
Abstract
Background
The contribution of genetic variants to congenital heart defects (CHDs) has been investigated in many postnatal cohorts but described in few prenatal fetus cohorts. Overall, specific genetic variants especially copy number variants (CNVs) leading to CHDs are somewhat diverse among different prenatal cohort studies. In this study, a total of 1118 fetuses with confirmed CHDs were recruited from three units over a 5-year period, composing 961 of singleton pregnancies and 157 of twin pregnancies. We performed chromosomal microarray analysis on all cases to detect numerical chromosomal abnormalities (NCAs) and pathogenic/likely pathogenic CNVs (P/LP CNVs) and employed whole-exome sequencing for some cases without NCAs and P/LP CNVs to detect P/LP sequence variants (P/LP SVs).
Results
Overall, NCAs and P/LP CNVs were identified in 17.6% (197/1118) of cases, with NCA accounting for 9.1% (102/1118) and P/LP CNV for 8.5% (95/1118). Nonisolated CHDs showed a significantly higher frequency of NCA than isolated CHD (27.3% vs. 4.4%, p < 0.001), but there was no significant difference in the frequency of P/LP CNVs between isolated and nonisolated CHD (11.7% vs. 7.7%). A total of 109 P/LP CNVs were identified in 95 fetuses, consisting of 97 (89.0%) de novo, 6 (5.5%) parental inherited and 6 (5.5%) with unavailable parental information. The 16p11.2 proximal BP4-BP5 deletion was detected in 0.9% (10/1118) of all cases, second only to the most common 22q11.21 proximal A-D deletion (2.1%, 23/1118). Most of the 16p11.2 deletions (8/10) detected were de novo, and were enriched in CHD cases compared with a control cohort from a previous study. Additionally, SV was identified in 12.9% (8/62) of cases without NCA and P/LP CNV, most of which were de novo with autosomal dominant inheritance.
Conclusions
Our cohort study provides a deep profile of the contribution of genetic variants to CHDs in both singleton and twin fetuses; NCA and P/LP CNV contribute to 9.1% and 8.5% of CHD in fetuses, respectively. We confirmed the 16p11.2 deletion as a CHD-associated hotspot CNV, second only to the 22q11.21 deletion in frequency. Most 16p11.2 deletions detected were de novo. Additionally, P/LP SV was identified in 12.9% (8/62) of fetuses without NCA or P/LP CNV.
Similar content being viewed by others


Background
Congenital heart defects (CHDs) are the most common congenital structural malformations in both Chinese and other populations [1,2,3,4]. Routine screening of fetal anatomical structures using improved ultrasound technology has partly increased prenatal detection of CHD in recent decades [3, 5]. The current scope for prenatal diagnosis of CHDs not only includes cardiovascular anatomical structure but also diagnosis or exclusion of genetic disorders, which may involve prenatally undetectable functional anomalies or neurodevelopmental disorders (NDDs). Extracardiac malformations (ECMs) and NDDs are estimated to occur in approximately 13% and 10% of patients with CHDs, respectively, with approximately 2% being caused by genetic syndromes [6, 7]. Although advances in perinatal care and medical interventions have led to drastically reduced mortality rates in neonates with CHDs, genetic disorders greatly influence the outcomes and medical management of CHD patients.
Genetic evaluation for CHDs in postnatal cohorts has been well described in recent years and contributes to most of the available genetic data associated with CHDs [8,9,10,11,12]. The contributions of numerical chromosomal abnormality (NCA), copy number variant (CNV) and monogenic sequence variant (SV) events to CHDs are estimated to be 13%, 10% and 12% respectively [13, 14], indicating that genetic variants play a significant role in CHD pathogenesis. Nevertheless, the frequencies of various genetic variants might be somewhat different between pre- and postnatal cohorts, mainly because prenatal cohorts include intrauterine demise, termination or selective reduction for twin fetuses as well as fetuses with many other ECMs in addition to CHDs. Although a few studies on prenatal CHD cohorts have been performed to evaluate the contribution of genetic variants [15,16,17,18,19,20,21], our study provides a comprehensive profile of genetic variants in a larger fetus cohort (including singleton and twin fetuses) from multiple centers in southern China.
