
Abstract
Background
Deletions and partial losses of chromosome 7 (chr7) are frequent in acute myeloid leukemia (AML) and are linked to dismal outcome. However, the genomic landscape and prognostic impact of concomitant genetic aberrations remain incompletely understood.
Methods
To discover genetic lesions in adult AML patients with aberrations of chromosome 7 [abn(7)], 60 paired diagnostic/remission samples were investigated by whole-exome sequencing in the exploration cohort. Subsequently, a gene panel including 66 genes and a SNP backbone for copy-number variation detection was designed and applied to the remaining samples of the validation cohort. In total, 519 patients were investigated, of which 415 received intensive induction treatment, typically containing a combination of cytarabine and anthracyclines.
Results
In the exploration cohort, the most frequently mutated gene was TP53 (33%), followed by epigenetic regulators (DNMT3A, KMT2C, IDH2) and signaling genes (NRAS, PTPN11). Thirty percent of 519 patients harbored ≥ 1 mutation in genes located in commonly deleted regions of chr7—most frequently affecting KMT2C (16%) and EZH2 (10%). KMT2C mutations were often subclonal and enriched in patients with del(7q), de novo or core-binding factor AML (45%). Cancer cell fraction analysis and reconstruction of mutation acquisition identified TP53 mutations as mainly disease-initiating events, while del(7q) or −7 appeared as subclonal events in one-third of cases. Multivariable analysis identified five genetic lesions with significant prognostic impact in intensively treated AML patients with abn(7). Mutations in TP53 and PTPN11 (11%) showed the strongest association with worse overall survival (OS, TP53: hazard ratio [HR], 2.53 [95% CI 1.66–3.86]; P < 0.001; PTPN11: HR, 2.24 [95% CI 1.56–3.22]; P < 0.001) and relapse-free survival (RFS, TP53: HR, 2.3 [95% CI 1.25–4.26]; P = 0.008; PTPN11: HR, 2.32 [95% CI 1.33–4.04]; P = 0.003). By contrast, IDH2-mutated patients (9%) displayed prolonged OS (HR, 0.51 [95% CI 0.30–0.88]; P = 0.0015) and durable responses (RFS: HR, 0.5 [95% CI 0.26–0.96]; P = 0.036).
Conclusion
This work unraveled formerly underestimated genetic lesions and provides a comprehensive overview of the spectrum of recurrent gene mutations and their clinical relevance in AML with abn(7). KMT2C mutations are among the most frequent gene mutations in this heterogeneous AML subgroup and warrant further functional investigation.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is characterized by aberrant proliferation of hematopoietic progenitor cells in the bone marrow, leading to suppression of normal hematopoiesis [1]. Chromosomal aberrations and gene mutations play a pivotal role during leukemogenesis and are essential for risk stratification and personalized treatment approaches [2, 3]. Aberrations of chromosome 7 [abn(7)], for which the DNA sequence and annotation were only described two decades ago[4], are common and found in ~ 10% of newly diagnosed AML [5, 6]. The most common abn(7) include complete loss of chromosome 7 [monosomy 7, (−7)] and deletions of the long arm of chromosome 7 [del(7q)] [7]. Both frequently appear in the context of a complex karyotype (CK) [6, 8], which is an established poor prognostic factor [2, 3]. Outside a CK, the current 2022 ELN risk classification assigns only −7 to the adverse risk category, while del(7q), in the absence of additional good or adverse genetic markers, belongs to the intermediate risk group [3]. With respect to co-occurring gene mutations, only a few studies have systematically investigated the landscape and clinical impact of concomitant aberrations in AML with abn(7) [9,10,11,12]. However, TP53 mutations frequently occur in AML with abn(7), especially in cases with CK, and are associated with dismal prognosis [13, 14]. TP53-status has been recently included as a new disease category in the International Consensus Classification of Myeloid Neoplasms and Acute Leukemias (ICC) [15].
Four commonly deleted regions (CDR) in chromosome 7 (chr7) have been reported: 7q21.2, 7q22.1, 7q34, and 7q35-36.1 [16]. These regions encode for several genes with a well-established functional role in hematopoiesis and are recurrent targets of somatic mutations in various hematologic malignancies such as EZH2, BRAF, or SAMD9 [17,18,19,20,21,22]. For example, mutations affecting EZH2 have been reported in up to 30% of follicular lymphoma and 5–10% of myelodysplastic syndromes, chronic myelomonocytic leukemia, and AML [2, 18, 23,24,25]. Also, deregulated mRNA expression of genes within these CDRs has been aligned with patient outcome, e.g., altered expression of the methyltransferase MLL5/KMT2E, located on chr7q22.3, associates with poor prognosis [26].
To date, studies focusing on AML with abn(7) were showing limitations regarding cohort size, sequencing technologies applied, and cohort heterogeneity, often investigating a wide variety of myeloid neoplasms with abn(7) precluding generalization for AML [9, 10, 12].
Therefore, we embarked on a comprehensive study to decipher the genomic landscape of AML with abn(7) in a large international cohort of 519 adult AML patients using a combination of whole-exome (WES) and targeted sequencing (TS). This workflow enabled the detection of previously underestimated somatic mutations and copy number variations (CNV) and allowed for the identification of genetic markers important for further risk refinement in this specific AML subgroup.
Methods
Patients
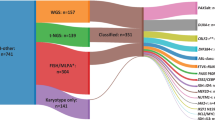
A total of 519 samples (bone marrow or peripheral blood) from first diagnosis of adult AML patients with abn(7) were collected from collaborating study groups in France, Germany, the Netherlands, Spain, Taiwan, and the United Kingdom. Patients had a median age of 59 years (range 15–89 years). For 60 patients, complete remission (CR) samples were used as non-tumor controls. Baseline karyotype information was obtained through conventional karyotyping at diagnosis. 225 patients (43%) had a chr7 aberration in the context of a CK (Fig. 1). Among the 294 patients without a CK (non-CK), 125 patients showed a −7 (−7sole) and 69 patients a deletion of 7q as sole aberrations [del(7q)sole]. Eighty percent of patients (n = 415) received intensive induction treatment, typically containing a combination of cytarabine and anthracyclines (“7 + 3”). Fifty-three percent of patients were treated within prospective trials of participating study groups. Furthermore, 33% of patients underwent allogeneic hematopoietic stem cell transplantation (allo-HSCT) in first CR (CR1, n = 124) or as salvage (n = 44) therapy. The characteristics and therapy specifications of patients are shown in Table 1 and supplemental Dataset 1. Written consent was obtained from all patients according to the Declaration of Helsinki, and the local ethics committee granted ethical approval.

