
Abstract
Background
Ferroptosis, characterized by iron-dependent lipid peroxidation, emerges as a promising avenue for hepatocellular carcinoma (HCC) intervention due to its tumor susceptibility. RNA N6-methyladenosine (m6A) modification has been involved in several types of regulated cell death. However, the roles and molecular mechanisms of m6A-related regulators in HCC cell ferroptosis remain unclear.
Methods
By examining a series of m6A modification enzymes upon ferroptosis induction or inhibition, we identified METTL16 as a novel ferroptotic repressor in HCC cells. The roles of METTL16 on ferroptosis and HCC development were investigated in multiple cell lines, human HCC organoids, subcutaneous xenografts and MYC/Trp53−/− HCC model in hepatocyte-specific Mettl16 knockout and overexpression mice. The underlying mechanism was elucidated with MeRIP/RIP-qPCR, luciferase assay, Co-IP assay and Mass Spectrometry. The clinical significance and relevance were evaluated in human samples.
Results
High METTL16 expression confers ferroptosis resistance in HCC cells and mouse models, and promotes cell viability and tumor progression. Mechanistically, METTL16 collaborates with IGF2BP2 to modulate SENP3 mRNA stability in an m6A-dependent manner, and the latter impedes the proteasome-mediated ubiquitination degradation of Lactotransferrin (LTF) via de-SUMOylation. Elevated LTF expression facilitates the chelation of free iron and reduces liable iron pool level. SENP3 and LTF are implicated in METTL16-mediated HCC progression and anti-ferroptotic effects both in vivo and in vitro. Clinically, METTL16 and SENP3 expression were positively correlated, and high METTL16 and SENP3 expression predicts poor prognosis in human HCC samples.
Conclusions
Our study reveals a new METTL16-SENP3-LTF signaling axis regulating ferroptosis and driving HCC development. Targeting this axis is a promising strategy for sensitizing ferroptosis and against HCC.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is a globally prevalent malignancy known for its significant incidence and mortality [1]. Systemic therapies utilizing Tyrosine Kinase Inhibitors (TKIs) have become fundamental in the management of advanced HCC for decades [2]. Sorafenib, the first pioneering TKI approved by the Food and Drug Administration (FDA) for treating malignant HCC, has been found to induce ferroptosis by inhibiting system Xc- and elevating intracellular iron levels, partly explaining its anti-cancer effects [3,4,5]. Studies have shown that it is the distinctive augmented metabolic activity of cancer cells observed within the hepatic tumor microenvironment, characterized by elevated oxidative stress, high reactive oxygen species (ROS) burden and increased reliance on iron, that enhances the efficacy of ferroptosis inducers such as sorafenib in eradicating tumor cells [6, 7].
The regulated cell death triggered by extensive iron-dependent lipid peroxidation, known as ferroptosis, has garnered significant attention as an intrinsic tumor suppressor with crucial roles in malignancies. Intriguingly, mesenchymal and dedifferentiated cancer cells, typically resistant to apoptosis and conventional treatments, exhibit a remarkable susceptibility to ferroptosis, indicating their promise as a therapeutic strategy for refractory cancers [8]. Recent research has unveiled the regulatory mechanisms and cancer implications of ferroptosis in HCC [9]. In terms of ferroptosis-associated oxidative-reductive system regulation, ATF4 and USP8 have been shown to upregulate the transcriptional expression of SLC7A11 and reduce SLC7A11 GlcNAcylation, thereby modulating the oxidative-reductive balance through the Xc--GSH-GPX4 axis to regulate ferroptosis [10, 11]. Regarding lipid metabolism modulation, unconventional prefoldin RPB5 interactor (URI) ameliorates TKI-induced ferroptosis by reprogramming lipid metabolism associated with stearoyl-CoA desaturase 1 (SCD1) [12]. However, research on the modulation of ferroptosis through iron metabolism, another crucial regulator, remains relatively limited.
N6-methyladenosine (m6A) methylation represents the predominant post-transcriptional modification in eukaryotic mRNA and is governed by m6A methyltransferases catalyzing m6A deposition (“Writer”), demethylases facilitating m6A removal (“Eraser”) and RNA-binding proteins recognizing and binding m6A motifs (“Reader”), playing significant roles in the regulation of mRNA splicing, decay, stability, translation and nuclear export. Aberrant expression of m6A-related regulators in cancer has been linked to tumor development and has recently attracted considerable attention. Interventions targeting RNA modifications have demonstrated efficacy in disrupting tumor cell survival, thus offering novel avenues for cancer treatment. Following the classical METTL3/14 methyltransferase, METTL16 was recently identified as the second m6A writer, demonstrating oncogenic or tumor-suppressive effects in various cancers by modifying specific RNA substrates [13]. Notably, in HCC, METTL16 has been implicated in oncogenesis through m6A modification of long non-coding RNAs (lncRNAs) [14, 15], influencing their transcriptional stability and degradation. Furthermore, METTL16 exhibited m6A-independent functions as a translation initiation factor, contributing to its pro-carcinogenic effects [16]. However, data remains scarce regarding the downstream mRNA targets regulated by METTL16 in an m6A-dependent manner in HCC.
