
Abstract
Background and objectives
Congenital heart defect (CHD) is one of the most common birth defects. The aim of this cohort study was to evaluate the prevalence of chromosomal abnormalities and the clinical utility of chromosomal microarray analysis (CMA) in fetuses with different types of CHD, aiming to assist genetic counseling and clinical decision-making.
Methods
In this study, 642 fetuses with CHD were enrolled from a single center over a six-year period (2017–2022). Both conventional karyotyping and CMA were performed simultaneously on these fetuses.
Results
The diagnostic yield of CMA in fetuses with CHD in our study was 15.3% (98/642). Our findings revealed a significant increase in the diagnostic yield of CMA compared to karyotyping in fetuses with CHD. Among CHD subgroups, the diagnostic yields were high in complex CHD (34.9%), conotruncal defects (28.6%), right ventricular outflow tract obstructive defects (RVOTO) (25.9%), atrioventricular septal defects (AVSD) (25.0%) and left ventricular outflow tract obstructive defects (LVOTO) (24.1%), while those in other CHD (10.6%) and septal defects (10.9%) were relatively low. The overall detection rate of clinically significant chromosomal abnormalities was significantly higher in the non-isolated CHD group compared to the isolated CHD group (33.1% vs. 9.9%, P < 0.0001). Interestingly, numerical chromosomal abnormalities were more likely to occur in the non-isolated CHD group than in the isolated CHD group (20.3% vs. 2.0%, P < 0.0001). The rate of termination of pregnancy (TOP)/Still birth in the non-isolated CHD group was significantly higher than that in the isolated CHD group (40.5% vs. 20.6%, P < 0.0001). Compared to the isolated CHD group, the detection rate of clinically significant chromosomal abnormalities was significantly higher in the group of CHD with soft markers (35.6% vs. 9.9%, P < 0.0001) and in the group of CHD with additional structural anomalies (36.1% vs. 9.9%, P < 0.0001).
Conclusions
CMA is a reliable and high-resolution technique that should be recommended as the front-line test for prenatal diagnosis of fetuses with CHD. The prevalence of chromosomal abnormalities varies greatly among different subgroups of CHD, and special attention should be given to prenatal non-isolated cases of CHD, especially those accompanied by additional structural anomalies or soft markers.
Similar content being viewed by others

Background
Congenital heart disease (CHD) is a structural anomaly of the heart and/or great vessels that arises due to disruptions in the normal developmental program of the cardiac system. CHD is one of the most common birth defects, affecting 8–9 babies out of every 1,000 live births and occurring in 10% of spontaneous miscarriages [1, 2]. It is estimated that over 130,000 babies are born with CHD each year in China, with annual treatment costs reaching up to $1.6 billion [3]. The etiology of CHD is complex, involving both genetic and non-genetic factors [4]. Despite significant advancements in the diagnosis and treatment of CHD, our understanding of its underlying causes has been limited until recently. Approximately 20% of cases with CHD have identifiable etiologies, while the majority remain sporadic with unknown origins. Genetic factors include chromosomal abnormalities, genetic syndromes, and single-gene disorders; whereas potential non-genetic factors comprise environmental teratogens, maternal exposures, and infectious agents [5,6,7].
With significant advancements in ultrasound technologies and improvements in equipment capabilities, the prenatal detection of cardiovascular malformations through ultrasound has witnessed a remarkable increase. Chromosomal abnormalities are recognized as a predominant genetic factor contributing to CHD, with their identification reported in over 20% of fetuses affected by this condition [8, 9]. Aneuploidies represent the earliest and most prevalent chromosomal abnormalities associated with cases of CHD, with trisomy 21 being the most frequently observed, followed by trisomy 18 and trisomy 13 [10]. Additionally, copy number variations (CNVs) are recognized as crucial chromosomal abnormalities in CHD cases. Previous studies have identified specific CNVs associated with CHD, including 22q11.2 deletion, 1q21.1 deletion and duplication, 1p36 deletion, 3p25.1 deletion, 7q11 deletion, 8p23 deletion, 11q24-25 deletion, 15q11.2 deletion and 16p13.1 duplication [11]. These CNVs typically encompass genes associated with CHD or genes known to play a crucial role in cardiac development. For example, the 22q11.2 deletion alters the dosage of a gene, TBX1, a T-box transcription factor facilitates cellular proliferation in the secondary heart field, which gives rise to the development of the outflow tract and right ventricle [2]. However, it is widely acknowledged that fetuses diagnosed with both CHD and chromosomal anomalies generally have a poor prognosis [12, 13]. Therefore, we believe that the genetic diagnosis of fetuses with CHD plays a pivotal role in prenatal counseling and prognosis evaluation, making it highly recommended.