Classification of the abn(7) cohort according to cytogenetic characteristics. Depiction of the classification of abn(7) patients into different groups according to the karyotype information available (n = 519). The first level separates a group of abn(7) samples with complex karyotype (CK) from the non-CK cases. A second division separates non-CK cases and CK cases according to the presence of monosomy 7 (−7/non-CK or −7/CK) or the presence of a del(7q) [del(7q)/non-CK or del(7q)/CK]. A group of samples not fitting these definitions was not included (other/non-CK, n = 10 or other/CK, n = 19). A third division separates samples by the presence or absence of another cytogenetic alteration in addition to the abnormality detected in abn(7)/non-CK. Samples with an accompanying alteration (non-complex karyotype non-sole, non-CKns) and samples with only a chr7 aberration (sole)
Whole-exome sequencing in the abn(7) exploration cohort
Analysis of single nucleotide variations, mutation signatures, and copy number variations from WES
Paired first diagnosis/CR samples were available from 60 patients for explorative mutation analysis through WES. Samples were prepared using the SureSelect XT-HS exome and SureSelect Human All Exon v7 library preparation kits (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed at the Genomic Core facility of the Berlin Institute of Health (BIH) on the NovaSeq 6000 platform (Illumina, San Diego, CA) with a median coverage of 211-fold. We used a detection cut-off for variant allele frequency (VAF) of ≥ 5%. Called variants were filtered thoroughly as described previously [27,28,29]. Analysis of mutational signatures and variant filtering are specified in the supplemental methods.
Targeted sequencing in the abn(7) extension cohort
Panel design and analysis of single nucleotide variations using TS
Based on our WES-results, literature research, and data from additional AML cases with abn(7) from the Cancer Genome Atlas database (TCGA) [2], we designed a unique gene panel covering 66 genes (Table S1) and genomic regions frequently affected by CNVs. A single nucleotide polymorphism (SNP)-backbone of 1217 probes was included to identify CNVs (supplemental dataset 2).
TS was performed on 467 diagnostic samples (= extension cohort) using a customized Twist Bioscience Gene Panel (Twist Bioscience, San Francisco, CA, USA). Samples were prepared with the Twist Bioscience Custom Prep Kits with unique molecular identifiers for error correction following the manufacturer's protocol. Sequencing was performed using a NovaSeq 6000-Sequencer (Illumina, San Diego, CA, USA) at the BIH Core Facility, with a mean coverage of 532-fold. Variants were called using an in-house pipeline [27] with a VAF cut-off at ≥ 2% after rigorous filtering criteria for quality and clinical scores (supplemental Methods, Figure S1).
Analysis of copy number variations from TS
Data obtained from the SNP-backbone was used to estimate copy number (CN) and B-allele-frequencies (BAF) for CNV and copy-neutral loss of heterozygosity (cnLOH) determination using the PureCN R-package [30]. For correction of assay-specific capture biases, we used 19 pooled healthy control samples. We restricted the analysis to samples with a ploidy between 1.5–3N. An alteration was considered a gain for log(CN) > 0.4 and a deletion with log(CN) < − 0.3. Upon curation, 342 samples passed our quality criteria, ensuring high reproducibility of the presented data. Manual curation was done to merge continuous segments and to rule out disagreements with BAF. We pursued internal validations (supplemental Methods) by calculation of CNV agreements with G-Banding information and with matching samples sequenced by long-read Oxford Nanopore Technology (n = 33, ONTseq, as previously described) [31] along with comparisons with WES-CNV-results.
Cancer cell fraction analysis and Bradley–Terry model
To unravel clonal hierarchies, we calculated cancer cell fractions (CCF) for mutations and CNVs from WES data as previously described [32]. To investigate the timing and order of mutation acquisition, we used similar calculations of CCFs [33] based on the PureCN algorithm data for the extension cohort. We applied a Bradley–Terry Model with pairwise comparison of the highest CCFs of a gene-sample pair for samples with available single nucleotide variant (SNV)/CNV/cnLOH data as previously reported[27] (supplemental Methods).
Statistical and survival analysis
All statistical analyses were performed using R statistical software packages (supplemental Methods). For survival analyses, only intensively treated patients were considered (n = 415) to ensure comparability and avoid potential treatment bias. The definitions of CR, overall survival (OS), and relapse-free survival (RFS) followed the recommended criteria [3]. Kaplan–Meier analysis was used to create survival curves. Log-rank tests were applied to evaluate differences between groups and were considered statistically significant if P < 0.05. Univariate and multivariable Cox proportional hazards models were used to investigate the association of variables for OS and RFS. The Cox proportionality assumption was guaranteed for all reported results.
Results
The genomic landscape of AML with abn(7)
Explorative WES of 60 paired diagnostic/CR samples revealed a total of 932 SNVs in 742 individual genes (median 12 SNVs/patient, supplemental dataset 3). Among these, we found 207 mutations in 59 genes known to be recurrently mutated in AML with a median of 2 mutated genes per patient. The most frequently mutated gene was TP53 (30%), followed by NF1 (20%), RUNX1 (20%), and DNMT3A (18.3%; Fig. 2A). Genes involved in epigenetic regulation were mutated in 58.3% of our exploration cohort and included DNMT3A (18.3%), ASXL1 (11.7%), TET2 (11.7%), IDH2 (10%), KMT2C (10%), EZH2 (8.3%), and IDH1 (8.3%, Fig. 2A). Of note, belonging to the group of genes located in the CDRs of chr7, KMT2C, and EZH2 showed a high mutation frequency.

Mutations and SBS signatures found by WES and Targeted Sequencing in the abn(7) exploration and extension cohorts. A Bar graph showing the frequency of mutations identified by WES in patients (n = 60) per gene for genes mutated in ≥ 2 patients. Bars colored dark blue signify genes of particular importance due to high mutation frequency or previously underestimated prevalence in AML. The fraction of mutated patients per gene is shown above each bar (%). B The graph shows the mutational profile extracted from WES, which represents the two main signatures of most samples from the exploration cohort. Sig-A had high cosine similarities to the signatures reconstituted from the components that expectation maximization extracted SBS1/SBS5 in COSMIC (0.939) and SBS1/SBSblood in normal blood cells (0.945)[34, 35]. The bars represent the relative contributions of clustered genome-wide substitutions (SBS, y-axis) for each 96 trinucleotide sequences (x-axis) and distributed across the six possible cytosine or thymine bases substitutions. C Oncoplot showing mutations found in 43 of the 64 genes included in the TS gene panel in 452 patients of the extension cohort (n = 467 patients). According to baseline karyotype information, patients were segregated into two major groups: abn(7)/non-complex karyotype (non-CK, grey) or abn(7)/complex karyotype (CK, red). The marks on the top rows correspond to patients classified by cytogenetic information into having −7 (dark blue) or del(7q) (yellow). FLT3 includes all FLT3 alterations. *In the TS panel, only exons 28–38 of NF1 were investigated. The left bar plot shows the frequency (%) of mutated patients per gene. On the right side, a boxplot shows the statistical difference between the number of mutations found for these groups, median 4 versus 2 respectively for abn(7)/non-CK versus abn(7)/CK (two-tailed t test, n = 467, ****P < 0.0001)
To dissect biological processes that result in the accumulation of somatic alterations during leukemogenesis, we investigated mutational signatures in our exploration cohort. One signature was extracted (Sig-A), which had high cosine similarities to SBS1/SBS5 in COSMIC (0.939) and SBS1/SBSblood in normal blood cells (0.945, Fig. 2B). The SBS1 and SBS5 (SBSblood) are age-related endogenous mutational signatures and have been reported as the predominant signatures in AML and myeloproliferative neoplasms [34] as well as in normal hematopoietic stem and progenitor cells [35].