In this study, we explored the involvement of m6A demethylases and methyltransferases in the regulation of ferroptosis and HCC development. METTL16 was then screened and identified as the key candidate. We also reported that METTL16 plays a critical role in ferroptosis and tumorigenesis through catalyzing mRNA m6A modification of SENP3, a redox-sensitive SUMO2/3-specific protease, and the latter modulates the stabilization of Lactotransferrin (LTF) within HCC cells, thereby inhibiting ferroptosis, contributing to disease progression and adverse clinical outcomes. These findings provide a new METTL16-SENP3-LTF regulatory axis that suppresses ferroptosis and facilitates HCC development, highlighting the importance of targeting this axis as a potential therapeutic strategy for this disease.
Materials and methods
Plasmids and reagents
The plasmid pLenti-CMV-METTL16-FLAG-GFP-Puro, p3*FLAG-CMV-SENP3 and pWPXLd-FLAG-SENP3 were sourced from the Public Protein/Plasmid Library (Nanjing, China). The pENTER-LTF was purchased from Vigene Biosciences (Shandong, China). LTF-GFP construction involved the insertion of the open reading frame (ORF) of LTF from the pENTER-LTF plasmid into the linearized pEX-1 vector (GenePharma, Shanghai, China). Mutated plasmids (METTL16-PPF185/186/187AAG, SENP3-C532A, LTF K57R, LTF K405R, LTF K118R and LTF 2KR) were generated via high-fidelity PCR-based site-directed mutagenesis using pLenti-CMV-METTL16-FLAG-GFP-Puro, p3*FLAG-CMV-SENP3 and LTF-GFP as templates (Vazyme, Nanjing, China), and mutations were confirmed via Sanger sequencing. pT3-EF1a-MYC (Addgene plasmid # 92046;http://n2t.net/addgene:92046;RRID:Addgene_92046) [17] and px330-sgp53 (Addgene plasmid # 59910;http://n2t.net/addgene:59910;RRID:Addgene_59910) [18] were acquired from Addgene. pLentiCRISPR-sgMettl16, px459-sgSenp3 and px459-sgLtf harboring two targets linked by gRNA scaffold were purchased from Tsingke Biotech Co., Ltd (Beijing, China). The MYC-SUMO3 and UBC9 were constructed by inserting the ORF of SUMO3 and UBC9 into the linearized pEX-3 vector (GenePharma). All of the cloning PCR primers and sgRNA sequences are provided in Supplementary Table S1. The plasmids were extracted using a NucleoBond Xtra Midi kit (MACHEREY-NAGEL, Germany).
RSL3 ((1 S,3R)-RSL3, HY-100218A, MedChemExpress, USA); Fer-1 (Ferrostatin-1, S7243, Selleck, TX, USA); DFO (Deferoxamine, D9533,Sigma-Aldrich, Merck KGaA, Germany), FAC (Ammonium iron(III) citrate, F5879, Sigma); MG132 (HY-13259, MedChemExpress); ActD (Actinomycin D, HY17559, MedChemExpress); CHX (Cycloheximide, S7418, Selleck); Leupeptin (HY-18234, MedChemExpress); NEM (N-Ethylmaleimide, HY-D0843, MedChemExpress); Sorafenib (S7397, Selleck); Geneticin (G418 Sulfate, 108321-42-2, Selleck).
Antibodies
Primary antibodies against the following proteins were obtained from Cell Signaling Technology (MA, USA): METTL16 (87538 S); SENP3 (5591 S); anti-rabbit IgG, HRP-linked Antibody (7074); anti-mouse IgG, HRP-linked Antibody (7076); Mouse (G3A1) mAb IgG1 Isotype Control (5415); Rabbit (DA1E) mAb IgG XP® Isotype Control (3900); from Sigma: MYC-Tag(SAB1305535); Anti-FLAG®M2 (F1804); from Abcam (Cambridge, UK): METTL16 (ab252420); IGF2BP2 (ab128175); LTF (ab109216); 4-HNE (ab46545); MDA (ab43066); GPX4 (ab125066); NRF2 (ab62352); from Proteintech (Wuhan, China): β-actin (66009-1-Ig); GFP-Tag (66002-1-Ig); HA-Tag (66006-2-Ig); METTL3 (15073-1-AP); METTL14 (26158-1-AP); ACSL4 (22401-1-AP); from ABclonal (Wuhan, China): LTF (A12902); FTH1 (A19544).
