
Abstract
Background
Conventional G-band karyotyping offers low-resolution detection of chromosome abnormalities and cannot provide information about the involved genomic content. On the other hand, array comparative genomic hybridization can offer a rapid and comprehensive detection of genomewide gains and losses with higher resolution, thus providing the genetic basis for prenatal diagnosis of fetal abnormalities.
Case presentation
A 35-year-old primigravid underwent cordocentesis at 28 weeks gestation due to the presence of polyhydramnios, intrauterine growth retardation, persistent right umbilical vein and mild stenosis of aortic arch at the ultrasound scan. Conventional G-band chromosome analysis revealed an apparently normal karyotype whereas the array CGH detected a de novo 8.97 Mb deletion at chromosome 11q22.3 → q23.3 and offered a precise characterization of the genetic defect.
Conclusions
The array CGH detected a de novo interstitial 11q deletion with its precise location and size which could be missed or confused by G-band chromosome analysis. The breakpoint was close to the folate sensitive rare fragile site FRA11B and the aphidicolin inducible common fragile site FRA11G, the co-localization fragile site could have caused instability and constitutional chromosomal breakage. This case study indicates that array CGH is a useful technique for detecting small unbalanced chromosomal abnormalities and should be an integral part of prenatal diagnosis for fetal malformations.
Similar content being viewed by others

Background
The incidence of major structural birth defects in China is approximately 5.6% [1] and is associated with inherited or de novo genetic rearrangements and mutations as well as with maternal risk factors, such as advanced age, exposure to teratogenic factors, illnesses and infections. The detailed second trimester ultrasound scan can detect major fetal malformations and is offered for routine prenatal care. The genetic basis of abnormal ultrasound findings is important for prenatal diagnosis and counseling.
Conventional Giemsa-band (G-band) karyotyping on metaphase cells, which is the standard procedures used for prenatal cytogenetic diagnosis for over 40 years, can detect aneuploidy, unbalanced and apparently balanced structural rearrangements, and deletions/duplications of at least 5-10 Mb. The culturing process usually takes several days to a few weeks in order to generate the number of metaphase chromosomes enough for a full karyotype report. Moreover, G-band karyotyping lacks the resolution to assess the involved genomic content. These limitations hinder the inference of karyotype-phenotype predictions and the identification of candidate genes associated with fetal anomalies.
As in the majority of cases with ultrasound abnormalities the karyotype in the fetus is normal, thus demonstrating the need for additional diagnostic techniques with higher diagnostic capacity [2]. Array comparative genomic hybridization (array CGH) has been introduced in prenatal diagnosis to rapidly detect genomewide gains and losses with higher resolution [3]. It is a high throughput method which can detect copy number changes to a resolution of even as low as 1 Kb. Array CGH is rapidly replacing conventional G-band karyotyping in postnatal diagnosis of children with developmental, intellectual, and physical disabilities [4]-[7], but its application in prenatal diagnosis is still limited. Several groups have demonstrated that by applying array CGH for prenatal diagnosis of fetal ultrasound abnormalities, there was an increased detection rate over G-band karyotyping or other molecular cytogenetic techniques [8]-[11].
In this paper, we demonstrate the application of array CGH in prenatal diagnosis, which permits the rapid identification of a de novo interstitial deletion of 11q (11q22.3 → q23.3) in a fetus with abnormal ultrasound findings.
Case presentation
A 35-year-old primigravid was referred to our hospital at 18 weeks of gestation for genetic counseling. The parents were healthy and nonconsanguineous. There was no family history of congenital malformations or genetic disorders. The mother was tested negative for toxoplasma, CMV, herpes, and rubella. No drugs or infections were reported during the course of the pregnancy. After informed about the possible risk of a chromosomal abnormality in the presence of advanced maternal age, the couple decided to receive noninvasive chromosomal aneuploidy screening. We performed sequencing analysis of the cell free DNA extracted from the maternal peripheral blood, and the result turned out to be negative for trisomy 13, 18 and 21.