Using TS in the extension cohort of 467 AML samples, we identified 1821 SNVs in 64 genes (median 3 SNVs/patient; Figure S2A, supplemental dataset 4). At least one gene mutation was detected in 452/467 investigated samples (96.7%). Consistent with our WES results, the most frequently mutated gene was TP53 (33.4%), followed by DNMT3A (18%), RUNX1 (16.7%), KMT2C (16.7%), ASXL1 (16.3%), NRAS (14.3%), TET2 (12.8%), PTPN11 (11.1%), EZH2 (10.3%) and IDH2 (9.4%) (Fig. 2C, Figure S2B). Mutations affecting genes of the splicing machinery (e.g. SRSF2 and U2AF1) were found in 25% of patients, while CHIP (clonal hematopoiesis of indeterminate potential)-associated gene mutations in ASXL1, DNMT3A, and TET2 [36, 37] were found in 40% of patients and associated with older age, as expected (P < 0.001). Of note, the high prevalence of KMT2C mutations found by WES was confirmed by TS (16.7%). In 78 patients a total of 98 KMT2C mutations were found with a median VAF of 5% (Fig. 3A, Figure S3A). 27% of mutations were indel or splice site alterations and spread over the entire coding region, while missense mutations showed a hotspot affecting codon A1685 (n = 15). This particular mutation (COSV51390875) has been mainly reported as somatic in various cancer entities, including non-Hodgkin lymphoma [22, 38, 39]. Although we could not independently confirm the somatic origin due to lack of non-tumor tissue, the observed subclonality of KMT2C A1685S mutations made a germline origin unlikely (Figure S3B). Of note, this particular KMT2C variant was not detected in the exploration cohort. 79% of the KMT2C-mutated patients harbored a concomitant deletion in the KMT2C locus (Fig. 3B). KMT2C mutations were enriched in patients with core-binding factor (CBF) AML [45%, inv(16): n = 3/8, and t(8;21): n = 2/3] but not in patients with a inv(3)/t(3;3) [14.8%, 4/27, Table S2]. KMT2C-mutated patients were significantly associated with de novo AML (P = 0.039), whereas mutations in splicing factor genes, SRSF2 and U2AF1, were significantly enriched in secondary AML (sAML; respectively P = 0.011 and P = 0.02 in the multivariate analysis, Fig. 3D, Figure S4A). Furthermore, we found both KMT2C and FLT3 mutations to be significantly enriched in del(7q) patients (P = 0.04 and P = 0.003 respectively, Fig. 3E), while patients with −7 were enriched for RUNX1 and PTPN11 mutations (P = 0.006 and P = 0.027 respectively; Fig. 3E, Figure S4B). A similar association was also observed for FLT3 and KMT2C mutations when comparing del(7q)/CK to –7/CK patients (P = 0.052 and P = 0.065 respectively, Figure S5).

Frequent low allele-burden mutations in KMT2C in AML with abn(7). A Lollipop plot depicts the localization and frequency of each KMT2C variant (n = 98, in 78/467 patients, from KMT2C refSeq NM_170606.3). Distribution in the context of Pfam domains was adapted from MutationMapper from cbioportal [64, 65] with information on the overlap of mutations to a statistically significant hotspot in cancer [66] or to reports of functional effects in the oncology knowledge base OncoKB™ [67, 68]. Color codes were used to distinguish types of observed mutations: 72 missense (blue), 16 truncating (red), 4 inframe (yellow), and 6 alterations affecting splice sites (light blue). B Frequency distribution of genomic events affecting the KMT2C locus: SNVs and CNVs lead to a multi-hit classification of 47 KMT2C-mutated patients from the extension cohort (n = 342) into groups according to genomic events present in the locus (1mut, > 1mut and mut + del). C Distribution of the VAF values of KMT2C mutations found for patients in the KMT2C multi-hit groups with at least one mutation present (n = 47). D, E The Odds ratio plot shows a multivariable binomial logistic regression fitted for the (D) ten genes with a P < 0.1 in univariate analysis (Figure S4A) and for (E) the six genes with a P < 0.1 in univariate analysis (Figure S4B). To the left, a bar plot diagram depicts the number of mutated patients: for each of the ten genes included in the multivariate model color-coded by (D) type of AML (de novo AML, blue; sAML, red) and (E) for each of the six genes included in the multivariate model color-coded by abn(7) group [−7, green; del(7q), grey]. To the right, logOR with confidence intervals of 95%, CI, and P values are shown
Additionally, we identified unexpected recurrent mutations in SETBP1 (7.7%), FAT1, and TACC2 (6.4% each), and a considerable low frequency of NPM1 (2.4%), the latter usually occurring in roughly 30% of adult AML cases [2]. TACC2 belongs to a conserved family of centrosome- and microtubule-interacting proteins and has been reported as a putative tumor suppressor in breast cancer [40, 41].
A significantly lower number of mutations was found in patients with abn(7)/CK as compared to abn(7) patients without CK (non-CK; median number of mutations/patient 2 vs. 4, P < 0.0001, Fig. 2C). We noted significant enrichment of TP53 and FAT1 mutations in patients with CK, while mutations affecting IDH1, IDH2, CBL, and RUNX1 were predominantly found in non-CK cases (Figure S6). 30% of abn(7) patients (n = 140) harbored at least one mutation in genes located within the CDRs of 7q, most frequently in KMT2C (16.7%), EZH2 (10.3%), and CUX1 (4.7%, Figure S7A). While the highest frequencies of EZH2 mutations were observed in patients with non-CK, KMT2C mutations were particularly enriched in del(7q)/CK patients (Figure S7B).
Within the non-CK group, we identified specific mutational patterns for −7 and del(7q). We found higher frequencies of KRAS and RUNX1 mutations in −7/non-CK patients and more frequent FLT3-mutations in del(7q)/non-CK cases (Figure S8). These mutational patterns were also confirmed in patients with −7 and del(7q) as sole aberrations (Figure S9). Next, we searched for pairwise gene associations to identify patterns of mutation co-occurrence or mutual exclusivity. We confirmed several significant co-occurrences reported in other AML subgroups and related myeloid malignancies, such as DNMT3A/IDH1 and BCOR/BCORL1 [2, 42,43,44]. With respect to mutual-exclusivity, TP53 mutations occurred exclusive of most other known AML mutations (Figure S10). Furthermore, exclusivities for SRSF2/EZH2 and previously reported TET2/IDH1/2 [45] were found. We noted a trend for mutual exclusivity for KMT2C mutations with JAK2 (P = 0.008, FDR = 0.042), ASXL1 (P = 0.009, FDR = 0.046) and DNMT3A mutations (P = 0.029, FDR = 0.12).