Results
Cohort characteristics
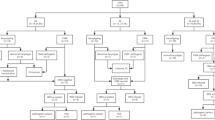
In total, 1118 fetuses including 961 from singleton pregnancies and 157 from twin pregnancies (n = 151), who underwent chromosomal microarray analysis (CMA) prenatally were included, while 61 fetuses including 55 from singleton pregnancies and 6 from twin pregnancies (n = 6), who did not undergo CMA prenatally were excluded. All parents of the 1118 fetuses were of Chinese Han ethnicity, and all parents declared their marriages were nonconsanguineous. Furthermore, prenatal CMA performed on the 1118 fetuses did not reveal large regions of homozygosity (ROHs) involving multiple chromosomes, confirming the lack of consanguineous marriage among the participating parents. Among the 151 twin pregnancies, 99 were dichorionic diamniotic, 44 monochorionic diamniotic and 8 monochorionic monoamniotic cases; both fetuses of a twin pair from 6 twin pregnancies had CHDs, and one fetus of a twin pair from 145 twin pregnancies had CHDs. CHDs were isolated in 887 fetuses (778 singleton; 109 twin) and nonisolated in 231 (183 singleton; 48 twin), with one or several ECMs (n = 192), fetal growth restriction (n = 27) or amniotic fluid volume abnormalities (n = 14). CHDs were classified into various types according to a previous study [22]. The primary cohort characteristics are summarized in Table 1. The study flowchart is illustrated in Fig. 1.

Flowchart diagram summarizing the genetic findings for singleton and twin fetuses with congenital heart defects. CHD, Congenital heart defect; CMA, Chromosomal microarray analysis; NCA, Numerical chromosomal abnormality; P/LP CNV, Pathogenic and likely pathogenic copy number variant; P/LP SV, Pathogenic and likely pathogenic sequence variant; ROH, Region of homozygosity; WES, Whole-exome sequencing. Other hits denoting cardiovascular phenotype-associated deleteriously rare variants in functionally intolerant genes
Contributions of NCA and pathogenic/likely pathogenic (P/LP) CNV to CHDs
In this study, CMA was performed for all 1118 fetuses, producing a diagnostic yield of 17.6% (197/1118), which included 9.1% (102/1118) for NCA and 8.5% (95/1118) for P/LP CNV.
As expected, NCA was commonly detected in this cohort. The most common was trisomy 18 (3.0%, 33/1118), followed by trisomy 21 (2.7%, 30/1118), trisomy 13 (1.0%, 11/1118), monosomy X (0.7%, 8/1118), mosaic monosomy X (0.4%, 4/1118), mosaic trisomy 13 (0.2% 2/1118), mosaic trisomy 14 (0.2% 2/1118), triploidy (0.2% 2/1118) and others (XYY and other rare mosaic trisomy/monosomy, at one each).
Another common genetic variant observed in the cohort was P/LP CNV. We present the top 19 kinds of CNVs with a frequency of more than once in our cohort in Table 2. The most common was 22q11.21 deletion/duplication with a frequency of 2.5% (28/1118), comprising proximal deletion A-D (including TBX1) (2.1%, 23/1118), central deletion B/C-D (including CRKL) (0.3%, 3/1118), proximal deletion A-B (including TBX1) (n = 1) and proximal duplication A-B (includes TBX1) (n = 1). Another relatively common 16p11.2 deletion/duplication was also detected, with a frequency of 1.0% (11/1118), consisting of proximal deletion BP4-BP5 (including TBX6) (0.9%, 10/1118) and proximal duplication BP4-BP5 (including TBX6) (0.1%, 1/1118). Other recurrently detected P/LP CNVs included 16p13.11 deletion/duplication BP2-BP3 (including MYH11) (0.4%), 2q37.3 terminal deletion (including HDAC4) (0.3%), 4p16.3 terminal deletion (0.3%), and 1q21.2 deletion/duplication (0.3%), among others.
In addition, CMA detected ROHs in 0.4% (4/1118) of the fetuses, including (5)×2 hmz, 5p15.33p15.32(113577_5240002)×2 hmz (5.13 Mb at 5pter), 19q13.11q13.41(34375323_51784153)×2 hmz (17.41 Mb) and 3p12.3p11.1(78679584_ 90,485,635)×2 hmz (11.81 Mb). The ROH (5)×2 hmz was uniparental disomy (UPD); the other three ROHs were not confirmed as UPD by parental CMA analysis and two of them did not show any P/LP SVs detected by whole-exome sequencing (WES).