Although karyotyping and fluorescence in situ hybridization (FISH) have been used for detecting chromosomal abnormalities in prenatal CHD cases over the past few decades, these approaches possess certain significant limitations. For example, karyotyping is time-consuming and has low resolution, while FISH has limited coverage; however, chromosomal microarray analysis (CMA), which is designed to identify microscopic and submicroscopic chromosomal abnormalities, has recently been widely used in the field of prenatal diagnosis. Therefore, we conducted a cohort study involving 642 fetuses with CHD at a single center to assess both the prevalence of chromosomal abnormalities and the clinical utility of CMA as a genetic diagnostic tool for prenatal CHD cases. Additionally, we categorized these cases into different CHD subgroups to gain better insights into the diagnostic yields of chromosomal abnormalities and provide guidance for genetic counseling and clinical decision-making in prenatal CHD cases.
Methods
Participants
During the period from January 2017 to June 2022, pregnant women were consecutively referred to the Medical Genetic Center of Jiangxi Maternal and Child Health Hospital for invasive prenatal testing due to factors such as advanced maternal age, a high risk of prenatal screening, a family history of hereditary disease or abnormal pregnancy, and fetuses with ultrasound abnormalities. Pregnant women diagnosed with fetal CHD were recruited for this study. Fetuses with CHD were identified through routine ultrasound anatomy scans in the first or second trimesters and subsequently confirmed by echocardiography. Karyotyping and CMA, which are routine diagnostic tests for chromosomal anomalies, were performed simultaneously in all participants. Prior to invasive prenatal testing, all participants were duly informed about the benefits and potential risks associated with CMA and provided with written informed consent. All fetal specimens were amniotic fluid and collected through amniocentesis. Soft markers refer to minor and transient structural changes that may indicate potential significant risks of fetal abnormalities, yet are often inconsequential in isolation. Based on the presence or absence of extracardiac ultrasound anomalies such as additional structural anomalies, soft markers or amniotic fluid volume abnormality, all cases were divided into an isolated group and a non-isolated group. Single umbilical artery, absent or shortened nasal bones, echogenic bowels, choroid plexus cysts, mild ventriculomegaly (10–15 mm), increased nuchal translucency (≥ 3.0 mm), enlarged cisterna magna, thickened nuchal folds (≥ 6.0 mm), persistent right umbilical veins and pyelectasis were considered as soft markers in this study. Additionally, all cases were categorized into a series of CHD groups and subgroups by Botto’s method [14]. After the birth of surviving fetuses, participants were recommended to undergo a comprehensive ultrasound examination. All participants were followed up through phone interview to inquire about the outcomes of their pregnancy. This study was approved by the Medical Ethics Committee of Jiangxi Maternal and Child Health Hospital.
CMA and karyotyping for routine prenatal diagnosis
The Affymetrix CytoScan 750 K array (Applied Biosystems, Affymetrix, Inc., Santa Clara, CA, USA) has been routinely utilized for prenatal genetic testing in our center. It encompasses a total of 750,000 probes distributed across the entire human genome. CMA was performed in all enrolled cases using the SNP-based platform of CytoScan 750 K. Fetal genomic DNA was extracted from amniotic fluid cells utilizing the commercially available QIAamp® DNA Mini Kit (Qiagen, Inc., Hilden, Germany). The experimental procedures were conducted in accordance with the manufacturer’s protocols. The data was analyzed using the Affymetrix® Chromosome Analysis Suite (ChAS) v4.2 Software, and the results were mapped to the Genome Reference Consortium Human Build 37 (GRCh37/hg19). After passing quality control standards, copy number variants (CNVs) were systematically evaluated by comparing them with scientific literature and consulting various public databases, including UCSC Genome Browser (http://genome.ucsc.edu/), ISCA (http://clinicalgenome.org/), Pubmed (http://www.ncbi.nlm.nih.gov/pubmed/), Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home), OMIM (http://omim.org/), DECIPHER (https://decipher.sanger.ac.uk/), and ClinGen Dosage Sensitivity Map (http://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/index.shtml). According to the standards and guidelines of the American College of Medical Genetics (ACMG), CNVs were classified into five categories: pathogenic CNVs, likely pathogenic CNVs, variants of uncertain significance (VUS), likely benign CNVs, and benign CNVs. In this study, clinically significant CNVs included pathogenic and likely pathogenic CNVs; however, likely benign and benign CNVs were not reported. Both aneuploidies and clinically significant CNVs were considered as clinically significant chromosomal abnormalities. G-banding karyotyping was performed in all cases following the standard procedure [15].