These data collectively indicate that distinct mutation profiles can be observed in patients with abn(7) and depend on both type of deletion (complete versus partial) and concurrent cytogenetic aberrations.
Concomitant copy number variations in AML patients with abn(7)
Based on conventional karyotyping techniques (e.g., G-banding, FISH probing), 63% of the cohort had −7 and 32% del(7q). The remaining patients (5%) had various chr7 aberrations [e.g., iso(7p), r(7), add(7)]. Many additional cytogenetic aberrations were reported. The five most common were deletions affecting chr5 [−5/del(5q) = 28%], chr17 [−17/del(17p)/del(17q) = 14.6%], chr18 [−18/del(18p)/del(18q) = 10.2%], chr16 [−16/del(16q) = 8.1%], and gains of chr8 [+ 8/add(8q) = 9.8%].
In the extension cohort, high-quality CNV data was generated for 342 of 467 patients (73%, supplemental datasets 5,6). TS-based CNV analysis revealed a high concordance with WES-CNV data and with the five most frequent aberrations detected by conventional karyotyping, assuming the standard limit of detection for G-banding of > 10 Mb (Table S3, Figure S11-12A,B).
Using TS-based CNV analysis, we identified a substantial number of genomic loci targeted by focal deletions/gains (≤ 10 Mb) that were missed by conventional G-banding due to its lower resolution. These focal events were most frequently observed as deletions in chr17p/q, chr12q, chr21q, and chr4q or gains in chr11p/q (Fig. 4A). In agreement with a recent study in TP53-mutated AML[46], most of these focal deletions affect known AML genes (Figure S12C, Table S4) such as TP53 (chr17p13.1, 8.5%), NF1 (chr17q11.2, 6.1%), or ETV6 (chr12p13.2, 5.3%, Figure S13). Furthermore, we noted several previously underappreciated small CNVs: gains of KMT2A (chr11q, 5%), U2AF1 (chr21q, 2.9%), and BCL11B (chr14q, 2%), and deletions of RUNX1 (chr21q, 3.5%) and TET2 (chr4q, 2.6%). ONTseq confirmed the presence of these focal CNVs in randomly chosen cases (Figure S14, Table S3). In fact, when comparing the presence of TS-based CNVs affecting chr17, we noted that 11 out of 78 patients (14% of patients) had no chr17 abnormality reported by conventional karyotyping and showed focal deletions affecting TP53 and/or NF1.

Positions and proportions of CNVs detected by TS. A Barplot illustrates the distribution of all 1376 manually curated CNVs derived from TS in the extension cohort (n = 342). Chromosomal CNVs spanning one arm or a whole chromosome are depicted on the left of each x-axis, while focal SNVs are shown to the right of the x-axis (according to arrows). The number of patients with a specific aberration is depicted according to the size of the event (yellow: large CNVs > 10 Mb; red: small CNVs ≤ 10 Mb). Chromosome 7 is displayed in blue. B To the right, genomic positions of chr7 covered by CNVs (deletions in blue, gains in red) identified in 188 AML patients (x-axis, excluding 154 cases with monosomy 7). Marked commonly deleted regions (CDRs)1–4 are adapted from Baeten et al. [16]. Potential genes of interest and their genomic positions are shown to the right. To the left, the frequency of deletions across the chromosome and recurrent breakpoint clusters, with the respective number of times a specific breakpoint occurred as a starting (black) or ending point of a CNV segment (red), are shown
Collectively, these data showed the detection advantages of next-generation-sequencing-(NGS)-based karyotyping versus conventional G-banding, as a high proportion of CNVs fell below the limit of detection through G-banding (Table S4). Furthermore, genomic regions enriched for CNVs were also common targets of somatic mutations in AML with abn(7), suggesting genetic convergence through CNVs and SNVs, a phenomenon typical for tumor suppressor genes.
Commonly deleted regions of chromosome 7 and clonal trajectories of genomic alterations in AML with abn(7)
Previously, four CDRs of chr7q have been defined using SNP-arrays, FISH, and mCGH-assays [16]. In our study, small focal deletions affecting chr7q were found in 17 patients (5%), predominantly in CDR2 (3.5%) and CDR4 (1.2%, Fig. 4B, Table S5). The most common focal deletions affected chr7:q21.2-q36.3 (n = 31) or chr7:q22.1-q36.3 (n = 26); the vast majority resulted from two recurrent breakpoints at the beginning of CDR1 or CDR2 and reached the telomere (Fig. 4B). Next, we investigated whether the chr7-CDRs showed differences in concurrent CNVs by co-occurrence and mutual-exclusivity analysis (Figure S15). Patients with the most frequent focal del(7q)(chr7:q21.2−q36.3) were mutually exclusive to deletions affecting chr7p (P = 0.05, FDR = 0.07), chr17p13.3−p13.1 (region spanning TP53-locus, n = 19) and chr17q11.2 (region spanning NF1-locus, n = 16). This focal del(7q) was instead correlated with gains affecting chr3q26.2−q29 encoding the MECOM/EVI1-locus (Figure S15).
To investigate the clonality and timing of genomic aberrations, we applied two complementary approaches. First, CCF analysis was performed to assign CNVs and SNVs to (sub-)clonality using our WES-data (n = 60, supplemental dataset 7). CCF revealed that deletions of chr 3, 5, and 17p were mainly clonal events, suggesting their role as disease-founding aberrations. Interestingly, del(7q) and −7 were subclonal in one-third of cases and thus might appear as both an early and late event in leukemogenesis (Fig. 5A). Regarding the main abn(7) groups, −7/CK, −7/non-CK, del(7q)/CK, and del(7q)/non-CK, −7 appeared more often as clonal (88%) in −7/CK cases, while in the other three groups, the clonality of −7 and del(7q) is more evenly distributed (Figure S16). Second, our TS-based Bradley–Terry model revealed that mutations in splicing genes, in TP53, and in CHIP-associated genes (DNMT3A and TET2) were likely initiating events in the development of AML with abn(7). In contrast, mutations in genes involved in RAS- and Tyrosine-kinase-signaling, such as KRAS, NRAS, and KIT, were consistently ranked among the later events. Notably, except for TP53, mutations in the genomic regions frequently targeted by both CNVs and SNVs (e.g., ETV6, KMT2C) occurred at later stages of leukemogenesis (Fig. 5B).