Cell lines
Huh7, PLC/PRF/5, Hepa1-6, Hep3B, HepG2, HCCLM3, MHCC97-H, SK-HEP-1 (used exclusively for METTL16 protein analysis due to its endothelial origin), and HEK293T cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Grand Island, NY, USA) and SNU387 cell lines were maintained in RPMI-1640 (Corning, USA). Both media were supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% penicillin/streptomycin (Gibco). Cells were incubated at 37 °C with 5% CO2. All cell lines were sourced from the Cell Bank, Chinese Academy of Sciences (Shanghai, https://www.cellbank.org.cn), and tested negative for mycoplasma contamination.
Cell transfection
Plasmids or siRNAs were transfected using Lipofectamine 3000 Reagents (Invitrogen, USA) following the manufacturer’s protocols. The small interference RNAs (siRNAs) and non-specific control (NC), provided by GenePharma, are listed in Supplementary Table S2. For stable HCC cell lines, lentiviruses designed for overexpressing METTL16 and SENP3, and for RNA interference against METTL16 and SENP3 (based on siMETTL16 and siSENP3 sequences) were prepared by GenePharma. HCC cells were infected with these lentiviruses in the presence of 4 µg/mL polybrene and selected with 2.5 µg/mL puromycin. To establish stable cell lines expressing LTF/LTF 2KR, cells were transfected with GFP-LTF/LTF 2KR plasmids and selected with 700 µg/mL G418 (Geneticin) to isolate single-cell-derived clones with successful LTF/LTF 2KR integration and overexpression.
Cell proliferation assays
Cell viability was measured using the Cell Counting Kit-8 reagent (CCK-8, Dojindo, Japan) according to the manufacturer’s instructions. Triplicate wells containing 3 × 103 cells each were plated in 96-well plates, followed by the addition of 10µL CCK-8 reagent per well and a 75-minute incubation at 37 °C. Absorbance was measured at 450 nm using a spectrophotometer. For colony formation assays, 1 × 103 cells per well were seeded in 6-well plates, with media changed every 4 days. Colonies, typically formed in approximately 10–17 days, were fixed with 4% paraformaldehyde, stained with 0.5% crystal violet, and subsequently photographed and counted.
Iron, MDA, and GSH level measurement
Following treatment, 2 × 106 HCC cells, mouse liver tissues, or subcutaneous xenografts were harvested and resuspended in PBS or saline for further analysis. Iron levels were determined using the Iron Assay Kit (A039-2-1, Jiancheng Bioengineering Institute, China), while MDA levels were assessed with the MDA Assay Kit (A003-4-1, Jiancheng Bioengineering Institute, China), and GSH levels were measured using the GSH Assay Kit (A006-2-1, Jiancheng Bioengineering Institute, China). Assays were conducted according to the manufacturer’s protocols.
ROS and lipid peroxidation measurement
Lipid peroxidation was assessed using BODIPY™ 581/591 C11 (D3861, Invitrogen). The intracellular ROS levels were measured using the ROS Assay Kit with the DCFH-DA probe (S0033M, Beyotime, China). BODIPY™ 581/591 C11 (10 mM) and DCFH-DA (10 mM) probes were diluted at ratios of 1:2000 and 1:1000, respectively, in a serum-free medium. Cells were incubated with the probes at 37 °C in the dark for 20–30 min. Following incubation, cells were washed with PBS, and fluorescent images were captured with an inverted microscope. HCC cells were subsequently resuspended and quantified with a CytoFLEX flow cytometer and reanalyzed using FlowJo software.
Labile iron pool and total iron assays
The labile iron pool within HCC cells was assessed utilizing Calcein-AM (HY-D0041, MedChemExpress). After collection, cells were washed twice with PBS and then incubated with 0.125µM Calcein-AM in either a serum-free medium or PBS for 15 min at 37 °C. Fluorescence measurements were performed using a fluorescence microscope and a CytoFLEX flow cytometer. The fluorescence intensity of Calcein-AM increases inversely with free iron content. Moreover, the presence of free Fe2+ in HCC cells was determined by employing FerroOrange (F374, Dojindo). Following three washes with Hank’s Balanced Salt Solution (HBSS), cells were exposed to 1 µM FerroOrange for 30 min at 37 °C. Fluorescence intensity was subsequently measured using both a fluorescence microscope and a CytoFLEX flow cytometer.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Cells and tissues were lysed for RNA extraction using Trizol (Invitrogen). 1 µg RNA was reverse transcribed into cDNA with the PrimeScript™ RT reagent kit (Takara Bio, Japan). qRT-PCR was carried out with the TB Green® Premix Ex Taq™ (Tli RNaseH Plus) kit (Takara Bio, Japan) on an ABI Q6 PCR system. Gene expression was quantified using the 2−ΔΔCT method with the human β-actin gene as the endogenous control. Primer sequences are provided in the Supplementary Table S3.
