Thr) variant of TUBB3, and multidirectional nystagmus was identified only in one patient (S16) with the c.1228G > A(p.Glu410Lys) variant of TUBB3.
The S2 and P12-III1 patients, who carried with the same variant of c.2860 C > T(p.Arg954Trp), were found to have aplasia of the sixth cranial nerve on MRI, although they had different types of CFEOM (S2, CFEOM1; and P12-III1, CFEOM3) (Fig. 5; Table 1). Unlike patients with the KIF21A mutation, those with the TUBB3 mutation exhibited more complex and varied findings on MRI (Fig. 6), even though they carried the same variant. Three patients with the Glu410Lys variant had hypoplasia of the corpus callosum and one had an enlarged temporal horn of the lateral ventricle (Fig. 7). Moreover, among the patients carrying the Glu410Lys variant, patient S16 exhibited hypoplasia of the third, six, and seventh cranial nerves and corpus callosum, whereas patient S35 only had hypoplasia of the third cranial nerve (Fig. 7). Because of the complex clinical findings observed in our patients, no correlations between genotype and phenotype were established.

Ocular and MRI findings of patients with CFEOM1 (a1–c2) and CFEOM3 (d1–d2) carrying KIF21A variants. a1, a2. Patient S2, harboring the Arg954Trp variant, displayed bilateral ptosis and absence of CN6 on MRI. b1,b2. Patient S4, harboring the Asp1302Glu variant, showed bilateral ptosis and hypoplasia of the bilateral CN3 on MRI. c1,c2. Patient S6, harboring the Arg954Gln variant, exhibited bilateral ptosis and hypoplasia of the bilateral levator palpebrae superioris-superior rectus on MRI. d1, d2. Patient P12-III1, with CFEOM3, harbored the Arg954Trp variant and showed bilateral ptosis and absence of left CN6 on MRI

Ocular and MRI findings of patients with CFEOM3 carrying TUBB3 variants. a1, a2 Patient P10-III1, harboring the Arg262Cys variant, displayed bilateral ptosis and hypoplasia of CN3 on MRI. b1, b2 Patient S21, harboring the Ala302Thr variant, showed bilateral ptosis and hypoplasia of the corpus callosum on MRI. c1, c2 Patient S24, harboring the Asp417Asn variant, exhibited left ptosis and absence of bilateral CN6 on MRI

Ocular and MRI findings of four patients with the TUBB3 Glu410Lys variant. a–d. Facial photographs of patients S16 (a), S17 (b), S26 (c), and S27 (d), showing bilateral blepharoptosis and facial weakness in patients S16, S17, and S27 (patient S26 had undergone correction of blepharoptosis). e. Enlargement of the lateral ventricle on its temporal horn in patient S16. f, g. Absence of the right CN7 in patient S16. h.i. Hypoplasia of the corpus callosum and the bilateral olfactory bulbs and sulci in patient S17. j. Hypoplasia of the bilateral CN6 in patient S26. k. Hypoplasia of the bilateral CN3 in patient S27
Discussion
In this study, we evaluated the clinical characteristics of 62 patients with CFEOM and identified six previously reported missense variants in the KIF21A and TUBB3 genes and a novel variant in the KIF21A gene in a patient with sporadic CFEOM1. We also found that patients with TUBB3 variants had more complex and variable clinical features that may be involved in multiple systemic disorders, in contrast to patients with KIF21A variants, who had few systemic disorders and only restrictive strabismus.
The KIF21A-encoded protein consists of an amino terminal motor domain, three central coiled-coil stalk domains, and a carboxy-terminal tail domain containing seven WD40 repeats. As a member of the motor kinesin family, KIF21A plays an important role in microtubule-dependent transport from the cell body to the developing growth cone and acts as an inhibitor of microtubule dynamics through interactions with microtubule-associated proteins [8, 24]. To date, at least 15 mutations have been identified in the KIF21A gene, most of which are missense mutations and cluster in the second coiled-coil and the motor domains, thus interfering with the function of the protein [12, 25,26,27,28,29,30,31]. The novel c.3906T > A(p.Asp1302Glu) variant identified here was located in the low-complexity regions (LCRs), downstream of the third coiled-coil domain. LCRs have been regarded as being involved in numerous aspects of cell biology, including transcriptional regulation, replication, genome stability, and protein–protein interactions [32]. Mutations in LCRs can lead to various human neurological disorders, such as frontotemporal dementia and Alzheimer’s disease [32, 33]. Because aspartate (Asp, D) has a shorter side chain compared with glutamate (Glu, E), it may establish a more stable connection with amino acids other than glutamate when constituting an activity center in the protein. A well-known example of this feature is that aspartate is commonly involved in the formation of protein activation centers via the Asp-His-Ser catalytic triad. Substitution of aspartate (Asp, D) at codon 1304 with glutamate (Glu, E) would destroy the stability of the protein and reduce its activity, which would be predicted to decrease its role as an inhibitor of microtubule dynamics and lead to the stalling of axon guidance [34, 35].