Clonal hierarchies of events in patients with aberrations in chromosome 7. A Clonality analysis of SNV and CNV events derived from the CCFs calculated using ASCAT [69] and CNACS [47] data in the exploration cohort (n = 60 paired diagnosis/remission samples). x/xLOH represents a chromosome or arm level CNV, and del(xp) or del(xq) represents the deletion of any part of the respective p or q arm. B Plot shows SNV acquisition order resulting from a Bradley–Terry model applied to mutation pairs, using the CCF calculated by correction of sample purity, ploidy, and CNV/cnLOH presence determined in pureCN (depicted on the left) in n = 342 patients. The number of mutations that entered the model is reported for each gene. To the right, the points correspond to point estimations, and the bars represent the 95% confidence intervals. Early mutations have high estimations, thus their points are arranged on the left. The median point estimation value is taken as a reference point for early versus late distinctions (grey arrow). Genes are color-coded by their assumed functional category
Survival analyses and clinico-biological correlations of abn(7) AML
Survival data were available for 518 patients. The median follow-up time for patients alive was 25 months (range, 0.2–160 months), with a median OS for all patients of 10.4 months. Due to therapeutic heterogeneity, we restricted subsequent survival analyses for clinico-biological correlations to intensively treated patients only (n = 415, Table 1). In the intensively treated cohort, the median OS was 11.9 months. While 61% of patients reached CR, the relapse rate was high at 67%.
Kaplan–Meier survival analyses revealed significant differences in RFS between all main abn(7) groups [del(7q)/non-CK, −7/non-CK, del(7q)/CK, and −7/CK], except for −7/non-CK versus del(7q)/CK patients (P = 0.72), which seem to have comparable outcomes (Fig. 6A). For OS, the results were similar (Fig. 6B). Focusing on −7/non-CK versus del(7q)/non-CK patients, RFS was significantly inferior in −7/non-CK patients (P = 0.048, Fig. 6A), with a trend toward an inferior OS in this patient group (P = 0.081, Fig. 6B). When looking at abn(7) as sole abnormalities, we found no differences in survival endpoints between −7sole and del(7q)sole patients (Figure S17).

Survival analysis according to the major abn(7) groups. Kaplan–Meier Curves showing the probability of A RFS for n = 230 and B OS for n = 389 intensively treated patients with available clinical data comparing the four major abn(7) groups: −7/CK versus del(7q)/CK versus −7/non-CK versus del(7q)/non-CK. Patients classified as "other/non-CK" or "other/CK" are not included (n = 25). P values derived from pairwise LogRank Test, *ns = not significant
To evaluate the prognostic importance of clinical and genetic variables on survival and response parameters for patients with abn(7), ten variables with a significance level of P < 0.1 in univariate were included in multivariate analyses (Table 2 and Table S6). We found older age (> median, 59 years) and higher white blood cell counts (WBC > median (9/nL) to be significant clinical predictors of worse OS (HR, 1.4 [95% CI 1.1–1.8], P = 0.007 and HR, 1.69, [95% CI 1.32–2.16], P < 0.001, respectively, Table 2A), and RFS (HR, 1.47, [95% CI 1.04–2.09], P = 0.030 and HR, 2.23, [95% CI 1.58–3.15], P < 0.001, respectively, Table 2B). Furthermore, we identified TP53abn (= mutations and/or deletions of the TP53 locus) and PTPN11mut as the strongest genomic predictors of inferior OS (HR, 2.53, [95% CI 1.66–3.86], and HR, 2.24, [95% CI 1.56–3.22], both P < 0.001, Table 2A, Figure S18A,B) and RFS (HR, 2.3, [95% CI 1.25–4.26], P = 0.008 and HR, 2.32, [95% CI 1.33–4.04], P = 0.003, respectively, Table 2B). The presence of a CK showed no significant influence on survival endpoints, likely due to its close association with the TP53abn status. Concerning response to therapy, we found TP53abn and PTPN11mut to associate with an inferior CR rate (OR, 0.44, [95% CI 0.21–0.92], P = 0.03 and OR, 0.45, [95% CI 0.23–0.88], P = 0.02, Table S6).
Of note, IDH2mut was associated with better OS and RFS in our cohort (OS: HR, 0.51, [95% CI 0.3–0.88], P = 0.015, and RFS: HR, 0.5, [95% CI 0.26–0.96], P = 0.036, Table 2, Figure S18C,D), and with a higher CR rate (OR, 3.5, [95% CI 1.38–10.78], P = 0.01, Table S6). While allo-HSCT in CR1 generally improved OS and RFS in this cohort, IDH2mut patients showed an exceptional benefit from allo-HSCT in CR1 (Figure S19A-D). For KMT2Cmut versus wild type (wt) patients, we detected no differences in clinical endpoints (Figure S20, Table S7). In fact, mutations in any of the most frequently affected genes located in the CDRs of chr7 (Figure S7A) did not influence survival endpoints (data not shown).
Focusing on TP53 and CK-status, abn(7) patients with TP53wt/CK showed similar probabilities of OS and RFS compared with TP53wt/non-CK patients, further indicating that CK-status provided no TP53abn-independent prognostic information (Figure S21).
According to its proposed impact on survival in myeloid malignancies [47], we investigated the effect of TP53 allelic status. Applying the current ICC-definition for TP53 multi-hit status [15], we discovered that the majority of TP53abn patients belonged to the TP53 multi-hit category (79%, Figure S22). Of note, in our cohort, there was no significant difference in survival between patients belonging to the TP53 single-versus multi-hit category.
Discussion
In this study, we deciphered the genomic landscape of adult AML with abn(7), including concomitant somatic and structural variants and their potential influence on prognosis in a large international cohort of 519 patients using a two-step NGS approach. As expected, for a large proportion of our patients, the chr7 aberration was embedded in a CK (43%), and CK-status was strongly associated with TP53 abnormalities (P < 0.001) [10, 48,49,50]. Accordingly, TP53 showed the highest mutation frequency (33%), and TP53mut appeared mostly mutually exclusive to other known gene mutations, further underscoring its importance in the current ICC-classification [15].