Dual-luciferase reporter assay
Dual-luciferase reporters, including SENP3-CDS WT-Luc, SENP3-CDS MUT-Luc, SENP3-3’UTR WT-Luc and SENP3-3’UTR MUT-Luc, were generated using the pGL3 vector by Sangon Biotech Co., Ltd (Shanghai, China). HCC cells, at 70–80% confluence in 24-well plates, were co-transfected with 500 ng of each plasmid and 10 ng of pRL-SV40 Renilla luciferase reporter plasmid. After 24–48 h, cells were harvested, and luciferase activity was measured using the dual-luciferase assay kit (Promega, Wisconsin, USA) and the Glomax-multi Luminometer.
MeRIP/RIP assay
The methylated RNA immunoprecipitation (MeRIP) assay was conducted using the Magna MeRIP m6A Kit (Millipore, USA) to quantify m6A modification in RNA. Initially, genomic DNA was eliminated using DNase, followed by RNA fragmentation. Magnetic beads were incubated with either 10 µg of anti-m6A antibody or IgG, then immunoprecipitated with RNA samples. Subsequently, m6A-enriched RNA fragments were eluted, reverse transcribed, and quantified via RT-qPCR using primers designed for m6A sites (see Supplementary Table S4). For the RNA immunoprecipitation (RIP) assay, the Magna RIP™ Kit (Millipore, USA) was utilized following the manufacturer’s instructions. Cell lysates from 4 × 106 cells were sonicated and subjected to immunoprecipitation at 4 °C overnight with primary antibodies against METTL16 or IGF2BP2, with a homologous IgG serving as the isotype control. The relative enrichment of m6A or SENP3 in each sample was determined by normalizing the Ct values obtained from samples immunoprecipitated with m6A/METTL16/IGF2BP2 antibodies against the Ct values from the corresponding input fractions. As described in the prior studies [19], the formulas used for analysis are presented as follows: ΔCTRIP = CTRIP – CTinput; ΔCTIgG = CTIgG – CTinput; ΔΔCT = ΔCTRIP – ΔCTIgG; fold enrichment = 2–ΔΔCT. The resultant relative fold enrichment values were normalized to those of the corresponding control group.
Western blot assay
Cells and tissues were cracked by RIPA lysis buffer (20–188, Merck Millipore, USA) supplemented with protease inhibitor cocktail (4693159001, Roche, Swiss), followed by centrifugation at 12,000 × rpm at 4 °C for 15 min. The resulting mixtures were then separated using SDS-PAGE and electro-transferred onto a nitrocellulose filter (NC) membrane. To block nonspecific binding, the membrane was incubated in a solution of 5% non-fat milk with 0.1% Tween-20 in PBS (PBST) for 1 h. After incubating with specific primary and corresponding secondary antibodies, the protein bands were visualized with the Odyssey Infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA) or Tanon 5200 chemiluminescent imaging system (Tanon, Shanghai, China).
mRNA and protein stability assays
Cells were treated by ActD and CHX for 0, 4, and 8 h. The total RNA and protein were extracted from the treated cells. The obtained results were normalized to the values measured at 0 h, serving as the baseline for comparison across various time points.
Co-immunoprecipitation (Co-IP)
The total proteins were lysed using IP lysis buffer (20 mM Tris, pH 8.0; 150 mM NaCl; 1.0% NP-40; 1 mM ethylenediaminetetraacetic acid (EDTA)) supplemented with protease inhibitor (4693159001, Roche) and phosphatase inhibitor (4906837001, Roche). The lysates were then centrifuged at 12,000 rpm at 4 °C for 15 min. After centrifugation, the supernatants were mixed with GFP-Nanoab-Agarose (LABLEAD Biotech, Beijing, China) overnight at 4 °C or incubated with IP antibody at 4 °C for 2 h, followed by mixed with Dynabeads™ Protein G beads (10003D, Thermo Fisher Scientific, Waltham, MA, USA) overnight at 4 °C. The beads were washed three times using pre-cooled IP buffer and then resuspended in 2 x SDS loading buffer (Beyotime). The analysis of protein-protein interactions was performed using western blot assay.
IP coupled with mass spectrometry
Total proteins were extracted from the cells, followed by immunoprecipitation (IP) using SENP3 antibodies and Dynabeads™ Protein G beads as previously described. The SENP3-associated proteins were separated by gel electrophoresis, and the gels were stained using a Fast Silver Stain kit (P0017S, Beyotime Biotech, Beijing, China) according to the manufacturer’s instructions. Subsequently, the silver-stained bands of interest were excised and processed for mass spectrometric analysis using an Ultimate 3000 nano ultra-performance liquid chromatography-tandem Q Exactive plus mass spectrometry system (LuMing Biotech, Shanghai, China). The information on SENP3-associated proteins was listed in Supplementary Table S5.