Comparison of NCA and P/LP CNV between isolated and nonisolated CHDs
The frequency of NCA and P/LP CNV in the nonisolated CHD group was significantly higher than that in the isolated CHD group (39.0% vs. 12.1%, p < 0.001) (Table 3), which was mainly due to the higher frequency of NCA in the former (nonisolated 27.3% vs. isolated 4.4%, p < 0.001). This difference was observed in both singleton and twin fetuses. The frequency of P/LP CNVs in the nonisolated CHD group was higher than that in the isolated CHD group but this difference did not reach significance (11.7% vs. 7.7%, p = 0.051). Additionally, the frequency of P/LP CNVs was higher than that of NCA in the isolated CHD group (7.7% vs. 4.4%) but not in the nonisolated CHD group (11.7% vs. 27.3%). This result indicates that P/LP CNVs were more frequently associated with isolated CHDs in this cohort and that NCAs were more frequently associated with nonisolated CHDs. The contributions of NCA and P/LP CNV among various CHD types are presented in Additional file 1: Table S1 and Additional file 2.
Comparison of NCA and P/LP CNV between singleton and twin fetuses
Of interest, the twin group showed a significantly higher frequency of NCA than the singleton group (14.0% vs. 8.3%, p = 0.022), but there was no significant difference in the frequency of P/LP CNVs between the groups (7.6% vs. 8.6%, p = 0.679) (Table 3). When only taking isolated CHDs into account, the frequency of NCA among twin fetuses was slightly higher than that among singleton fetuses (6.4% vs. 4.1%) but P/LP CNVs were similar (7.3% vs. 7.7%).
In twin fetuses with CHDs, the frequency of NCA was higher than that of P/LP CNV (14.0% vs. 7.6%); in particular, the frequency of NCA reached to 31.3% (15/48) in twin fetuses with nonisolated CHDs. However, when only considering isolated CHDs, the contribution of NCA and P/LP CNVs to CHDs showed little difference.
Contribution of P/LP SVs in fetuses without NCAs and P/LP CNVs
For the 921 fetuses without NCAs and P/LP CNVs (including 917 fetuses with negative CMA results and 4 fetuses with ROHs detected by CMA), fetus-only WES or fetus-parent WES (trio-WES) was conducted for further investigation in 62 fetuses (including 59 fetuses from singleton pregnancies and 3 fetuses from 3 twin pregnancies): 41 with isolated CHDs and 21 with nonisolated CHDs. Compared with CMA, WES produced an incremental diagnostic yield of 12.9% (8/62) for P/LP SVs. These P/LP SVs were detected in 8 fetuses from singleton pregnancies. These SVs, consisting of 5 frameshift variants, 1 splicing variant and 2 missense variants, were detected in 8 genes known to be associated with CHD, including KMT2D, WAC, CHD7, RAF1, EP300, GDF1, PQBP1 and LZTR1 (Table 4).
This study identified 8 P/LP SVs, 4 of which are novel variants and the others are known variants reported previously. Six of the eight variants are de novo. However, for two parental inherited P/LP SVs in two affected fetuses in this study, one is an X linked recessive PQBP1 gene, and it is understandable that the maternal inherited PQBP1 variant resulted in CHDs in a male fetus; however, another variant with an autosomal dominant inheritance in the LZTR1 gene (c.851G > A, p.Arg284His) was identified in both the affected fetus and healthy father, which led to a prenatal counseling dilemmas, although the variant was classified as LP variants according to American College of Medical Genetics and Genomics (ACMG) and Clinical Genome Resource consensus recommendation (ClinGen) guidelines (Additional files 2 and 3). This kind of phenomenon is usually attributed to incomplete penetrance, which is not uncommon in CHD [10, 13]. The prenatal counseling dilemmas is due to the fact that the cause of incomplete penetrance for this kind of variant is not currently known, and understanding the genotype–phenotype association is difficult.