As a novel finding, we identified a high frequency of mutations in KMT2C (16.7%). KMT2C belongs to the KMT2 family of histone methyltransferases, which catalyze the methylation of lysine 4 at histone 3 (H3) and function as epigenetic regulators. The KMT2 genes are among the most frequently altered genes in various cancer types, and KMT2C mutations have been mainly reported in solid malignancies to date [51, 52]. A recent study investigating the mutational spectrum of patients suffering from a wide variety of myeloid malignancies with abn(7) did not report recurrent KMT2C mutations [10]. Several lines of argumentation might explain this discrepancy: (1) KMT2C encodes for a large protein of 4911 amino acids and is characterized by a high GC-content and repetitive elements, which are both well-known factors for sequencing artifacts. We observed a similar prevalence of KMT2C mutations with WES and error-corrected TS and confirmed the somatic nature of these mutations using CR samples when available. (2) KMT2C mutations were mainly subclonal with a median VAF of 5%. Thus, studies using sequencing technologies without incorporation of error-correction are likely to miss the majority of KMT2C mutations. (3) Studies focusing on pediatric AML [53] and adult AML (CBF [54] and elderly [55] AML patients) reported KMT2C mutations at low frequencies, providing independent evidence of the true nature of these variants. Very recently, a high prevalence of KMT2C mutations was reported in adult blastic plasmacytoid dendritic cell neoplasm (BPDCN), affecting 48% of investigated cases [56]. With a prevalence of 45%, we found the highest KMT2C mutation prevalence in patients with CBF AML and abn(7). The majority of the KMT2C variants in our cohort were categorized as multi-hit (= concomitant mutation and deletion at the KMT2C locus). Considering that our clonality analyses revealed TP53 and CHIP-associated gene mutations as disease-initiating and mutations of genes involved in signaling pathways and KMT2C as late events, KMT2C mutations seem to appear predominantly as the "second hit" driven by clonal selection pressure in abn(7) leukemia evolution. Collectively, these data suggest a rather supporting than initiating role for KMT2C mutations during leukemogenesis and disease progression. This conclusion would be backed by the report from Chen et al., where TP53-deficient mice only developed leukemia after transplantation of hematopoietic stem cells with concomitant Kmt2c and Nf1-knockdown [19], pointing to a collaboration of several pathways promoting leukemogenesis in a multistep manner. Therefore, it will be of interest to systematically study KMT2C mutations over time and investigate mutation stability and evolution in relapsing AML with abn(7). Furthermore, future studies are needed to validate the pathogenicity of the detected KMT2C variants.
Overall, our NGS-based CNV analysis revealed a very high concordance with diagnostic karyotyping based on G-banding/FISH analyses. In addition, this technology enabled us to calculate CN-adjusted VAFs, an essential prerequisite for investigations on clonal hierarchies, and to define precise breakpoints. Thereby, we uncovered a previously underappreciated association between the most frequent focal deletion of chr7q (chr7:q21.2–q36.3) with gains of the MECOM/EVI1 locus (chr3q26.2–q29). Moreover, we identified a substantial number of focal lesions < 10 MB, which were likely missed by conventional karyotyping. Most of these small lesions covered loci of genes like TP53, NF1, or other recurrent AML genes, pointing toward the possibility of an even higher proportion of TP53-altered AML patients than currently expected.
The large size of our cohort allowed for a systematic investigation of genetic markers with patient outcome in AML with abn(7). In addition to the confirmation of the well-known detrimental outcome for patients harboring a TP53 abnormality, we report for the first time a similar dismal prognosis for patients with PTPN11 mutations in AML with abn(7). The prevalence of 11% in our study is slightly higher than in studies not focusing on distinct AML subgroups [57, 58]. In AML, PTPN11 mutations mainly affect residues on the interacting surfaces of the Src-homology 2 domain and a tyrosine phosphatase domain, leading to a gain of function and activation of this proto-oncogene. This results in downstream activation of numerous pathways, including RAS/ERK1/2, FLT3, JAK/STAT, PI3K/AKT, and NF-κB, immune-evasion mediated by PD-1, and overexpression of anti-apoptotic proteins [59,60,61]. Furthermore, PTPN11 and other RAS/RTK-pathway gene mutations have been associated with resistance-development to venetoclax/azacitidine treatment and poor outcome [62], indicating the high medical need for novel treatment approaches for PTPN11-mutant AML.
Furthermore, we observed that IDH2-mutant patients had a higher CR rate and longer RFS and OS than IDH2wt patients with AML abn(7). While being aware of the limitations of the retrospective nature of our analyses, we additionally observed a benefit of allo-HSCT in CR1 in patients with abn(7), which was especially accentuated in IDH2-mutant cases. The outcome of these IDH2-mutant patients somewhat resembled the outcome of patients with ELN 2022 intermediate-risk [3]. Importantly, this superior outcome is at least partially explained by a higher responsiveness to induction therapy and a higher usage of allo-HSCT in CR1 for IDH2mut as compared to IDH2wt patients (41% vs. 28%). These data are in line with a recent study investigating the role of IDH1/2 mutations in 4930 AML patients that reported a significantly better RFS and OS for patients with IDH2 mutations affecting residue R172 [63] as compared with IDHwt patients of the ELN 2017 intermediate and high-risk subgroups. In our cohort, both commonly affected codons (R140 and R172) showed similar superior survival. Collectively, it will be of special interest to see whether the incorporation of IDH2-inhibition leads to further outcome improvement in IDH2-mutant AML with abn(7).
To conclude, we retrospectively investigated the genomic landscape of adult AML with abn(7), revealing the nature of this genetically multifaceted leukemia subgroup. Its distinct clinical outcomes and genetic patterns depend on the type of abn(7) and the co-occurrence of other chromosomal aberrations. Although AML with −7 is classified into one ELN adverse-risk group, it should be noted that survival among these clinically poorly performing patients may still differ with a potential survival benefit in IDH2mut patients but at the same time, even poorer outcomes in the presence of TP53 and PTPN11 aberrations. Additional studies will be essential to elaborate on the functional consequences of KMT2C alterations in AML with abn(7).
Availability of data and materials
Data for the WES and TS cohorts are summarized in the supplementary information files. All first diagnosis and complete remission WES data have been uploaded on EGA (Accession ID: EGAD50000000621).
Abbreviations
- abn(7):
-
Aberrations of chromosome 7
- Allo-HSCT:
-
Allogeneic stem cell transplantation
- AML:
-
Acute myeloid leukemia
- BIH:
-
Berlin Institute of Health
- BPDCN:
-
Blastic plasmacytoid dendritic cell neoplasm
- CBF AML:
-
Core binding factor acute myeloid leukemia
- CCF:
-
Cancer cell fraction
- CDR:
-
Commonly deleted region
- CHIP:
-
Clonal hematopoiesis of indeterminate potential
- Chr7:
-
Chromosome 7
- CK:
-
Complex karyotype
- CNV:
-
Copy number variation
- CR:
-
Complete remission
- del(7q):
-
Deletion of the long arm of chromosome 7
- ELN:
-
European LeukemiaNet
- FISH:
-
Fluorescence in situ hybridization
- HR:
-
Hazard ratio
- ICC:
-
International Consensus Classification of Myeloid Neoplasms and Acute Leukemias
- LOH:
-
Loss of heterozygosity
- NGS:
-
Next-generation sequencing
- Non-CK:
-
Non-complex karyotype
- ONT:
-
Oxford Nanopore Technology
- OR:
-
Odds ratio
- OS:
-
Overall survival
- RFS:
-
Relapse-free survival
- sAML:
-
Secondary acute myeloid leukemia
- SNP:
-
Single nucleotide polymorphism
- SNV:
-
Single nucleotide variant
- tAML:
-
Therapy-related acute myeloid leukemia
- TCGA:
-
The Cancer Genome Atlas database
- TS:
-
Targeted sequencing
- VAF:
-
Variant allele frequency
- WES:
-
Whole-exome sequencing
- WT:
-
Wild type
- −7:
-
Monosomy 7
References
Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140(12):1345–77.