Interestingly, we also found that two patients with the Arg954Trp variant exhibited hypoplasia not only in the third, but also in the six cranial nerve on MRI. Previous studies have shown that hypoplasia of the third and sixth cranial nerves can occur simultaneously in patients with CFEOM [10]. However, specific CFEOM types and their associated genotypes were not mentioned. Definitely, we lack sufficient evidence to support the contention that the hotspot mutation of Arg954Trp is responsible for the hypoplasia of abducens. However, in a recent case report, the syndromic CFEOM phenotype caused by the c.2015T > C(p.Leu672Pro) variant of the KIF21A gene illustrates that KIF21A mutation may result in multiple damage to the nerve system, rather than only involvement in the ocular motor nerve [12].
The TUBB3 gene encodes a neuron-specific Class III β-tubulin isotype, which plays a critical role in neuronal migration and axonal guidance [36, 37]. Heterozygous missense mutations in TUBB3 may cause two distinct phenotypes, isolated or syndromic CFEOM3 or malformations of cortical development, and occasionally both of these conditions [5, 7, 38]. The possible underlying mechanism is that amino acid substitutions in TUBB3 may disturb the equilibrium of microtubule dynamics and the interaction of microtubules with kinesin motors [7, 39, 40]. In our cohort, patients with the Arg262Cys, Ala302Thr, and Asp417Asn variants of TUBB3 exhibited isolated CFEOM3 phenotypes. Unlike that reported previously, our patient with the Asp417Asn variant did not have atrophy of the intrinsic foot muscles and an elevated arch [7]. In agreement with the previous reports of the severe clinical findings commonly detected in isolated patients with CFEOM3 with Arg262Cys, one of our patients (P10-III1) with the Arg262Cys variant had a large exodeviation angle of more than 90 PD at the horizontal direction and extremely tight extraocular muscles. Although she has undergone three operations to correct her large exodeviation, she still exhibits recurrent exotropia. Instead of a de novo mutation, she inherited the Arg262Cys variant from her affected father. In addition, all four patients with sporadic CFEOM3 carrying the Glu410Lys variant displayed complex clinical manifestations, including facial weakness, developmental delays, and additional systemic disorders, such as microphallus and cryptorchidism. In addition, two patients with the Glu410Lys and Asp417Asn variants had hypoplasia of the third and sixth cranial nerves. In a previous report, nystagmus was present in patients with TUBB3 mutations [5]; however, in our patients, nystagmus was present not only in patients with TUBB3 mutations, but also in patients with KIF21A mutations. At one point, it was suggested that nystagmus might be related to reduced visual acuity or sensory deficits. However, we suggest that nystagmus would be associated with dysgenesis of the ocular motor control system caused by underdevelopment of the central nervous system.
Conclusions
We have identified a novel variant in the KIF21A gene that will extend the mutation spectrum of KIF21A and supply some of the novel phenotypes induced by the KIF21A and TUBB3 mutations. In addition, we described a large cohort of Chinese patients with CFEOM with detailed phenotypes and genotypes.
Data availability
The original contributions presented in the study are included in the Supplementary Material, part in https://www.biosino.org/download/node/data/public/OED908528, further inquiries can be directed to the corresponding author.
References
Reck AC, Manners R, Hatchwell E. Phenotypic heterogeneity may occur in congenital fibrosis of the extraocular muscles. Br J Ophthalmol. 1998;82(6):676–9.
Whitman MC, Engle EC. Ocular congenital cranial dysinnervation disorders (CCDDs): insights into axon growth and guidance. Hum Mol Genet. 2017;26(R1). https://doi.org/10.1093/hmg/ddx168. R37-37R44.
Whitman MC, Jurgens JA, Hunter DG, Engle EC. Congenital fibrosis of the extraocular muscles: overview. 2021. Seattle (WA).