Statistical analyses
Chi-square test or Fisher exact test was used to compare the detection rate of chromosomal abnormalities in different CHD groups. P ≤ 0.05 was considered statistically significant.
Results
Characteristics of patients
From January 2017 to June 2022, a total of 16,362 fetuses underwent prenatal diagnosis at our center. Among them, 642 fetuses with CHD were included in this study, accounting for 3.9% (642/16,362) of the entire population that was diagnosed. The demographic characteristics of all enrolled cases are summarized in Table 1. The overall mean age of the pregnant women was 28.0 ± 5.0 years, and the mean gestational age at invasive testing was 24.3 ± 2.9 weeks. The overall rates of Ongoing/Live birth and termination of pregnancy (TOP)/Still birth in all fetuses with CHD were 74.8% (480/642) and 25.2% (162/642), respectively. The rate of TOP/Still birth in the non-isolated CHD group was significantly higher than that in the isolated CHD group (40.5% vs. 20.6%, P < 0.0001).
Diagnostic yield of karyotyping vs. CMA
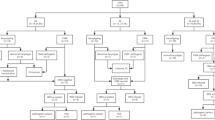
A total of 642 fetuses with CHD underwent karyotyping and CMA. Chromosomal abnormalities were detected in 53 fetuses by conventional karyotyping, resulting in a detection rate of 8.3% (53/642). Meanwhile, CMA identified clinically significant chromosomal abnormalities in 98 fetuses, increasing the detection rate to 15.3% (98/642). Among them, 2 fetuses exhibited triploidy, 38 fetuses displayed aneuploidies, and 58 fetuses manifested pathogenic/likely pathogenic (P/LP) CNVs. Notably, compared to karyotyping, CMA not only identified all numerical chromosomal abnormalities and deletions/duplications detected by karyotyping but also revealed submicroscopic chromosomal abnormalities (Fig. 1).

Flowchart of prenatal testing performed in 642 fetuses with CHD and the detection rate of karyotyping vs. CMA in this study. CHD: Congenital heart defects; CMA: chromosomal microarray analysis; CNVs: copy number variants; P/LP: pathogenic/likely pathogenic; DR: detection rate
Chromosomal abnormalities in fetuses with CHD
After prenatal testing, numerical chromosomal abnormalities were detected in 40 (6.2%) fetuses, including 16 fetuses with trisomy 18 (T18), 15 fetuses with trisomy 21 (T21), 4 fetuses with trisomy 13 (T13), 3 fetuses with monosomy X (45, XO), 1 fetus with a karyotype of 69, XXX and another fetus with a karyotype of 69, XXY. Therefore, T18, T21 and T13 were the most prevalent numerical chromosomal abnormalities observed among fetuses with CHD in our study. Additionally, clinically significant CNVs were identified in a total of 58 (9.0%) fetuses, including 52 fetuses with pathogenic CNVs and 6 fetuses with likely pathogenic CNVs. Among these findings, the most common CNV was 22q11.2 deletion, accounting for 48.2% (28/58) of all the clinically significant CNVs detected. The other three recurrent CNV loci were 7q11.23 (involving ELN), 1q21.1q21.2 (involving GJA5) and 16p11.2 (involving TXB6). There were a total of 29 cases involving the 22q11.2 region (28 deletions and 1 duplication), 4 cases involving the 7q11.23 region (2 deletions and 2 duplications), 4 cases involving the 1q21.1q21.2 region (1 deletion and 3 duplications), and 2 cases involving the 16p11.2 region (1 deletion and 1duplication). The remaining clinically significant CNVs were detected only once, including 1q31.2q32.1 deletion (involving CDC73), 2q13 deletion (involving BCL2L11), 2q37.1q37.3 deletion (involving HDAC4), 4p16.3q12 duplication, 4q32.1q35.2 duplication, 5q22.3q35.3 duplication (involving NSD1), 6q16.3q21 duplication, 6q25.3q27 deletion (involving ARID1B and DLL1), 8p23.3p23.1 deletion, 8p23.1p22 deletion (involving GATA4), 8q21.11q24.13 duplication, 8q24.3 duplication, 9p24.3p24.2 deletion, 9p24.3p13.1 duplication, 9q34.11q34.3 duplication, 9p24.2q13 duplication, 10q23.2q23.31 deletion (involving PTEN), 14q24.1q32.33 mosaic duplication, 16p13.12p13.11 deletion (involving MYH11), 17p12 duplication, 22q13.2q13.33 deletion (involving TCF20), Xp22.31q28 duplication and Xq22.2 duplication(involving PLP1). The cardiac ultrasound findings and pregnancy outcomes of cases with four recurrent CNV loci associated with CHD are summarized in Table 2. The details of 19 cases with CNVs detected only once are shown in supplement Table S1. Additionally, 32 (5.0%) fetuses were detected to harbor variants of uncertain significance (VUS), including 17 (2.6%) duplications, 9 (1.4%) deletions, and 6 (0.9%) regions of allelic homozygosity (ROHs). The details of these cases are listed in supplementary Table S2.