Schiffer CA, Lee EJ, Tomiyasu T, Wiernik PH, Testa JR. Prognostic impact of cytogenetic abnormalities in patients with de novo acute nonlymphocytic leukemia. Blood. 1989;73(1):263–70.
Scherer SW, Cheung J, MacDonald JR, Osborne LR, Nakabayashi K, Herbrick JA, et al. Human chromosome 7: DNA sequence and biology. Science. 2003;300(5620):767–72.
Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–65.
Hussain FT, Nguyen EP, Raza S, Knudson R, Pardanani A, Hanson CA, et al. Sole abnormalities of chromosome 7 in myeloid malignancies: spectrum, histopathologic correlates, and prognostic implications. Am J Hematol. 2012;87(7):684–6.
Mrózek K. Cytogenetic, molecular genetic, and clinical characteristics of acute myeloid leukemia with a complex karyotype. Semin Oncol. 2008;35(4):365–77.
Eisfeld A-K, Kohlschmidt J, Mrózek K, Volinia S, Blachly JS, Nicolet D, et al. mutational landscape and gene expression patterns in adult acute myeloid leukemias with monosomy 7 as a sole abnormality. Can Res. 2017;77(1):207–18.
Mori M, Kubota Y, Durmaz A, Gurnari C, Goodings C, Adema V, et al. Genomics of deletion 7 and 7q in myeloid neoplasm: from pathogenic culprits to potential synthetic lethal therapeutic targets. Leukemia. 2023;37(10):2082–93.
Hasle H, Alonzo TA, Auvrignon A, Behar C, Chang M, Creutzig U, et al. Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood. 2007;109(11):4641–7.
McNerney ME, Brown CD, Peterson AL, Banerjee M, Larson RA, Anastasi J, et al. The spectrum of somatic mutations in high-risk acute myeloid leukaemia with −7/del(7q). Br J Haematol. 2014;166(4):550–6.
Grob T, Al Hinai ASA, Sanders MA, Kavelaars FG, Rijken M, Gradowska PL, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2022;139(15):2347–54.
Short NJ, Montalban-Bravo G, Hwang H, Ning J, Franquiz MJ, Kanagal-Shamanna R, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–9.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200–28.
Baeten JT, Liu W, Preddy IC, Zhou N, McNerney ME. CRISPR screening in human hematopoietic stem and progenitor cells reveals an enrichment for tumor suppressor genes within chromosome 7 commonly deleted regions. Leukemia. 2022;36(5):1421–5.
Pastor VB, Sahoo SS, Boklan J, Schwabe GC, Saribeyoglu E, Strahm B, et al. Constitutional SAMD9L mutations cause familial myelodysplastic syndrome and transient monosomy 7. Haematologica. 2018;103(3):427–37.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–6.
Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014;25(5):652–65.
Chen R, Okeyo-Owuor T, Patel RM, Casey EB, Cluster AS, Yang W, et al. Kmt2c mutations enhance HSC self-renewal capacity and convey a selective advantage after chemotherapy. Cell Rep. 2021;34(7):108751.
McNerney ME, Brown CD, Wang X, Bartom ET, Karmakar S, Bandlamudi C, et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood. 2013;121(6):975–83.
Sarkozy C, Hung SS, Chavez EA, Duns G, Takata K, Chong LC, et al. Mutational landscape of gray zone lymphoma. Blood. 2021;137(13):1765–76.
Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014;28(1):44–9.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–506.
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–36.
Damm F, Oberacker T, Thol F, Surdziel E, Wagner K, Chaturvedi A, et al. Prognostic importance of histone methyltransferase MLL5 expression in acute myeloid leukemia. J Clin Oncol. 2011;29(6):682–9.
Christen F, Hoyer K, Yoshida K, Hou HA, Waldhueter N, Heuser M, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood. 2019;133(10):1140–51.
Damm F, Mylonas E, Cosson A, Yoshida K, Della Valle V, Mouly E, et al. Acquired initiating mutations in early hematopoietic cells of CLL patients. Cancer Discov. 2014;4(9):1088–101.
Mylonas E, Yoshida K, Frick M, Hoyer K, Christen F, Kaeda J, et al. Single-cell analysis based dissection of clonality in myelofibrosis. Nat Commun. 2020;11(1):73.
Riester M, Singh AP, Brannon AR, Yu K, Campbell CD, Chiang DY, et al. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol Med. 2016;11(1):13.
Heuser M, Damrah N, Gabdoulline R, Lai CK, Sträng E, Salim M, et al. Rapid and reproducible karyotyping with nanopore sequencing in AML patients. Blood. 2022;140(Supplement 1):6070–2.
Teixeira VH, Pipinikas CP, Pennycuick A, Lee-Six H, Chandrasekharan D, Beane J, et al. Deciphering the genomic, epigenomic, and transcriptomic landscapes of pre-invasive lung cancer lesions. Nat Med. 2019;25(3):517–25.
Noerenberg D, Briest F, Hennch C, Yoshida K, Hablesreiter R, Takeuchi Y, et al. Genetic characterization of primary mediastinal B-cell lymphoma: pathogenesis and patient outcomes. J Clin Oncol. 2024;42(4):452–66.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101.
Machado HE, Mitchell E, Øbro NF, Kübler K, Davies M, Leongamornlert D, et al. Diverse mutational landscapes in human lymphocytes. Nature. 2022;608(7924):724–32.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–8.
Arends CM, Galan-Sousa J, Hoyer K, Chan W, Jager M, Yoshida K, et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia. 2018;32(9):1908–19.
Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2011;44(1):17–9.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13.
Cheng S, Douglas-Jones A, Yang X, Mansel RE, Jiang WG. Transforming acidic coiled-coil-containing protein 2 (TACC2) in human breast cancer, expression pattern and clinical/prognostic relevance. Cancer Genom Proteom. 2010;7(2):67–73.
Ito Y, Terao Y, Noma S, Tagami M, Yoshida E, Hayashizaki Y, et al. Nanopore sequencing reveals TACC2 locus complexity and diversity of isoforms transcribed from an intronic promoter. Sci Rep. 2021;11(1):9355.
Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29(21):2889–96.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–27; quiz 99.
Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–77.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–67.
Abel HJ, Oetjen KA, Miller CA, Ramakrishnan SM, Day RB, Helton NM, et al. Genomic landscape of TP53-mutated myeloid malignancies. Blood Adv. 2023;7(16):4586–98.