Hotspot P/LP CNVs contributing to CHDs
A total of 109 P/LP CNVs were identified in 95 (8.5%) fetuses with CHDs, of which 97 (89.0%) were de novo CNVs and 6 (5.5%) parental inherited CNVs; 6 (5.5%) had unknown parental information. The two most common P/LP CNVs were 22q11.21 deletion/duplication (2.5%, 28/1118) and 16p11.2 deletion/duplication (1.0%, 11/1118); the former showed a frequency of 92.9% (26/28) for de novo events, and the latter showed a frequency of 81.8% (9/11) (Table 2). More specifically, a de novo frequency of 91.3% (21/23) was observed for the 22q11.21 proximal deletion A-D (including TBX1); the 16p11.2 proximal deletion BP4-BP5 (including TBX6) showed 80% (8/10). Table 5 presents ultrasound findings, clinical characteristics and CNV descriptions for the 11 CHD fetuses carrying 16p11.2 deletion (n = 10) or duplication (n = 1).
We considered the 16p11.2 proximal deletion BP4-BP5 (including TBX6) as a potential hotspot CNV for CHD. First, most of the 16p11.2 deletions in our study are de novo occurrences consistent with the perspectives of previous studies that genetic variants originating from de novo events confer a critical contribution to CHD [9, 10, 13]. The deletion has been reported sporadically in other previous cohorts, case reports or the DECIPHER database (ID: 251,630, 278,277, 288,280, 357,605, 359,216) [15, 17, 19,20,21, 23,24,25,26,27,28,29], but it was repeatedly observed in our large cohort. Second, comparison of the frequency of the deletion in our cohort with that in a previous control cohort (6/22246) indicated apparent enrichment of the deletion (p < 0.001) in our fetal CHD cohort [30]. Third, WES analysis performed for the ten fetuses carrying the deletion excluded causative P/LP SVs or other cardiovascular phenotype-associated rare variants of functionally intolerant genes considered to be modifying factors for interpretation of incomplete penetrance and variable expressivity of CNV [31].
Discussion
Although there have been some prenatal cohort studies on associations between genetic variants and CHDs [16, 17, 19,20,21, 32], to the best of our knowledge, this study is the largest cohort to investigate the contribution of genetic variants to CHDs in fetuses in southern China. The contributions of the diverse genetic variants mentioned above in this study include some few differences from previous studies [16, 19,20,21], but our cohort recruited fetuses with CHDs of both singleton pregnancies and twin pregnancies and this might be expected to be highly representative of the current practice of prenatal diagnosis. This work mainly shows that NCA and P/LP CNV are important genetic variants contributing to CHDs in fetuses, most of which are de novo variants. We also present a variety of recurrent CNVs related to CHDs in fetuses. These CNVs may be worthy of further study to investigate their pathogenic mechanism in CHD.
This study confirmed that the 16p11.2 deletion is a hotspot CHD-associated CNV (explained in Results section). The deletion is one of the most common CNVs for NDDs (such as autism spectrum disorder) widely identified in postnatal cases and less commonly recognized in cases with congenital anomalies involving the spine, kidney and urinary tract and brain, rarely garnering intensive attention in CHDs [24, 33,34,35,36,37,38]. The high frequency of the deletion has not been revealed in previous prenatal cohorts but only reported in a postnatal study [24]. This discordance may be attributed to the inclusion criteria of case types (pre- or postnatal cases), sample sizes, CHD types, CHD concomitant extracardiac anomaly types or region and population differences. Of note, NDDs frequently occur in patients with CHDs, in 10% of patients with CHDs and in 50% of patients with severe CHDs [7]. For example, for the 16p11.2 deletion, the penetrance of CHD phenotypes in individuals with the deletion is estimated to be no more than 10% in postnatal cohorts [24, 39]. Our previous study also showed that CHDs are not uncommon in fetuses (4/12) carrying 16p11.2 deletions [38]. However, the penetrance of 16p11.2 deletion-associated NDDs is much higher than that of CHDs [24]. Identification of the 16p11.2 deletion in fetuses regardless of mild or severe CHDs would be a potential reminder of NDDs and have significant value for prenatal diagnosis and genetic counseling. In general, medical management and prognosis between CHDs with P/LP CNV and CHDs without P/LP CNV are apparently different, as demonstrated by a study on the 22q11.2 deletion [40].
In addition, several studies with either relatively small or large sample sizes have reported an incremental yield of P/LP SVs ranging from 4.5 to 12.7% in CHD fetuses without NCAs and CNVs [17,18,19,20,21, 41]. Although a limited number of patients underwent WES in our study, WES also showed an incremental yield of 12.9% for P/LP SVs. Importantly, de novo P/LP SVs with autosomal dominant inheritance are the main type of causative variants in this study. This finding is consistent with previous studies showing that ~ 80% of P/LP SVs contributing to CHDs originate from de novo occurrence [8, 10, 18].