Subgroup analysis of different types of CHD
According to prenatal ultrasound findings and Botto’s anatomical classification [14], all cases were categorized into 10 groups and 26 subgroups. Among them, ventricular septal defects (VSD) accounted for the most common heart defects, representing 44.5% (286/642), followed by right aortic arch (RAA) at 19.5%, multiple complex heart anomalies at 6.2%, coarctation of the aorta at 3.0%, pulmonic stenosis at 2.2%, and tetralogy of Fallot at 2.2%. The diagnostic yields were high in complex CHD (34.9%), conotruncal defects (28.6%), right ventricular outflow tract obstructive defects (RVOTO) (25.9%), atrioventricular septal defects (AVSD) (25.0%), and left ventricular outflow tract obstructive defects (LVOTO) (24.1%). On the other hand, the diagnostic yields were relatively low in other CHD (10.6%) and septal defects (10.9%). The types of CHD and CMA diagnostic yield of fetuses with CHD in this cohort are summarized in Table 3 and the distribution of chromosomal abnormalities in different CHD subgroups are listed in Table 4.
Diagnostic yield of CMA in isolated vs. non-isolated CHD
In this study, a total of 494 cases presenting solely with cardiac ultrasound anomalies were classified as the isolated CHD group; meanwhile, 148 cases accompanied by extracardiac ultrasound anomalies were classified as the non-isolated CHD group. Extracardiac ultrasound anomalies included soft markers (n = 73), additional structural anomalies (n = 61), intrauterine growth retardation (IUGR) (n = 7), and polyhydramnios (n = 7). The frequencies of clinically significant chromosomal abnormalities were found to be 9.9% (49/494), 35.6% (26/73), 36.1% (22/61), 14.3% (1/7) and 0.0% (0/7) in fetuses with isolated CHD, CHD with soft markers, CHD with additional structural anomalies, CHD with IUGR, and CHD with polyhydramnios, respectively. Compared to the isolated CHD group, the detection rate of clinically significant chromosomal abnormalities was significantly higher in the CHD with soft markers group (35.6% vs. 9.9%, P < 0.0001) and the CHD with additional structural anomalies group (36.1% vs. 9.9%, P < 0.0001); however, no highly significant differences were observed between these two groups (35.6% vs. 36.1%, P = 0.956). The CHD group with soft markers consisted of 34 cases of VSD with soft markers and 39 cases of other cardiac malformations with soft markers. The detection rate of clinically significant chromosomal abnormalities in the group with other cardiac malformations and soft markers was higher than that in the group with VSD and soft markers, but the difference was not statistically significant (43.6% vs. 26.5%, P = 0.148). However, compared to the isolated VSD group, the detection rate was significantly higher in the VSD group with soft markers (26.5% vs. 6.4%, P < 0.05). In the CHD with soft markers group, there were 73 fetuses with a single soft marker and 3 fetuses with multiple soft markers. The detection rate of clinically significant chromosomal abnormalities in fetuses with a single soft marker was lower than that in fetuses with multiple soft markers, but this difference was not statistically significant (34.3% vs. 66.7%, P = 0.287). Among this group, the detection rates of clinically significant chromosomal abnormalities were high in fetuses exhibiting increased nuchal translucency (4/4, 100.0%), pyelectasis (1/1, 100.0%), mild ventriculomegaly (3/4, 75.0%), and absent or shortened nasal bone (12/19, 63.2%). Similarly, in the CHD with additional structural anomalies group, 40 fetuses had a single structural anomaly and 21 fetuses had multiple structural anomalies. The detection rate of clinically significant chromosomal abnormalities in fetuses with multiple structural anomalies was higher than that in fetuses with a single structural anomaly; however, there were no statistically significant differences (52.4% vs. 27.5%, P = 0.091). The detection rates of clinically significant chromosomal abnormalities were high in fetuses with exomphalos (3/3, 100.0%), diaphragmatic hernia (1/1, 100.0%), nuchal cystic hygroma (1/2, 50.0%), urinary tract system anomalies (3/9, 33.3%), and facial abnormalities (2/6, 33.3%) within this group. The detection rates of chromosomal abnormalities in fetuses with isolated CHD and non-isolated CHD are presented in Table 5.