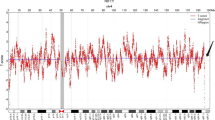
At 28 weeks of gestation the couple consulted our hospital again due to the presence of polyhydramnios, intrauterine growth retardation, persistent right umbilical vein and mild stenosis of aortic arch at the ultrasound scan. Interventional prenatal diagnosis was referred to the couple. A cordocentesis was carried out for prenatal diagnosis. Chromosome analysis was performed on cultured cord blood lymphocytes by Giemsa banding at approximately 350 band resolution. The cytogenetics revealed an apparently normal karyotype 46,XX (Figure 1) with the limited banding resolution. Array CGH using Agilent's 8 × 60 K commercial arrays (Agilent Technologies, Santa Clara, CA, USA) was performed on DNA extracted from uncultured cord blood and a 8.97 Mb deletion was detected at chromosome 11q22.3-11q23.3 or arr 11q22.3q23.3(107,686,511-116,660,613)x1 (Figure 2). The molecular karyotyping refer to the Human Genome February 2009 (versions GRCh37, hg19) assembly. The array CGH analysis of the parental blood did not reveal any deletion at chromosome 11q, no balanced chromosomal rearrangements or inversions were found by G-band karyotyping.

G-banded karyotype of the fetus indicated an apparently normal 46,XX.

Array CGH analysis of the fetus revealed a 8.97 Mb deletion at chromosome 11q22.3-11q23.3 or arr 11q22.3q23.3 (107,686,511-116,660,613)x1 (hg19; NCBI build 37).
After genetic counseling, termination of pregnancy was performed at parent's request at 30 weeks of gestation. A female fetus was delivered with no apparent phenotypic abnormalities. Autopsy was rejected by the parents.
Conclusion
The majority of chromosome 11 long arm deletions are at the terminal region, which is associated with a clinically well-described phenotype, called Jacobsen syndrome. Whereas the interstitial deletions of chromosome 11q are rarely observed, it may being resulted from a direct de novo deletion or familial inheritance [12]-[14]. To our knowledge, only 33 cases with interstitial 11q deletion have been reported in literature and previous reports of such cases have been associated with a wide variability of phenotypic findings. Most of these cases had not been analyzed with a molecular method, thus the phenotype-genotype correlation was not very clear.
Among the 33 cases previously described, only six were characterized with a molecular definition (Table 1). Danijela Krgovic reported a deletion of chromosome 11q22.3 in a 5-year-old girl with mild mental retardation, developmental and speech delay, facial dysmorphism, ptosis, hypertelorism, low-set dysplastic ears, prominent forehead and hypoplastic corpus callosum [15]. Peining Li reported a deletion of chromosome 11q14.1 → q23.2 involving the FZD4 gene in a patient with growth retardation, facial anomalies, exudative vitreoretinopathy (EVR), cleft palate, and minor digital anomalies [16]. Daniela Melis reported a deletion of chromosome 11q13.5 → q14.2 in a 5-year-old boy with low frontal hairline, flat profile, round face, full cheeks, periorbital fullness, hypertelorism, broad nasal bridge, down-turned corners of the mouth and developmental delay [17]. Rebecca L. Sparkes reported a maternally inherited 11q14.3 → q22.3 deletion of a male fetus, the ultrasound scan revealed choroid plexus cysts, echogenic intracardiac foci, mild polyhydramnios, a relatively enlarged right atrium with abnormal cardiac axis, small cerebellum and left talipes equinovarus. The 38-year-old mother with a 17, 3 Mb deletion (from nt 89,492,818 to nt 106,832,040) and a 0.9 Mb duplication (from nt 88,258,744 to nt 89,103,489) in 11q21 → q23 was of normal intellect and healthy only with a surgically repaired bilateral club foot and high myopia [14]. Josephine Wincent reported a deletion of chromosome 11q13.4 → q14.3 in a boy with microcephaly, ptosis and developmental delay [18]. Renata Nacinovich followed up a boy with 11q14.3 → q22.3 deletion from early infancy to adolescence, the proband was born with hypotonia and minor dysmorphisms (submucous cleft palate), his height growth and cognitive development were at the lower limit during childhood [19].
As we mentioned above, due to the heterogeneity in size and position of the deletions, it is not easy to define a distinctive genotype/phenotype of the interstitial 11q deletion. Most of the patients with an overlapping deletion of this region had mild to severe mental retardation and developmental delay, usually depending on the size and position of the deletion. However, because our case was diagnosed in uterus, it was not possible to investigate the mental and developmental delay. In the only prenatal diagnosed report, mild polyhydramnios and a suspected structural cardiac malformation was described [14], our case also shows the similar ultrasound results.