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549–56.
Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22(8):1539–41.
Stirewalt DL, Kopecky KJ, Meshinchi S, Appelbaum FR, Slovak ML, Willman CL, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97(11):3589–95.
Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–21.
Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15(6):334–46.
Kinnersley B, Sud A, Everall A, Cornish AJ, Chubb D, Culliford R, et al. Analysis of 10,478 cancer genomes identifies candidate driver genes and opportunities for precision oncology. Nat Genet. 2024. https://doi.org/10.1038/s41588-024-01785-9.
Umeda M, Ma J, Westover T, Ni Y, Song G, Maciaszek JL, et al. A new genomic framework to categorize pediatric acute myeloid leukemia. Nat Genet. 2024;56(2):281–93.
Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48(12):1551–6.
Jahn E, Saadati M, Fenaux P, Gobbi M, Roboz GJ, Bullinger L, et al. Clinical impact of the genomic landscape and leukemogenic trajectories in non-intensively treated elderly acute myeloid leukemia patients. Leukemia. 2023;37(11):2187–96.
Künstner A, Schwarting J, Witte HM, Xing P, Bernard V, Stölting S, et al. Genome-wide DNA methylation-analysis of blastic plasmacytoid dendritic cell neoplasm identifies distinct molecular features. Leukemia. 2024;38(5):1086–98.
Sargas C, Ayala R, Larráyoz MJ, Chillón MC, Carrillo-Cruz E, Bilbao-Sieyro C, et al. Molecular landscape and validation of new genomic classification in 2668 adult AML patients: real life data from the PETHEMA registry. Cancers (Basel). 2023;15(2):438.
Alfayez M, Issa GC, Patel KP, Wang F, Wang X, Short NJ, et al. The Clinical impact of PTPN11 mutations in adults with acute myeloid leukemia. Leukemia. 2021;35(3):691–700.
Chen L, Chen W, Mysliwski M, Serio J, Ropa J, Abulwerdi FA, et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia. 2015;29(6):1290–300.
Nabinger SC, Li XJ, Ramdas B, He Y, Zhang X, Zeng L, et al. The protein tyrosine phosphatase, Shp2, positively contributes to FLT3-ITD-induced hematopoietic progenitor hyperproliferation and malignant disease in vivo. Leukemia. 2013;27(2):398–408.
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–33.
Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. 2020;1(12):1176–87.
Middeke JM, Metzeler KH, Röllig C, Krämer M, Eckardt JN, Stasik S, et al. Differential impact of IDH1/2 mutational subclasses on outcome in adult AML: results from a large multicenter study. Blood Adv. 2022;6(5):1394–405.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):1.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34(2):155–63.
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;1:1–16.
Suehnholz SP, Nissan MH, Zhang H, Kundra R, Nandakumar S, Lu C, et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 2024;14(1):49–65.
Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci USA. 2010;107(39):16910–5.
Acknowledgements
We thank all participating patients and their families, the ALFA, AMLCG, AMLSG, CETLAM, HOVON/SAKK, UK NCRI study groups, and all participating centers. We thank Dr. Tatiana Borodina and Jeannine Wilde for their technical assistance and acknowledge the assistance of the Core Unit Genomics.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported in part by grants from the Deutsche José Carreras Leukämie-Stiftung (03 R/2020), Deutsche Krebshilfe (70113643), the Berliner Krebsgesellschaft (DAFF201907), and the DKTK awarded to FD, as well as by the Bundesministerium für Bildung und Forschung (ERA PerMed project MEET-AML 01KU2014) awarded to LB, the Deutsche Forschungsgemeinschaft (FOR 2674) awarded to LB, KD, and HD, and National Cancer Center Research and Development Funds (2022-A-1) awarded to KY. AH received support from the Berlin Institute of Health clinician-scientist program. MT was supported by a fellowship from the Berliner Krebsgesellschaft and the José Carreras-DGHO-fellowship.
Author information
Authors and Affiliations
Contributions
F.D. designed the research; A.H., M.T., P.S., N.E., R.A., E.J., A.D., K.K., M.S., and K.Y. performed the research experiments and/or bioinformatics. M.H., H.-A.H., M.P., R.K.H., K.H.M., L.F., C.T., O.B., F.C., J.K., L.V., B.L., J.E., P.J.M.V., M.D., W.-C.C., D.C.L., H.D., R.E.G., K.D., L.B. and F.D. contributed patient samples and clinical data; A.H., M.T., P.S., K.Y., and F.D. analyzed the data; A.H., M.T., P.S., and F.D. wrote the paper. All authors read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
All study protocols were approved by the Institutional Review Boards at each participating center in accordance with the Declaration of Helsinki. Each patient provided written informed consent for the research use of their specimens before enrollment.
Competing interests
F.D. reports personal fees from AbbVie, Amgen, Astra Zeneca, BeiGene, Gilead, Incyte, Novartis, and Roche outside the submitted work. L.B. reports advisory roles for Abbvie, Amgen, Astellas, Bristol-Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Sanofi, Servier; as well as research funding from Bayer, Jazz Pharmaceuticals. A.H. reports travel fees outside of the submitted work from Jazz Pharmaceuticals. M.T. and P.S. declare no COI related to this work. E.J. reports current employment at AstraZeneca. H.D. declares consultancy with honoraria from AbbVie, AstraZeneca, Gilead, Janssen, Jazz, Pfizer, Servier, Stemline, Syndax and Clinical Research funding to the Institution from AbbVie, Astellas, Bristol Myers Squibb, Celgene, Jazz Pharmaceuticals, Kronos Bio and Servier. J.E. reports participation in Advisory Boards of Abbvie, Novartis, Astellas, Jazz Pharmaceuticals, BMS-Celgene, Pfizer, Amgen and Research grants from Novartis and Jazz Pharmaceuticals. J.K. reports personal fees from BMS/Celgene, Takeda, Janssen, Abbvie, Sanofi, Pfizer and Jazz Pharmaceuticals outside the submitted work. M.H. reports research funding to institution from Abbvie, Servier, Astellas, BergenBio, Glycostem, Jazz Pharmaceuticals, Karyopharm, Loxo Oncology, Novartis, PinotBio, honoraria by Abbvie, Bristol Myers Squibb, Janssen, Jazz Pharmaceuticals, Pfizer, Qiagen, Servier, Sobi, and consultancy fees by AvenCell, Abbvie, Astellas, Glycostem, Janssen, LabDelbert, Miltenyi, Novartis, Pfizer, PinotBio and Servier.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Halik, A., Tilgner, M., Silva, P. et al. Genomic characterization of AML with aberrations of chromosome 7: a multinational cohort of 519 patients. J Hematol Oncol 17, 70 (2024). https://doi.org/10.1186/s13045-024-01590-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01590-1