The overall detection rate of clinically significant chromosomal abnormalities was significantly higher in the non-isolated group compared to the isolated group (33.1% vs. 9.9%, P < 0.0001). Additionally, within the CHD subgroups, there was a significantly higher detection rate observed in the non-isolated VSD group compared to the isolated VSD group (23.9% vs. 6.4%, P < 0.0001). Interestingly, numerical chromosomal abnormalities were more likely to occur in the non-isolated group than the isolated group (20.3% vs. 2.0%, P < 0.0001). However, there were no significant differences in the incidence of pathogenic/likely pathogenic CNVs (12.8% vs. 7.9%, P = 0.065) and VUS (6.8% vs. 4.5%, P = 0.258) between these two groups. The comparison of the distribution of chromosomal abnormalities between different CHD groups is summarized in Table 6.
Discussion
In this study, we conducted a cohort study on fetuses with various types of CHD to evaluate the prevalence of chromosomal abnormalities and the clinical utility of CMA. Our results demonstrated a significant increase in diagnostic yield through CMA testing among fetuses with CHD. The overall detection rate achieved by CMA was 15.3%, which represents a 7% incremental diagnostic yield over karyotype analysis, consistent with several previous studies [9, 16]. In recent years, CMA has been widely used in prenatal diagnosis, including fetal cardiac ultrasound anomalies. Numerous studies have reported varying detection rates ranging from 10.1 to 24.5% when using CMA for fetal CHD diagnosis [16,17,18,19,20,21,22]. However, in comparison to previous studies [16,17,18,19,20,21,22], our cohort exhibited relatively lower rates of diagnosing chromosomal abnormalities, particularly aneuploidies. The observed discrepancy can be attributed to several factors, including potential biases in case selection, differences in the proportion of CHD subgroups, and patients’ preference for noninvasive prenatal strategies. Nevertheless, it is indisputable that CMA should be recommended as a first-tier diagnostic technique for prenatal cases of CHD.
As the first recognized genetic cause of CHD, aneuploidy accounts for approximately 14% of all genetic causes of syndromic CHD [2, 23]. T18 was identified as the most prevalent form of aneuploidy detected in fetuses with CHD in this study, followed by T21 and T13. Additionally, CNVs have emerged as significant genetic contributors to CHD in recent years [24,25,26]. In our study, a total of fifty-two pathogenic CNVs and six likely pathogenic CNVs were identified. The most common CNV was the 22q11.2 deletion, accounting for 50.0% of all clinically significant CNVs detected. Other recurrent CNV loci included 7q11.23 (involving ELN), 1q21.1q21.2 (involving GJA5) and 16p11.2 (involving TXB6). These four recurrent CNV loci have previously been reported to be associated with CHD; however, patients with these CNVs exhibit highly variable clinical phenotypes [27,28,29,30,31,32]. Moreover, nineteen rare CNVs were detected only once in our study, which might either be the genetic cause for CHD or a secondary finding. Nevertheless, their relationship to prenatal ultrasound phenotype remains unclear and requires further confirmation. Given that a large portion of cardiovascular anomalies in fetuses with CHD are undetectable even with advanced ultrasound equipment and well-trained sonographers [33], it is extremely challenging for clinicians to accurately assess their true condition based solely on ultrasound findings. Therefore, precise genetic diagnosis is crucial for prenatal genetic counseling and prognosis evaluation of cases with CHD. Furthermore, this study identified thirty-two variants of uncertain significance, which posed great challenges to clinicians during genetic counseling due to the ambiguity surrounding their association with clinical phenotypes. Through postnatal follow-up analysis, we observed that a large proportion of participants whose fetuses were diagnosed with VUS chose to terminate their pregnancies, especially when the source of these variants was not confirmed. It was undeniable that the genetic diagnosis of fetuses with CHD had some influence on the outcome of their pregnancies, so clinicians should exercise extreme caution in genetic counseling for cases involving VUS.