In the deleted region of this case, about 30 genes with already known or unknown functions are mapped [http://www.ncbi.nlm.nih.gov/]. In a proportion of patients with Jacobsen syndrome (terminal 11q deletions) the breakpoints cluster in chromosomal subband 11q23.3 [20]-[22], a breakage-prone region which encompasses both the folate sensitive rare fragile site FRA11B and the aphidicolin inducible common fragile site FRA11G[20],[23]-[25]. In our case, the deleted region maps at 11q22.3 → q23.3, the breakpoint is on the proximal side of FRA11G and FRA11B, indicating that the co-localization fragile site could have caused instability and constitutional chromosomal rearrangements in vivo [23],[26]. Aphidicolin inducible common fragile sites do not break at defined sequences but in breakage-prone regions up to 10 Mb where the break is most likely to appear [25],[27],[28]. The molecular basis of common fragile site fragility has not been fully clarified. Common fragile sites contain more areas of high DNA torsional flexibility with more highly AT-dinucleotide-rich islands than neighbouring non-fragile regions. The inconsistency in DNA replication progression between fragile and flanking non-fragile regions might contribute to occurrence of breaks at these fragile sites.
Folate sensitive fragile sites are caused by expansive mutations of the normally occurring CCG/CGG trinucleotide repeat sequences adjacent to a CpG island [29],[30]. In majority of normal individuals this CCG/CGG repeat is present in 8-80 copies [23]. The repeat can expand to 85-100 copies as a premutation, without cytogenetic expression of the fragile site. During the premutation phase, the repeats become highly unstable when transmitted to the next generation. The offspring may then have a longer or a shorter extension of the repeat sequence than the parent [31]. In individuals with cytogenetic expression of the FRA11B the repeat is expanded to several hundred copies. This expansion is unstable and dependent upon the length of the repeat tract: the longer the tract, the higher the instability and probability of further expansion. It is hypothesized that hypermethylation of the expanded CCG/CGG trinucleotide on chromosome 11 could delay DNA replication of this fragile site, resulting in a break and/or impaired DNA replication [32].
The chromosome band 11q23 is often involved in multiple tumor associated rearrangements. On the distal side of FRA11B lies the MLL gene. The MLL gene regulates the HOX gene expression by directly binding to the promoter sequences. Translocations involving the MLL gene have been found in acute myeloid leukemia (AML), acute lymphoblastic leukemia, or mixed linkage leukemia (MLL) [33]. The region could be considered as a hotspot of various tumors and chromosomal recombination or breakage.
We report the prenatal diagnosis of a de novo interstitial deletion of 11q (11q22.3 → q23.3) performed by array CGH in a fetus with polyhydramnios, intrauterine growth retardation, persistent right umbilical vein and minor cardiac abnormalities at 28 weeks of gestation. In this study, we used combinations of classic G-band karotyping, with array CGH methods to undertake a genome-wide screening for chromosome aberrations. The array CGH successfully detected the 8.9 Mb deletion with its precise location which could be missed or confused by G-band chromosome analysis. It provides valuable information for genetic counselors to achieve molecular diagnosis of prenatal anomalies and to make more accurate predictions about the clinical outcomes. The results indicate that array CGH is a useful technique for detecting small unbalanced chromosomal abnormalities and should be an integral part of prenatal diagnosis for fetal malformations.
Consent
Written informed consent was obtained from the parents of the proband for publication of this Case Report and any accompanying images. The consent form was approved by the ethical committee of Hubei Maternal and Child Health Hospital, China. A copy of the written consent is available for review by the editor of this journal.
Authors' contributions
NL contributed to design the study, analyzed the result, collected the literature data and drafted the manuscript. JY contributed to design the study and revised the manuscript. XC carried out the ultrasound scan and collected the related data. JS participated in genetic counseling. BW carried out array CGH and the analysis of the results. YY carried out chromosome analysis. All authors read and approved the final manuscript.
References
The Ministry of Health: Report on birth defects prevention of China. 2012.
Lichtenbelt KD, Knoers NV, Schuring-Blom GH: From karyotyping to array-CGH in prenatal diagnosis. Cytogenet Genome Res 2011,135(3-4):241–250. 10.1159/000334065
ACOG Committee Opinion No. 446: array comparative genomic hybridization in prenatal diagnosis. Obstet Gynecol 2009, 114(5):1161-1163.