Furthermore, previous CHD cohort studies included only singleton fetuses but not twin fetuses [16, 17, 19,20,21, 32]. For twin pregnancies, although cardiac hemodynamic changes from vascular anastomoses in the placenta and some influence from the special intrauterine environment of twins might have a secondary impact on fetal cardiovascular morphogenesis, the contributions cannot be quantitatively estimated [42]. Nonetheless, previous studies have indicated that a portion of CHDs in twin fetuses result from NCA and P/LP CNV [43, 44]. Our study also reveals that approximately 21.7% of CHD in twin fetuses result from NCA and CNV, with subtle differences between singleton and twin fetuses regarding the contributions of NCA and CNV, as mentioned in Table 3.
However, the primary limitation of our study is the small sample size of fetuses who underwent WES, limiting deep insight into the associations between SVs and CHDs. It was also not possible to estimate the cost-effectiveness and clinical performance of whether CMA and WES should be performed in parallel or sequentially, as prenatal genetic testing strategies are worthy of attention in the current genomic era.
Currently, definitive causal genetic variants contributing to CHDs have been identified in no more than 40% of patients with CHD [14]. In addition, the mean rate of CHD recurrence is approximately 3.1% in the offspring of patients with CHDs [45], indicating that de novo variants only explain a proportion of CHDs. In general, increased understanding of genotype-CHD phenotypes has led to new insight into the molecular pathogenesis of CHDs, and other genetic variants, such as oligogenic or polygenic risk factors, noncoding variants and structural variants, have been recognized as potential mechanisms for CHD cases lacking definitive causes. We believe that accumulating comprehensive whole-genome sequencing genotype and phenotype datasets is essential to exploring the residual etiology of CHDs, which will improve our understanding of the contributions of currently undefined genetic variants to the pathogenesis.
Conclusions
In conclusion, our cohort study provides a deep profile of the contribution of genetic variants to CHDs in both singleton and twin fetuses; NCA and P/LP CNV contribute to 9.1% and 8.5% of CHD in fetuses, respectively. Furthermore, we confirmed the 16p11.2 deletion as a CHD-associated hotspot CNV, second only to the 22q11.21 deletion in frequency. Most 16p11.2 deletions detected were de novo. Additionally, P/LP SV was identified in 12.9% (8/62) of fetuses without NCA or P/LP CNV.
Methods
Subjects
This study reviewed singleton and twin pregnancies with fetal CHD registered in prenatal diagnosis databases of the First Affiliated Hospital of Sun Yat-sen University, the First Affiliated Hospital of Jinan University and Guangdong Women and Children Hospital from March 2016 to June 2022. The inclusion criteria were as follows: (1) Prenatal ultrasound examination, echocardiography and follow-up information were available; (2) Fetuses were diagnosed with CHD by prenatal ultrasound examination and echocardiography; (3) Fetuses with confirmed CHD underwent CMA prenatally; (4) for some fetuses with negative CMA results or ROH, prenatal WES was offered based on parents’ informed consent and willingness. The exclusion criteria included the following: (1) Prenatal follow-up ultrasound examination or echocardiography excluding a diagnosis of CHDs; (2) Isolated cardiovascular ultrasound soft markers or common structural variations including isolated right aortic arch, persistent left superior vena cava, aberrant right subclavian artery, left venous catheter and atrial septal aneurysm; (3) Monochorionic twins with specific complications including twin-to-twin transfusion syndrome, selective intrauterine growth restriction, twin reversed arterial perfusion sequence, twin anemia-polycythemia sequence and conjoined twins.
CHDs were classified as isolated or nonisolated CHDs. The latter included CHDs with one or multiple ECMs, fetal growth restriction or amniotic fluid volume abnormalities.
In all cases, consultation with both a genetic counselor and a prenatal diagnostician was carried out before invasive prenatal puncture sampling, genetic testing and other medical management. Fetal specimens included amniotic fluid or umbilical cord blood. This study was approved by the Medicine Ethical Committee of three units, and the parents of all fetuses provided written informed consent for prenatal puncture sampling and genetic testing.