SUMOylation and ubiquitination assays
To assess SUMOylation, HCC cells were cultured in 10 cm plates, transfected with the indicated plasmids, and lysed using SUMO lysis buffer (20 mM Tris, pH 8.0; 150 mM NaCl; 1.0% NP-40; 1 mM EDTA; 1.0% SDS; 20 µM NEM) with protease inhibitors and PMSF. Following a 30-minute incubation on a 4 °C shaker and subsequent sonication until the lysate became fluid, the protein samples were centrifuged. The supernatant was then incubated with GFP-Nanoab-Agarose beads overnight at 4 °C. Afterward, the beads were washed five times with SUMO lysis buffer without SDS and boiled for 10 min in SDS loading buffer. The eluted proteins were subjected to Western blot analysis. For ubiquitination, transfected HCC cells were treated with MG132 (40 µM for 4 h) before lysis to inhibit proteasomal degradation. Whole-cell lysates were extracted using UB lysis buffer (20 mM Tris, pH 8.0; 150 mM NaCl; 1.0% NP-40; 1 mM EDTA) and processed as described for the SUMOylation assay.
Mouse models
The mice were housed in a controlled environment with air filtration, temperature maintained at 22–24 °C, lighting controlled, and humidity between 40% and 70%. They were provided with unlimited access to a standard diet.
Male athymic nude mice (5–6 weeks old) were obtained from GemPharmatech Laboratories (Shanghai, China) for establishing a xenograft model of HCC. The mice were randomly divided into different groups before injection. 1.0 × 106 stable HCCLM3 shNC, shMETTL16-1 or shMETTL16-2 cells per mouse were subcutaneously injected into nude mice (12 mice per group). All mice in each group received intraperitoneal injections of either DMSO or RSL3 (5 mg/kg) every 2 days. Additionally, 2.0 × 106 Hep3B stable LV-VEC + shNC, LV-METTL16 + shNC, LV-VEC + shSENP3 or LV-METTL16 + shSENP3 cells per mouse were subcutaneously injected into nude mice (5 mice per group). Male C57BL/6 mice (5–6 weeks old) obtained from GemPharmatech Laboratories received injections of 6.0 × 105 stable Hepa1-6 cells sgNC or sgMettl16 (10 mice per group). Similarly, all mice in each group were administered intraperitoneal injections of either DMSO or RSL3 (10 mg/kg) every 2 days. Furthermore, 1.0 × 106 stable HCCLM3 shNC + LV-VEC, shMETTL16 + LV-VEC, shNC + LV-SENP3 or shMETTL16 + LV-SENP3 per mouse were subcutaneously injected into nude mice (6 mice per group). Tumor volume was monitored every two days using the formula: Tumor volume = (Length × Width2)/2. The tumors were resected for weighting and photographing after the mice were euthanized.
Hepatocyte-specific Mettl16 conditional knockout mice (designated Mettl16f/f, Alb-Cre mice) were generated using CRISPR/Cas9 technology by mating Mettl16-floxed (Mettl16f/f) mice (loxP sequences inserted at the ends of exons 2 and 5, GemPharmatech Co., Ltd) with Alb-Cre transgenic mice (Model Organisms Center, Inc, Shanghai, China). Additionally, Mettl16f/f mice were injected with 100 µL AAV suspension in saline (AAV8-TBG-Cre/Ctrl virus titer > 1.5 × 1012 vg/mL, Vigene Biosciences Co., Ltd) in 4 weeks via the tail vein to generate the Mettl16f/f, AAV8-TBG-Cre and Mettl16f/f, AAV8-TBG-Ctrl mice.
For detecting Mettl16 function in de novo mice HCC model, a sterile solution containing 12 µg of pT3-EF1a-MYC (MYC), 10 µg of px330-sgp53 (sgp53) and 3 µg of transposon SB13 transposase-encoding plasmid was prepared in 2 mL 0.9% NaCl solution. For assessing Mettl16/Senp3/Ltf axis function, the solution included 12 µg of pT3-EF1a-MYC-IRES-Mettl16 (or 12 µg of the control vector pT3-EF1a-MYC), 10 µg of sgp53, 3 µg of transposon SB13 transposase-encoding plasmid and 10 µg of px459-sgSenp3(sgSenp3) or 10 µg of px459-sgLtf (sgLtf) (or 10 µg of the control vector px459) in 2 mL 0.9% NaCl. Each mouse was injected with a volume equivalent to 10% of its body weight, administered within 5–7 s.
The serum biochemistry assay
At the end of the experiment, all mice were euthanized. Blood samples were collected and centrifuged at 2500 rpm for 15 min. The serum was then separated and stored at -80 °C until analysis. Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglycerides (TG), cholesterol (CHO), cholinesterase (CHE), and γ-glutamyltransferase (γ-GGT) were quantified using commercial assay kits (Jiancheng Bioengineering Institute, China), following the manufacturer’s protocols.
Immunohistochemistry (IHC), Hematoxylin and Eosin (H&E)
Tissues from mouse models were fixed in 4% paraformaldehyde (Beyotime) and embedded in paraffin. For the IHC staining assay, sections were deparaffinized, rehydrated, and boiled for antigen retrieval. Blocking was done with 5% BSA, followed by overnight incubation with primary antibodies at 4 °C, and a 1-hour room temperature incubation with horseradish peroxidase-conjugated secondary antibodies. Antigen detection was enhanced using a diaminobenzidine (DAB)-based chromogenic system. H&E staining was performed according to the standard procedures.