Our study revealed significant differences in the overall detection rate of clinically significant chromosomal abnormalities between the non-isolated CHD group and the isolated CHD group (33.1% vs. 9.9%, P < 0.0001), which is consistent with findings from several previous studies [16, 17, 19, 21]. Among subgroups of CHD, fetuses with complex CHD had the highest detection rate at 34.9%, followed by conotruncal defects at 28.6%, RVOTO at 25.9%, AVSD at 25.0%, and LVOTO at 24.1%. The detection rate for fetuses with VSD was relatively low, only reaching 10.1%; however, within the VSD subgroup, non-isolated cases had a significantly higher detection rate compared to isolated cases (23.9% vs. 6.4%, P < 0.0001). Maya et al. found that the risk of chromosomal abnormalities in isolated VSD showed no difference from the background risk, but the risk was higher in non-isolated VSD [34], which is consistent with our findings. Several recent studies have reported that the risk of chromosomal abnormalities in pregnancies with isolated VSD is not significant [17, 34, 35], thus it is considered unreasonable to perform invasive prenatal testing for isolated VSD. To better guide clinical decision-making in cases of prenatal isolated VSD, the correlation between chromosomal abnormalities and isolated VSD are yet to be further investigated.
In line with previous studies [16, 19,20,21, 36], our study discovered that the detection rate in the group of CHD with additional structural anomalies was significantly higher than that in the isolated CHD group (36.1% vs. 9.9%, P < 0.0001). However, the comparison of detection rates between the group of CHD with soft markers and the isolated CHD group was controversial. Wang et al. found no significant differences in the detection rates between these two groups (19.8% vs. 14.3%, ns) [16], while our study observed significant differences (35.6% vs. 9.9%, P < 0.0001). These inconsistent findings could be partially explained by differences in cohort size, inclusion criteria, and proportion of CHD subgroups. The soft markers, although they may be transient and self-resolve later in pregnancy, are acknowledged as underlying risk factors for fetal aneuploidy [37]. Multiple studies have reported that the risk of chromosomal abnormalities increases when multiple soft markers appear or when soft markers are combined with structural abnormalities [37,38,39,40]. Therefore, we suggest paying special attention to prenatal cases of CHD combined with soft markers, as well as cases of CHD combined with additional structural anomalies.
The remarkable strength of this study lies in the large size of our cohort from a single center, which enabled us to compare the detection rate of chromosomal abnormalities among different CHD subgroups and investigate the correlation between chromosomal abnormalities and prenatal cardiac ultrasound phenotypes. However, our study had several limitations. These included the potential selection bias caused by patients having to pay for invasive prenatal testing and some patients refusing further testing. Additionally, CMA could not detect sequence variants; moreover, most patients declined subsequent whole-exome sequencing which resulted in incomplete genetic diagnoses. Finally, although our cohort was large enough overall, the small sample sizes in certain CHD subgroups limited the analysis.
Conclusions
We conducted one of the largest cohort studies to assess the clinical value of CMA in prenatal genetic diagnoses of fetuses with CHD from a single center. CMA, being a reliable and high-resolution technique, should be recommended as the front-line test for prenatal diagnosis of fetuses with CHD. Our findings revealed that the prevalence of chromosomal abnormalities varied greatly among different subgroups of CHD, and special attention should be given to prenatal non-isolated cases of CHD, especially those accompanied by additional structural anomalies or soft markers.
Data availability
All relevant data generated or analyzed during this study are included in this published article. If further datasets are requested, these are available on request from the corresponding author.
Abbreviations
- CHD:
-
Congenital heart defects
- CMA:
-
Chromosomal microarray analysis
- CNVs:
-
Copy number variants
- P:
-
Pathogenic
- LP:
-
Likely pathogenic
- VUS:
-
Variants of uncertain significance
- ASD:
-
Atrial septal defect
- VSD:
-
Ventricular septal defect
- d-TGA:
-
d-Transposition of the great arteries
- DORV:
-
Double outlet right ventricle
- LVOTO:
-
Left ventricular outflow tract obstructive defects
- HLHS:
-
Hypoplastic left heart syndrome
- RVOTO:
-
Right ventricular outflow tract obstructive defects
- AVSD:
-
Atrioventricular septal defects
- APVR:
-
Anomalous pulmonary venous return
- L-TGA:
-
L-Transposition of the great arteries
- TOP:
-
Termination of pregnancy
References
van der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–7.
Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707–20.