Chromosomal microarray analysis
Genomic DNA was extracted from amniotic fluid, umbilical cord blood or tissues of induction labor with a QIAamp DNA Blood Mini kit (Qiagen, Germany). CNVs and regions of homozygosity (ROHs) were detected using a CytoScan HD or 750K chip on the single-nucleotide polymorphism array platform according to the manufacturer's standard operating procedures (Thermo Fisher Scientific, USA). Chromosome Analysis Suite software (Thermo Fisher Scientific, USA) was applied to analyze data based on genome version GRCh37/hg19. A threshold of resolution at least 100 kb called by ≥ 50 contiguous probes for CNV and at least 5 Mb for ROH was established. CNV and ROH were classified according to the ACMG and ClinGen guidelines. Pathogenic (P) and likely pathogenic (LP) CNVs are abbreviated to P/LP CNVs. In this study, the scope of CNVs includes deletion/duplication of fragments ≥ 100 kb in size and chromosomal segment deletion/duplication of fragments ≥ 10 Mb in size; numerical chromosomal abnormality (NCA) refers to aneuploidy, polyploidy and mosaic NCA.
Whole-exome sequencing
Using genomic DNA extracted from amniotic fluid, umbilical cord blood or tissues of induction labor with a QIAamp DNA Blood Mini kit (Qiagen, USA), exome sequences were captured with an Agilent SureSelect Human All Exon capture kit v6 (Agilent, USA). Genomic DNA was fragmented randomly, purified, and enriched to construct DNA libraries and then sequenced using the NextSeq500 platform according to the manufacturer's protocols (Illumina, USA). The sequencing reads were aligned to the reference genome sequence (GRCh37/hg19) using BWA software. After alignment, SAMtools software was used to create, sort, and index bam files. Duplicate reads and multiple mapped reads in the exome were removed using Picard software. Calling and annotation of single-nucleotide variants and small insertions/deletions were performed using GATK and ANNOVAR software, respectively. Data quality control criteria reached to an average sequencing depth of ≥ 150× and a minimum coverage of 20× for ≥ 98% of targeted regions. Variant filtering and selection mainly utilized the following criteria: variants with sequencing depth ≥ 20 and alternate allele proportion ≥ 0.3; variants in known disease-causing genes; absent or rare variants (minor allele frequency < 0.01); deleterious variants as predicted by computational prediction tools; variants fulfilling disease inheritance models or family cosegregation; and variants with previously reported cases and/or supported by experimental evidence through searching public literature and databases. The minor allele frequencies of all detected variants were determined according to their frequencies in public population databases, including the gnomAD, dbSNP, 1000 Genomes Project and ESP6500 databases. Computational prediction tools, including SIFT, PolyPhen-2, Mutation Taster, PROVEAN, CADD, Revel, SpliceAI and MaxEntScan, were used to predict whether a variant had a deleterious effect on the gene. Databases such as OMIM, ClinVar, HGMD, LOVD and PubMed were used to assist in the interpretation of variant pathogenicity. Variant pathogenicity classification followed ACMG and ClinGen guidelines. P/LP SVs associated with CHDs are considered causative SVs.
Prenatal WES was performed for some fetuses with negative CMA results (including 59 fetuses from singleton pregnancies and 3 fetuses from 3 twin pregnancies) to detect causative P/LP SVs.
In addition, because 16p11.2 proximal deletion BP4-BP5 (including TBX6) was less frequently identified in cases with CHDs and their correlation was not reliably confirmed in previous studies, prenatal WES was also performed for fetuses carrying the 16p11.2 deletion detected in this study to exclude causative P/LP SVs and cardiovascular phenotype-associated deleteriously rare variants in functionally intolerant genes (“other hits”) [31].
Statistical analysis
SPSS statistics software (IBM SPSS statistics version 22.0) was used for statistical analysis. The statistical analysis methods performed included the chi-square test or Fisher’s exact test with p < 0.05 considered statistically significant.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CHD:
-
Congenital heart defect
- CMA:
-
Chromosomal microarray analysis
- CNV:
-
Copy number variant
- ECM:
-
Extracardiac malformation
- LP:
-
Likely pathogenic
- NDD:
-
Neurodevelopmental disorder
- P:
-
Pathogenic
- ROH:
-
Region of homozygosity
- SV:
-
Sequence variant
- WES:
-
Whole-exome sequencing
References
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American heart association. Circulation. 2022;145(8):e153–639.