Human HCC tissue specimens and human tissue microarray (TMA)
The study utilized tumors and matched adjacent normal control samples from 37 patients with HCC, sourced from Shanghai East Hospital, Tongji University School of Medicine, China, with approval from the Ethics Committee of Shanghai East Hospital, and all participating patients provided informed consent. Upon collection, the tissues were promptly preserved in liquid nitrogen and stored at -80 °C until required for analysis.
A human HCC microarray (TMA, #HLivH180Su09) containing 90 paired HCC tissues and their corresponding matched normal control samples, was obtained from Shanghai Outdo Biotech (Shanghai, China). Two independent pathologists, blinded to the clinical data, evaluated the immunostaining of the samples. They assessed each cancer specimen based on staining intensity (0 = no staining, 1 = weak staining, 2 = moderate staining, 3 = strong staining) and the percentage of stained cells (0, positive cells ≤ 10%; 1, 11–25%; 2, 26–50%; 3, 51–75%; and 4, > 75%). Expression scores, calculated by multiplying intensity and percentage scores, were classified into low (negative (expression scores ≤ 2) and weak positive (2 < expression scores ≤ 4)) and high expression levels (moderate positive (4 < expression scores ≤ 6) and strong positive (expression scores > 6)).
Isolation and culture of tumor organoids
The noncancerous portion of hepatic carcinoma tissue was excised, and hepatic carcinoma cells were isolated utilizing a combination of mechanical disruption and enzymatic digestion. Briefly, the HCC tissue of appropriate dimensions underwent mincing, washing, and subsequent incubation at 37 °C with Tumor Tissue Digestion Solution (K601003, BioGenous Technologies, Suzhou, China) on an orbital shaker for 1–2 h. The resultant suspension was filtered through a 100 μm nylon cell strainer to remove tissue fragments. After centrifugation and resuspension, cells were seeded onto Matrigel Organoid Culture ECM (M315066, BioGenous Technologies) within 24-well plates and overlaid with the Hepatocellular Carcinoma Organoid Kit (K2105-HCC, BioGenous Technologies). The culture medium was replenished every 4 days, and organoids were passaged every 1–4 weeks. Following treatment with RSL3 (2.5µM) or Sorafenib (10µM) for 10 days, organoids were examined, photographed and quantified. Organoid formation capacity was assessed based on the number of viable organoids with diameters exceeding 50 μm and quantified utilizing ImageJ software.
Bioinformatics analysis
Expression levels of METTL16, SENP3, and LTF, along with survival analysis in cancer, were conducted utilizing gene expression and clinical information from The Cancer Genome Atlas (TCGA) project (https://portal.gdc.cancer.gov) and the supplementary information provided by Gao, Q., et al [20]. The public datasets of ICGC (https://dcc.icgc.org/projects/LIRI-JP), GSE104462, GSE109211, GSE104580, GSE45436, GSE182607, GSE181515, GSE182593, GSE248769 and GSE262114 were employed. Similar genes to METTL16 were analyzed using GEPIA2 (http://gepia2.cancer-pku.cn/#similar) and are listed in the Supplementary table S6. METTL16 and SENP3 expression in the TCGA-LIHC cohort from different cancer stages was analyzed using UALCAN (https://ualcan.path.uab.edu/). scRNA-seq data from GSE181515 were analyzed through scLiverDB (https://guolab.wchscu.cn/liverdb). Drug activity data and mRNA expression levels of METTL16 were assessed via CTRP (https://portals.broadinstitute.org/ctrp/). Ferroptosis genes were obtained from FerrDb V1(http://www.zhounan.org/ferrdb) and Ferroptosis signature genes were provided by Wu, A., et al. (Supplementary table S6) [21]. To predict potential SUMOylated residues in LTF, bioinformatic platforms including GPS-SUMO (http://sumosp.biocuckoo.org), SUMOplot™ (https://www.abcepta.com/sumoplot), and JASSA (http://www.jassa.fr/index.php?m=jassa) were used. Raw data for determining the genetic dependence of METTL family members in cancers were obtained from https://depmap.org/portal/ and https://score.depmap.sanger.ac.uk/.
Statistical analysis
Data are expressed as mean ± SD. Statistical significance was determined using the Student t-test or Mann-Whitney test. The χ2 test was employed for categorical data comparisons. ANOVA or Kruskal-Wallis test was used for comparisons among multiple groups. Statistical significance for survival was calculated using the Log-rank test. Spearman and Pearson’s correlation analyses were performed for correlation analysis. GraphPad Prism 9 software was used to create the graphs and for the statistical analysis (GraphPad Software, Inc.). Significance values were set at *, P < 0.05; **, P < 0.01; ***, P < 0.001, n.s., not significant.