de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal S, Nillesen WM, Huys EH, Leeuw N, Smeets D, Sistermans EA, Feuth T, van Ravenswaaij-Arts CM, van Kessel AG, Schoenmakers EF, Brunner HG, Veltman JA: Diagnostic genome profiling in mental retardation. Am J Hum Genet 2005,77(4):606–616. 10.1086/491719
Xiang B, Li A, Valentin D, Nowak NJ, Zhao H, Li P: Analytical and clinical validity of whole-genome oligonucleotide array comparative genomic hybridization for pediatric patients with mental retardation and developmental delay. Am J Med Genet A 2008,146a(15):1942–1954. 10.1002/ajmg.a.32411
Shaffer LG, Bejjani BA, Torchia B, Kirkpatrick S, Coppinger J, Ballif BC: The identification of microdeletion syndromes and other chromosome abnormalities: cytogenetic methods of the past, new technologies for the future. Am J Med Genet C Semin Med Genet 2007,145c(4):335–345. 10.1002/ajmg.c.30152
Shaffer LG, Coppinger J, Alliman S, Torchia BA, Theisen A, Ballif BC, Bejjani BA: Comparison of microarray-based detection rates for cytogenetic abnormalities in prenatal and neonatal specimens. Prenat Diagn 2008,28(9):789–795. 10.1002/pd.2053
Shaffer LG, Rosenfeld JA, Dabell MP, Coppinger J, Bandholz AM, Ellison JW, Ravnan JB, Torchia BS, Ballif BC, Fisher AJ: Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat Diagn 2012,32(10):986–995. 10.1002/pd.3943
Tyreman M, Abbott KM, Willatt LR, Nash R, Lees C, Whittaker J, Simonic I: High resolution array analysis: diagnosing pregnancies with abnormal ultrasound findings. J Med Genet 2009,46(8):531–541. 10.1136/jmg.2008.065482
Valduga M, Philippe C, Bach Segura P, Thiebaugeorges O, Miton A, Beri M, Bonnet C, Nemos C, Foliguet B, Jonveaux P: A retrospective study by oligonucleotide array-CGH analysis in 50 fetuses with multiple malformations. Prenat Diagn 2010,30(4):333–341.
D'Amours G, Kibar Z, Mathonnet G, Fetni R, Tihy F, Desilets V, Nizard S, Michaud JL, Lemyre E: Whole-genome array CGH identifies pathogenic copy number variations in fetuses with major malformations and a normal karyotype. Clin Genet 2012,81(2):128–141. 10.1111/j.1399-0004.2011.01687.x
Goumy C, Gouas L, Tchirkov A, Roucaute T, Giollant M, Veronese L, Francannet C, Vago P: Familial deletion 11q14.3-q22.1 without apparent phenotypic consequences: a haplosufficient 8.5 Mb region. Am J Med Genet A 2008,146a(20):2668–2672. 10.1002/ajmg.a.32511
Li L, Moore P, Ngo C, Petrovic V, White SM, Northrop E, Ioannou PA, McKinlay Gardner RJ, Slater HR: Identification of a haplosufficient 3.6-Mb region in human chromosome 11q14.3→q21. Cytogenet Genome Res 2002,97(3-4):158–162. 10.1159/000066612
Sparkes RL, Shetty S, Chernos JE, Mefford HC, Micheil Innes A: Interstitial deletion of 11q in a mother and fetus: implications of directly transmitted chromosomal imbalances for prenatal genetic counseling. Prenat Diagn 2009,29(3):283–286. 10.1002/pd.2209
Krgovic D, Marcun Varda N, Zagorac A, Kokalj-Vokac N: Submicroscopic interstitial deletion of chromosome 11q22.3 in a girl with mild mental retardation and facial dysmorphism: case report. Mol Cytogenet 2011, 4: 17. 10.1186/1755-8166-4-17
Li P, Zhang HZ, Huff S, Nimmakayalu M, Qumsiyeh M, Yu J, Szekely A, Xu T, Pober BR: Karyotype-phenotype insights from 11q14.1-q23.2 interstitial deletions: FZD4 haploinsufficiency and exudative vitreoretinopathy in a patient with a complex chromosome rearrangement. Am J Med Genet A 2006,140(24):2721–2729. 10.1002/ajmg.a.31498
Melis D, Genesio R, Cozzolino M, Del Giudice E, Mormile A, Imperati F, Ronga V, Della Casa R, Nitsch L, Andria G: An emerging phenotype of proximal 11q deletions. Eur J Med Genet 2010,53(5):340–343. 10.1016/j.ejmg.2010.07.010
Wincent J, Schoumans J, Anderlid BM: De novo deletion of chromosome 11q13.4-q14.3 in a boy with microcephaly, ptosis and developmental delay. Eur J Med Genet 2010,53(1):50–53. 10.1016/j.ejmg.2009.10.003
Nacinovich R, Villa N, Redaelli S, Broggi F, Bomba M, Stoppa P, Scatigno A, Selicorni A, Dalpra L, Neri F: Interstitial 11q deletion: genomic characterization and neuropsychiatric follow up from early infancy to adolescence and literature review. BMC Res Notes 2014,7(1):248. 10.1186/1756-0500-7-248
Jones C, Slijepcevic P, Marsh S, Baker E, Langdon WY, Richards RI, Tunnacliffe A: Physical linkage of the fragile site FRA11B and a Jacobsen syndrome chromosome deletion breakpoint in 11q23.3. Hum Mol Genet 1994,3(12):2123–2130. 10.1093/hmg/3.12.2123
Penny LA, Dell'Aquila M, Jones MC, Bergoffen J, Cunniff C, Fryns JP, Grace E, Graham JM Jr, Kousseff B, Mattina T, Syme J, Voullaire L, Zelante L, Zenger-Hain J, Jones OW, Evans GA: Clinical and molecular characterization of patients with distal 11q deletions. Am J Hum Genet 1995,56(3):676–683.