Mamasoula C, Addor MC, Carbonell CC, Dias CM, Echevarria-Gonzalez-de-Garibay LJ, Gatt M, et al. Prevalence of congenital heart defects in Europe, 2008–2015: a registry-based study. Birth Defects Res. 2022;114(20):1404–16.
Liu Y, Chen S, Zuhlke L, Black GC, Choy MK, Li N, et al. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48(2):455–63.
Triedman JK, Newburger JW. Trends in congenital heart disease: the next decade. Circulation. 2016;133(25):2716–33.
Findley TO, Northrup H. The current state of prenatal detection of genetic conditions in congenital heart defects. Transl Pediatr. 2021;10(8):2157–70.
Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann Pediatr Cardiol. 2014;7(2):86–91.
Marino BS, Lipkin PH, Newburger JW, Peacock G, Gerdes M, Gaynor JW, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126(9):1143–72.
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498(7453):220–3.
Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350(6265):1262–6.
Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49(11):1593–601.
Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. 2012;91(3):489–501.
Liu Y, Chang X, Glessner J, Qu H, Tian L, Li D, et al. Association of rare recurrent copy number variants with congenital heart defects based on next-generation sequencing data from family trios. Front Genet. 2019;10:819.
Zaidi S, Brueckner M. Genetics and genomics of congenital heart disease. Circ Res. 2017;120(6):923–40.
Yasuhara J, Garg V. Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl Pediatr. 2021;10(9):2366–86.
Zhu X, Li J, Ru T, Wang Y, Xu Y, Yang Y, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. 2016;36(4):321–7.
Wang Y, Cao L, Liang D, Meng L, Wu Y, Qiao F, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. 2018;218(2):244e1–17.
van Nisselrooij AEL, Lugthart MA, Clur SA, Linskens IH, Pajkrt E, Rammeloo LA, et al. The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet Med. 2020;22(7):1206–14.
Li R, Fu F, Yu Q, Wang D, Jing X, Zhang Y, et al. Prenatal exome sequencing in fetuses with congenital heart defects. Clin Genet. 2020;98(3):215–30.
Qiao F, Wang Y, Zhang C, Zhou R, Wu Y, Wang C, et al. Comprehensive evaluation of genetic variants using chromosomal microarray analysis and exome sequencing in fetuses with congenital heart defect. Ultrasound Obstet Gynecol. 2021;58(3):377–87.
Lu F, Xue P, Zhang B, Wang J, Yu B, Liu J. Estimating the frequency of causal genetic variants in foetuses with congenital heart defects: a Chinese cohort study. Orphanet J Rare Dis. 2022;17(1):2.
Xing Y, Zhang Y, Chen J, Wu F, Yuan M, Zou G, et al. Prenatal diagnosis for fetuses with isolated and non-isolated congenital heart defects using chromosomal microarray and exome sequencing. Prenat Diagn. 2022;42(7):873–80.
Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A, National Birth Defects Prevention S. Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol. 2007; 79(10):714–27.
Vysotskiy M, Zhong X, Miller-Fleming TW, Zhou D, Autism Working Group of the Psychiatric Genomics C, Bipolar Disorder Working Group of the Psychiatric Genomics C, et al. Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes. Genome Med. 2021; 13(1):172.
Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, et al. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012;49(10):660–8.
Gatti M, Tolva G, Bergamaschi S, Giavoli C, Esposito S, Marchisio P, et al. Mayer-Rokitansky-Kuster-Hauser Syndrome and 16p11.2 Recurrent microdeletion: a case report and review of the literature. J Pediatr Adolesc Gynecol. 2018;31(5):533–5.
Shen Y, Chen X, Wang L, Guo J, Shen J, An Y, et al. Intra-family phenotypic heterogeneity of 16p11.2 deletion carriers in a three-generation Chinese family. Am J Med Genet B Neuropsychiatr Genet. 2011;156(2):225–32.
Dell’Edera D, Dilucca C, Allegretti A, Simone F, Lupo MG, Liccese C, et al. 16p11.2 microdeletion syndrome: a case report. J Med Case Rep. 2018;12(1):90.
Puvabanditsin S, Nagar MS, Joshi M, Lambert G, Garrow E, Brandsma E. Microdeletion of 16p11.2 associated with endocardial fibroelastosis. Am J Med Genet A. 2010;152A(9):2383–886.