Result
METTL16 is a critical repressor of ferroptosis in HCC cells
To investigate the potential involvement of m6A regulatory factors in ferroptosis, we performed qRT-PCR to evaluate changes in the expression of a series of m6A demethylases and methyltransferases in HCC cell lines in the presence of RSL3 (ferroptosis inducer), Fer-1 (ferroptosis inhibitor), or a combination of the two reagents. Among these factors, only METTL16 demonstrated concordant alterations during ferroptosis, with increased expression upon inhibition and decreased expression upon induction (Fig. 1A). Furthermore, we found that METTL16 expression was decreased in a dose-dependent manner following RSL3 treatment at both mRNA and protein levels (Fig. 1B).

METTL16 has been screened out and identified as the crucial repressor of ferroptosis in HCC. (A) The heatmap of relative mRNA fold change of m6A regulatory factors after ferroptotic stimulation (RSL3:1µM, Fer-1:10µM). (B) The levels of METTL16 following different doses of RSL3. (C-D) Intracellular ROS content of (C) Huh7 (RSL3:1µM, Fer-1:10µM) and (D) HCCLM3 (RSL3:2µM, Fer-1:10µM) were observed by fluorescence microscope (left) and quantified by flow cytometry (right). (E-F) Intracellular lipid peroxidation content of (E) Huh7 (RSL3:1µM, Fer-1:10µM) and (F) HCCLM3 (RSL3:2µM, Fer-1:10µM) were analyzed by fluorescence microscope (left) and quantified by flow cytometry (right). (G-H) Intracellular GSH levels, Iron content, and MDA levels of Huh7 (G) and HCCLM3 (H) were measured by corresponding assay kits. (I-J) Cell growth capacities of Huh7 (I) (RSL3:0.1µM, Fer-1:1µM) and HCCLM3 (J) (RSL3:0.5µM, Fer-1:1µM) were measured by the colony formation assay. Scale bar: 100 μm. Statistical significance was analyzed using one-way ANOVA with Tukey’s test (B, C, D, E and F, n = 3) and two-way ANOVA with Tukey’s test (G, H, I and J, n = 3)
Next, we evaluated the effect of METTL16 on typical ferroptosis-associated phenotypes, including ROS, lipid peroxidation, glutathione (GSH), malondialdehyde (MDA) and iron levels. Based on the endogenous expression level of METTL16 in each HCC cell line (Fig. S1A), Huh7 cells were used to observe overexpression effects and HCCLM3 cells were employed for knockdown effects. The final results indicated that METTL16 significantly decreased ROS generation and lipid peroxidation (Fig. 1C-F). As well, METTL16 reduced GSH depletion, intracellular iron overload and MDA (an intracellular product of lipid peroxidation) induced by RSL3 (Fig. 1G-H). Functionally, METTL16 overexpression attenuated RSL3-induced growth and clonogenicity inhibition, whereas METTL16 knockdown showed the opposite effects (Fig. S1B, I-J). To avoid the off-target effect of RNA interference, ferroptotic functional assays were also conducted in cells stably expressing shMETTL16-2 (shMETTL16 referred to cells expressing shMETTL16-1) (Fig. S1C-D). Notably, these effects were effectively reversed by Fer-1, confirming that the observed changes after METTL16 alteration were primarily driven by the induction of ferroptosis. In addition, multiple key ferroptosis-related markers were also analyzed. Upon treatment with RSL3, the expression levels of ferroptosis-suppressors, namely NRF2, GPX4 and FTH1, were increased in METTL16 overexpression cells compared to control cells, while the decreased expression levels of these factors were exhibited in METTL16 knockdown cells. Conversely, the ferroptosis-promoting markers ACSL4 and PTGS2 displayed inverse expression patterns (Fig. S1E-F).
Additionally, data from GSE104462 revealed that out of 24 m6A regulatory factors, including readers, only METTL16 displayed a significant negative correlation with ferroptosis, while others exhibited variable responses (Fig. S2A). The analysis of Ferroptosis-correlation ratios utilizing the Ferroptosis Signature [21] identified that METTL16 emerged as the top m6A regulatory factor displaying a negative correlation with ferroptosis (Fig. 2B). Gene Set Enrichment Analysis (GSEA_KEGG_FERROPTOSIS) further revealed enrichment of ferroptosis-related genes in HCC samples with low METTL16 expression (Fig. S2C). Subsequent analysis employing the Cancer Therapeutics Response Portal (CTRP) database suggested a positive correlation between METTL16 expression and resistance to several ferroptosis inducers, including ML162, RSL3 and ML210 (Fig. S2D). Interestingly, elevated METTL16 expression was also observed across different ferroptosis-resistant cell clones (Fig. S2E). Notedly, even in ferroptosis-resistant samples, METTL16 showed strong positive associations with ferroptosis suppressors, such as GPX4 and SLC40A1, while displaying a negative correlation with ferroptosis drivers, like NCOA4 (Fig. S2F). Taken together, these results suggested that METTL16 might serve as a critical negative regulator of ferroptosis in HCC.