Khan AO, Shaheen R, Alkuraya FS. The ECEL1-related strabismus phenotype is consistent with congenital cranial dysinnervation disorder. J AAPOS. 2014;18(4):362–7. https://doi.org/10.1016/j.jaapos.2014.03.005
Whitman MC, Andrews C, Chan WM, Tischfield MA, Stasheff SF, Brancati F, et al. Two unique TUBB3 mutations cause both CFEOM3 and malformations of cortical development. Am J Med Genet A. 2016;170A(2):297–305. https://doi.org/10.1002/ajmg.a.37362
Chew S, Balasubramanian R, Chan WM, Kang PB, Andrews C, Webb BD, et al. A novel syndrome caused by the E410K amino acid substitution in the neuronal β-tubulin isotype 3. Brain. 2013;136(Pt 2):522–35. https://doi.org/10.1093/brain/aws345
Tischfield MA, Baris HN, Wu C, Rudolph G, Van Maldergem L, He W, et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140(1):74–87. https://doi.org/10.1016/j.cell.2009.12.011
Yamada K, Andrews C, Chan WM, McKeown CA, Magli A, de Berardinis T, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type 1 (CFEOM1). Nat Genet. 2003;35(4):318–21. https://doi.org/10.1038/ng1261
Puri D, Barry BJ, Engle EC. TUBB3 and KIF21A in neurodevelopment and disease. Front Neurosci. 2023;17:1226181. https://doi.org/10.3389/fnins.2023.1226181
Demer JL, Clark RA, Engle EC. Magnetic resonance imaging evidence for widespread orbital dysinnervation in congenital fibrosis of extraocular muscles due to mutations in KIF21A. Invest Ophthalmol Vis Sci. 2005;46(2):530–9. https://doi.org/10.1167/iovs.04-1125
Nakano M, Yamada K, Fain J, Sener EC, Selleck CJ, Awad AH, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29(3):315–20. https://doi.org/10.1038/ng744
Soliani L, Spagnoli C, Salerno GG, Mehine M, Rizzi S, Frattini D, et al. A Novel De Novo KIF21A variant in a patient with congenital fibrosis of the extraocular muscles with a syndromic CFEOM phenotype. J Neuroophthalmol. 2021;41(1):e85–8588. https://doi.org/10.1097/WNO.0000000000000921
Aubourg P, Krahn M, Bernard R, Nguyen K, Forzano O, Boccaccio I, et al. Assignment of a new congenital fibrosis of extraocular muscles type 3 (CFEOM3) locus, FEOM4, based on a balanced translocation t(2;13) (q37.3;q12.11) and identification of candidate genes. J Med Genet. 2005;42(3):253–9. https://doi.org/10.1136/jmg.2004.021899
Tukel T, Uzumcu A, Gezer A, Kayserili H, Yuksel-Apak M, Uyguner O, et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. J Med Genet. 2005;42(5):408–15. https://doi.org/10.1136/jmg.2004.026138
Shinwari JM, Khan A, Awad S, Shinwari Z, Alaiya A, Alanazi M, et al. Recessive mutations in COL25A1 are a cause of congenital cranial dysinnervation disorder. Am J Hum Genet. 2015;96(1):147–52. https://doi.org/10.1016/j.ajhg.2014.11.006
Han P, Wei G, Cai K, Xiang X, Deng WP, Li YB, et al. Identification and functional characterization of mutations in LPL gene causing severe hypertriglyceridaemia and acute pancreatitis. J Cell Mol Med. 2020;24(2):1286–99. https://doi.org/10.1111/jcmm.14768
Zhang R, Chen S, Han P, Chen F, Kuang S, Meng Z, et al. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24(2):1906–16. https://doi.org/10.1111/jcmm.14887
Dai Y, Liang S, Dong X, Zhao Y, Ren H, Guan Y, et al. Whole exome sequencing identified a novel DAG1 mutation in a patient with rare, mild and late age of onset muscular dystrophy-dystroglycanopathy. J Cell Mol Med. 2019;23(2):811–8. https://doi.org/10.1111/jcmm.13979
Zheng Y, Xu J, Liang S, Lin D, Banerjee S. Whole exome sequencing identified a Novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front Genet. 2018;9:129. https://doi.org/10.3389/fgene.2018.00129
Talevich E, Shain AH, Botton T, Bastian BC, CNVkit. Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12(4):e1004873. https://doi.org/10.1371/journal.pcbi.1004873
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535–e4824. https://doi.org/10.1016/j.cell.2018.12.015
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30
Rigsby RE, Parker AB. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol Biol Educ. 2016;44(5):433–7. https://doi.org/10.1002/bmb.20966
van der Vaart B, van Riel WE, Doodhi H, et al. CFEOM1-associated kinesin KIF21A is a cortical microtubule growth inhibitor. Dev Cell. 2013;27(2):145–60.