Tyson C, Qiao Y, Harvard C, Liu X, Bernier FP, McGillivray B, Farrell SA, Arbour L, Chudley AE, Clarke L, Gibson W, Dyack S, McLeod R, Costa T, Vanallen MI, Yong SL, Graham GE, Macleod P, Patel MS, Hurlburt J, Holden JJ, Lewis SM, Rajcan-Separovic E: Submicroscopic deletions of 11q24–25 in individuals without Jacobsen syndrome: re-examination of the critical region by high-resolution array-CGH. Mol Cytogenet 2008, 1: 23. 10.1186/1755-8166-1-23
Jones C, Penny L, Mattina T, Yu S, Baker E, Voullaire L, Langdon WY, Sutherland GR, Richards RI, Tunnacliffe A: Association of a chromosome deletion syndrome with a fragile site within the proto-oncogene CBL2. Nature 1995,376(6536):145–149. 10.1038/376145a0
Fechter A, Buettel I, Kuehnel E, Savelyeva L, Schwab M: Common fragile site FRA11G and rare fragile site FRA11B at 11q23.3 encompass distinct genomic regions. Genes Chromosomes Cancer 2007,46(1):98–106. 10.1002/gcc.20389
Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A: Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 2010,36(4):929–940.
Liehr T, Kosayakova N, Schroder J, Ziegler M, Kreskowski K, Pohle B, Bhatt S, Theuss L, Wilhelm K, Weise A, Mrasek K: Evidence for correlation of fragile sites and chromosomal breakpoints in carriers of constitutional balanced chromosomal rearrangements. BJMG 2011,14(2):13–16.
Curatolo A, Limongi ZM, Pelliccia F, Rocchi A: Molecular characterization of the human common fragile site FRA1H. Genes Chromosomes Cancer 2007,46(5):487–493. 10.1002/gcc.20432
Zhu Y, McAvoy S, Kuhn R, Smith DI: RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene 2006,25(20):2901–2908. 10.1038/sj.onc.1209314
Sutherland GR: Rare fragile sites. Cytogenet Genome Res 2003,100(1-4):77–84. 10.1159/000072840
Sutherland GR, Richards RI: The molecular basis of fragile sites in human chromosomes. Curr Opin Genet Dev 1995,5(3):323–327. 10.1016/0959-437X(95)80046-8
Lukusa T, Fryns JP: Human chromosome fragility. Biochim Biophys Acta 2008,1779(1):3–16. 10.1016/j.bbagrm.2007.10.005
Michaelis RC, Velagaleti GV, Jones C, Pivnick EK, Phelan MC, Boyd E, Tarleton J, Wilroy RS, Tunnacliffe A, Tharapel AT: Most Jacobsen syndrome deletion breakpoints occur distal to FRA11B. Am J Med Genet 1998,76(3):222–228. 10.1002/(SICI)1096-8628(19980319)76:3<222::AID-AJMG5>3.0.CO;2-S
Dolan M, McGlennen RC, Hirsch B: MLL amplification in myeloid malignancies: clinical, molecular, and cytogenetic findings. Cancer Genet Cytogenet 2002,134(2):93–101. 10.1016/S0165-4608(01)00602-1
Acknowledgements
We thank proband's family for the cooperation. We thank the lab technician Huai Guo, Shuqin Xu, Hui Zhang and Ji Huang for their valuable help.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Liu, N., Yan, J., Chen, X. et al. Prenatal diagnosis of a de novo interstitial deletion of 11q (11q22.3 → q23.3) associated with abnormal ultrasound findings by array comparative genomic hybridization. Mol Cytogenet 7, 62 (2014). https://doi.org/10.1186/s13039-014-0062-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-014-0062-y