Yue F, Xi Q, Zhang X, Jiang Y, Zhang H, Liu R. Molecular cytogenetic characterization of 16p11.2 microdeletions with diverse prenatal phenotypes: four cases report and literature review. Taiwan J Obstet Gynecol. 2022;61(3):544–50.
Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15(6):478–81.
Pizzo L, Jensen M, Polyak A, Rosenfeld JA, Mannik K, Krishnan A, et al. Rare variants in the genetic background modulate cognitive and developmental phenotypes in individuals carrying disease-associated variants. Genet Med. 2019;21(4):816–25.
Yi T, Hao X, Sun H, Zhang Y, Han J, Gu X, et al. Genetic aetiology distribution of 398 foetuses with congenital heart disease in the prenatal setting. ESC Heart Fail. 2022.
Verbitsky M, Westland R, Perez A, Kiryluk K, Liu Q, Krithivasan P, et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet. 2019;51(1):117–27.
Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–75.
Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry. 2015;77(9):785–93.
Chung WK, Roberts TP, Sherr EH, Snyder LG, Spiro JE. 16p11.2 deletion syndrome. Curr Opin Genet Dev. 2021;68:49–56.
Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015;372(4):341–50.
Lin S, Shi S, Zhou Y, Ji Y, Huang P, Wu J, et al. Intrauterine phenotypic features associated with 16p11.2 recurrent microdeletions. Prenat Diagn. 2018;38(6):381–9.
Ehrlich L, Prakash SK. Copy-number variation in congenital heart disease. Curr Opin Genet Dev. 2022;77:101986.
O’Byrne ML, Yang W, Mercer-Rosa L, Parnell AS, Oster ME, Levenbrown Y, et al. 22q11.2 Deletion syndrome is associated with increased perioperative events and more complicated postoperative course in infants undergoing infant operative correction of truncus arteriosus communis or interrupted aortic arch. J Thorac Cardiovasc Surg. 2014;148(4):1597–605.
Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393(10173):747–57.
Glinianaia SV, Rankin J, Wright C. Congenital anomalies in twins: a register-based study. Hum Reprod. 2008;23(6):1306–11.
Li L, He Z, Huang X, Lin S, Wu J, Huang L, et al. Chromosomal abnormalities detected by karyotyping and microarray analysis in twins with structural anomalies. Ultrasound Obstet Gynecol. 2020;55(4):502–9.
Zhang Y, Huang L, Huang X, He Z, Lin S, Wang Y, et al. Chromosomal aberrations and CNVs in twin fetuses with cardiovascular anomalies: comparison between monochorionic diamniotic and dichorionic diamniotic twins. Prenat Diagn. 2018;38(5):318–27.
Yokouchi-Konishi T, Yoshimatsu J, Sawada M, Shionoiri T, Nakanishi A, Horiuchi C, et al. Recurrent congenital heart diseases among neonates born to mothers with congenital heart diseases. Pediatr Cardiol. 2019;40(4):865–70.
Acknowledgements
We thank all the patients and their family members for their participation in this study. We also thank the National Natural Science Foundation of China and Guangdong Basic and Applied Basic Research Foundation for supporting this work.
Funding
This work was supported by the National Natural Science Foundation of China (82001564) and Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515110752).
Author information
Authors and Affiliations
Contributions
Study concepts: YL, YZ; Study design: SL, SS, JL, YL, YZ; Chromosomal microarray analysis: SL, SS, JL; Whole-exome sequencing: SL, SS, JL; Data collection and collation: ZH, DL, LH, XH; Statistical analysis and manuscript preparation: SL, ZH, DL, LH, XH. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Medicine Ethical Committee of The First Affiliated Hospital of Sun Yat-sen University, The First Affiliated Hospital of Jinan University and Guangdong Women and Children Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
The frequencies of NCA, P/LP CNV and ROH among different types of CHD.
Additional file 2.
Contributions of NCA and P/LP CNV among various CHD types and interpretation for classification of LZTR1 variant.

Additional file 3.
Homology analysis of LZTR1 proteins among various species (A) and protein structural models of LZTR1 with the Arg284His variant (B).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Lin, S., Shi, S., Lu, J. et al. Contribution of genetic variants to congenital heart defects in both singleton and twin fetuses: a Chinese cohort study. Mol Cytogenet 17, 2 (2024). https://doi.org/10.1186/s13039-023-00664-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-023-00664-y