Pei L, Kang Y, Zhao Y, Yan H. Prevalence and risk factors of congenital heart defects among live births: a population-based cross-sectional survey in Shaanxi province, Northwestern China. BMC Pediatr. 2017;17:18.
Zaidi S, Brueckner M. Genetics and Genomics of congenital heart disease. Circ Res. 2017;120:923–40.
Blue GM, Kirk EP, Sholler GF, Harvey RP, Winlaw DS. Congenital heart disease: current knowledge about causes and inheritance. Med J Aust. 2012;197:155–9.
Pierpont ME, Basson CT, Benson DW, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–38.
Lalani SR, Belmont JW. Genetic basis of congenital cardiovascular malformations. Eur J Med Genet. 2014;57:402–13.
Yan Y, Wu Q, Zhang L, Wang X, Dan S, Deng D, et al. Detection of submicroscopic chromosomal aberrations by array-based comparative genomic hybridization in fetuses with congenital heart disease. Ultrasound Obstet Gynecol. 2014;43:404–12.
Luo S, Meng D, Li Q, Hu X, Chen Y, He C, et al. Genetic testing and pregnancy outcome analysis of 362 fetuses with congenital heart Disease identified by prenatal Ultrasound. Arq Bras Cardiol. 2018;111:571–7.
Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, et al. The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol. 2011;32:1147–57.
Simmons MA, Brueckner M. The genetics of congenital heart disease… understanding and improving long-term outcomes in congenital heart disease: a review for the general cardiologist and primary care physician. Curr Opin Pediatr. 2017;29:520–8.
van Nisselrooij AEL, Lugthart MA, Clur S-A, Linskens IH, Pajkrt E, Rammeloo LA, et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet Med. 2020;22:1206–14.
Russell MW, Chung WK, Kaltman JR, Miller TA. Advances in the understanding of the genetic determinants of congenital heart Disease and their impact on clinical outcomes. J Am Heart Assoc. 2018;7:e006906.
Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A, National Birth Defects Prevention Study. Seeking causes: classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res Clin Mol Teratol. 2007;79:714–27.
Zhang R, Chen S, Han P, Chen F, Kuang S, Meng Z, et al. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24:1906–16.
Wang Y, Cao L, Liang D, Meng L, Wu Y, Qiao F, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. 2018;218:e2441–24417.
Salzer-Sheelo L, Polak U, Barg A, Kahana S, Yacobson S, Agmon-Fishman I, et al. Prenatal and postnatal chromosomal microarray analysis in 885 cases of various congenital heart defects. Arch Gynecol Obstet. 2022;306:1007–13.
Zhang Z, Hu T, Wang J, Hu R, Li Q, Xiao L, et al. Pregnancy outcomes of fetuses with congenital heart disease after a prenatal diagnosis with chromosome microarray. Prenat Diagn. 2022;42:79–86.
Lu F, Xue P, Zhang B, Wang J, Yu B, Liu J. Estimating the frequency of causal genetic variants in foetuses with congenital heart defects: a Chinese cohort study. Orphanet J Rare Dis. 2022;17:2.
Zhu X, Li J, Ru T, Wang Y, Xu Y, Yang Y, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. 2016;36:321–7.
Qiao F, Wang Y, Zhang C, Zhou R, Wu Y, Wang C, et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet Gynecol. 2021;58:377–87.
Mademont-Soler I, Morales C, Soler A, Martínez-Crespo JM, Shen Y, Margarit E, et al. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: evaluation of chromosomal microarray-based analysis. Ultrasound Obstet Gynecol. 2013;41:375–82.
Wilde AAM, Semsarian C, Márquez MF, Sepehri Shamloo A, Ackerman MJ, Ashley EA, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for Cardiac diseases. Heart Rhythm. 2022;19:e1–60.
Li P, Chen W, Li M, Zhao Z, Feng Z, Gao H, et al. Copy number variant analysis for syndromic congenital heart disease in the Chinese population. Hum Genomics. 2022;16:51.
Ehrlich L, Prakash SK. Copy-number variation in congenital heart disease. Curr Opin Genet Dev. 2022;77:101986.
Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, Zelenika D, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012;91:489–501.
Putotto C, Pugnaloni F, Unolt M, Maiolo S, Trezzi M, Digilio MC, et al. 22q11.2 deletion syndrome: impact of Genetics in the treatment of Conotruncal Heart defects. Child (Basel). 2022;9:772.
Gavril E-C, Popescu R, Nucă I, Ciobanu C-G, Butnariu LI, Rusu C, et al. Different types of deletions created by Low-Copy repeats sequences location in 22q11.2 deletion syndrome: genotype-phenotype correlation. Genes (Basel). 2022;13:2083.