Xia CR, Shi LH, Nan J, Hao YZ, Jia Y. Identification of a novel KIF21A gene mutation in a Chinese family with congenital fibrosis of the extraocular muscles. Zhonghua Yan Ke Za Zhi. 2022;58(3):213–4.
Ali Z, Xing C, Anwar D, et al. A novel de novo KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Möbius syndrome. Mol Vis. 2014;20:368–75.
Wang P, Li S, Xiao X, Guo X, Zhang Q. KIF21A novel deletion and recurrent mutation in patients with congenital fibrosis of the extraocular muscles-1. Int J Mol Med. 2011;28(6):973–5.
Lu S, Zhao C, Zhao K, Li N, Larsson C. Novel and recurrent KIF21A mutations in congenital fibrosis of the extraocular muscles type 1 and 3. Arch Ophthalmol. 2008;126(3):388–94.
Chan WM, Andrews C, Dragan L, et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet. 2007;8:26.
Yamada K, Hunter DG, Andrews C, Engle EC. A novel KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn jaw-winking phenomenon. Arch Ophthalmol. 2005;123(9):1254–9.
Yamada K, Chan WM, Andrews C, et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraocular muscles type 3 (CFEOM3). Invest Ophthalmol Vis Sci. 2004;45(7):2218–23.
Lee J, Cho H, Kwon I. Phase separation of low-complexity domains in cellular function and disease. Exp Mol Med. 2022;54(9):1412–22.
Zhou X, Sumrow L, Tashiro K, et al. Mutations linked to neurological disease enhance self-association of low-complexity protein sequences. Science. 2022;377(6601):eabn5582.
Polgár L. The catalytic triad of serine peptidases. Cell Mol Life Sci. 2005;62(19–20):2161–72. https://doi.org/10.1007/s00018-005-5160-x
Gutteridge A, Thornton JM. Understanding nature’s catalytic toolkit. Trends Biochem Sci. 2005;30(11):622–9. https://doi.org/10.1016/j.tibs.2005.09.006
Garcin C, Straube A. Microtubules in cell migration. Essays Biochem. 2019;63(5):509–20.
Chakraborti S, Natarajan K, Curiel J, Janke C, Liu J. The emerging role of the tubulin code: from the tubulin molecule to neuronal function and disease. Cytoskeleton (Hoboken). 2016;73(10):521–50.
Poirier K, Saillour Y, Bahi-Buisson N, et al. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet. 2010;19(22):4462–73.
Ti SC, Pamula MC, Howes SC, et al. Mutations in human tubulin proximal to the kinesin-binding site alter dynamic instability at microtubule plus- and minus-ends. Dev Cell. 2016;37(1):72–84.
Minoura I, Takazaki H, Ayukawa R, et al. Reversal of axonal growth defects in an extraocular fibrosis model by engineering the kinesin-microtubule interface. Nat Commun. 2016;7:10058.
Acknowledgements
We thank all patients and family members for their participation in this study.
Funding
The study was supported by the National Natural Science Foundation of China (No.81670883), Fujian Provincial Health Technology Project (No.2023GGA047) and Natural Science Foundation of Fujian Province (No.2023J01724).
Author information
Authors and Affiliations
Contributions
JW, LH and NL designed the study. JW and LH performed the study. JW, LH, YZ and YX managed the data. JW, LH, YZ, YX and TM analysed and interpreted the data. JW and LH wrote the initial draft. NL revised the manuscript. NL supervised the study. All authors provided a final review and approved the manuscript before submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The human ethics committees at Beijing Children’s Hospital approved the protocol ([2022]-E-213-R). All protocols adhered to the tenets of the Declaration of Helsinki. All participants provided written informed consent, including the consent of usage of photos.
Consent for publication
Written consent was obtained for all involved participants.
Competing interests
The authors have no proprietary or commercial interest in any materials discussed in this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, J., Huang, L., Zhou, Y. et al. Clinical and genetic characteristics of Chinese patients with congenital fibrosis of the extraocular muscles. Orphanet J Rare Dis 19, 300 (2024). https://doi.org/10.1186/s13023-024-03206-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-024-03206-w