Xue J, Shen R, Xie M, Liu Y, Zhang Y, Gong L, et al. 22q11.2 recurrent copy number variation-related syndrome: a retrospective analysis of our own microarray cohort and a systematic clinical overview of ClinGen curation. Transl Pediatr. 2021;10:3273–81.
Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet. 2012;21:1513–20.
Li F-F, Chen W-J, Yao D, Xu L, Shen J-Y, Zeng Y, et al. Clinical phenotypes study of 231 children with Williams syndrome in China: a single-center retrospective study. Mol Genet Genomic Med. 2022;10:e2069.
Wang Y, Zhou H, Fu F, Cheng K, Yu Q, Huang R, et al. Prenatal diagnosis of chromosome 16p11.2 Microdeletion. Genes (Basel). 2022;13:2315.
Stoll C, Garne E, Clementi M, EUROSCAN Study Group. Evaluation of prenatal diagnosis of associated congenital heart diseases by fetal ultrasonographic examination in Europe. Prenat Diagn. 2001;21:243–52.
Maya I, Singer A, Yonath H, Reches A, Rienstein S, Zeligson S, et al. What have we learned from 691 prenatal chromosomal microarrays for ventricular septal defects? Acta Obstet Gynecol Scand. 2020;99:757–64.
Vedel C, Rode L, Jørgensen FS, Petersen OB, Sundberg K, Tabor A, et al. Prenatally detected isolated ventricular septum defects and the association with chromosomal aberrations-A nationwide register-based study from Denmark. Prenat Diagn. 2021;41:347–53.
Donnelly JC, Platt LD, Rebarber A, Zachary J, Grobman WA, Wapner RJ. Association of copy number variants with specific ultrasonographically detected fetal anomalies. Obstet Gynecol. 2014;124:83–90.
Hu T, Tian T, Zhang Z, Wang J, Hu R, Xiao L, et al. Prenatal chromosomal microarray analysis in 2466 fetuses with ultrasonographic soft markers: a prospective cohort study. Am J Obstet Gynecol. 2021;224:e5161–51616.
Pan L, Liang H, Meng Z, Wang J, Zhang R, Wu Y. Assessing the value of second-trimester nasal bone hypoplasia in predicting chromosomal abnormalities: a retrospective chromosomal microarray analysis of 351 fetuses. Arch Gynecol Obstet. 2022.
Xu X, Wang L, Cheng X, Ke W, Jie S, Lin S, et al. Machine learning-based evaluation of application value of the USM combined with NIPT in the diagnosis of fetal chromosomal abnormalities. Math Biosci Eng. 2022;19:4260–76.
Ekin A, Gezer C, Taner CE, Ozeren M. The effect of associated structural malformations in the prediction of chromosomal abnormality risk of fetuses with echogenic bowel. J Matern Fetal Neonatal Med. 2016;29:41–5.
Acknowledgements
We would like to thank all the patients and their families for participation in this study.
Funding
This study was supported by Jiangxi Provincial Key Laboratory of Birth Defect for Prevention and Control (No. 2024SSY06201). Jiangxi Provincial Clinical Research Center for Birth Defects (No. 20223BCG74002), Jiangxi Province Key Research and Development Project (Grant No. 20232BBG70023), Provincial Health Commission Program of Jiangxi (No. 202410428 to QL), Provincial Health Commission Program of Jiangxi (No. 202210062 to KX).
Author information
Authors and Affiliations
Contributions
QL and LPL collected the clinical data, analyzed the data, and wrote the manuscript. XJW and LJQ carried out ultrasound anatomy scans and clinical diagnosis. BTZ, HYL, YY and CXF conducted chromosomal microarray analysis. JHZ, PPM, TH and KX conducted G-banding karyotyping. TTH, HZY and SHH carried out clinical consultations and provided genetic counseling. YLH participated in follow-up visits. BCY, YYZ and YQL conceived and designed the study. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study design and protocol were reviewed and approved by the Medical Ethics Committee of Jiangxi Maternal and Child Health Hospital.
Consent for publication
The parents of the patients participating in this research provided written informed consent.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Q., Luo, L., Zeng, B. et al. Prenatal chromosomal microarray analysis in a large Chinese cohort of fetuses with congenital heart defects: a single center study. Orphanet J Rare Dis 19, 307 (2024). https://doi.org/10.1186/s13023-024-03317-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-024-03317-4