METTL16 deletion facilitates ferroptosis and attenuates the hepatic tumorigenesis
To further elucidate the role of METTL16 in ferroptosis and liver cancer, we constructed xenograft tumor models by subcutaneously injecting HCCLM3 and Hepa1-6 cells with lentivirus-mediated METTL16 downregulation, assisted by an intraperitoneal injection of RSL3 or DMSO (Figs. 2A and S3A). The final data demonstrated that METTL16 downregulation hindered tumor growth (Figs. 2B-D and S3B-D) and enhanced the inhibitory effect of RSL3 on HCC xenograft tumors. Moreover, METTL16 downregulation exacerbated the RSL3-induced escalation in MDA levels in subcutaneous xenograft tumors (Fig. S3E). Immunohistochemistry (IHC) staining demonstrated that the RSL3-induced expression of 4-hydroxynonenal (4-HNE), a lipid peroxidation product of ferroptosis, was further amplified by METTL16 knockdown (Fig. 2E), revealing enhanced ferroptotic activity.

METTL16 deletion facilitates ferroptosis and attenuates hepatic tumorigenesis. (A) Schematic diagram of HCCLM3 subcutaneous xenograft tumor model establishment. (B-D) Representative images (B) and statistical data of weights (C) and volumes (D) of xenografts from HCCLM3 with or without the treatment of RSL3 (n = 6 mice per group). (E) Representative images of HE staining and METTL16, 4-HNE IHC staining of xenografts. Scale bar, 100 μm. (F) Schematic diagram of hepatocyte-specific Mettl16 knockout (Mettl16f/f, Alb-Cre) mouse construction. (G) Representative images of livers (n = 9 mice per group) from the murine MYC/Trp53−/− HCC model. Scale bar, 1 cm. (H) The liver weight (Mann-Whitney test), liver weight/body weight ratio, tumor number per liver and max tumor diameter (Mann-Whitney test) in the indicated mice. (I) AST (Mann-Whitney test) and ALT levels in mice serum. (J) The MDA concentration in mice livers. (K) Representative images of HE staining and Mettl16, 4-HNE IHC staining from mice livers. Scale bar, 2.5 mm (left) and 100 μm (right). Statistical significance was analyzed using one-way ANOVA with Tukey’s test (C), two-way ANOVA with Tukey’s test (D) and two-tailed Student’s t test (H, I and J)
Additionally, we established liver-specific conditional Mettl16 knockout mice (Mettl16f/f, Alb-Cre, Mettl16CKO) by crossing Mettl16-floxed (Mettl16f/f) mice with Alb-Cre mice (Fig. 2F). A substantial reduction in Mettl16 expression in the liver was confirmed (Fig. S3F). The Mettl16CKO mice exhibited normal liver function and morphology (Fig. S3G), with no disruption of liver homeostasis compared to Mettl16f/f (Mettl16WT) mice. This was evidenced by comparable body and liver weights, along with no significant differences in serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), triglycerides (TG), cholesterol (CHO), cholinesterase (CHE), and γ-glutamyltransferase (γ-GGT) (Fig. S3H-I). Using a murine HCC model featuring MYC overexpression and Trp53 knockout (MYC/Trp53-/-) via hydrodynamic tail vein injection (HDTVi) [22], we explored the effects of METTL16 deletion on HCC progression within a context highly relevant to human hepatocellular carcinoma [23]. Compared with Mettl16CKO mice, Mettl16WT mice displayed increased liver size and higher liver weight/body weight ratios, along with increased tumor lesion numbers and tumor diameters (Fig. 2G-H). Furthermore, Mettl16CKO mice showed milder liver damage (Fig. 2I) and enhanced ferroptotic activity in hepatic tumors, as characterized by increased levels of MDA concentration (Fig. 2J) and 4-HNE staining (Fig. 2K). In addition, we also introduced liver-specific conditional Mettl16 knockout (Mettl16f/f, AAV8-Cre) and control mice (Mettl16f/f, AAV8-Ctrl) utilizing AAV8-TBG-Cre/Ctrl virus in the MYC/Trp53-/- HCC model and conducted comparative evaluations, yielding consistent outcomes (Fig. S3J-N). Collectively, our findings revealed that METTL16 deficiency attenuates hepatic tumorigenesis and promotes ferroptosis, and its inhibitory effect on ferroptosis partially elucidates the oncogenic role of METTL16.
METTL16/IGF2BP2 facilitates the mRNA stability of SENP3 in an m6A-dependent manner
To explore the molecular mechanism by which METTL16 regulates ferroptosis and HCC, we initially predicted METTL16 co-expressed genes in the TCGA database through the GEPIA platform, with SENP3 emerging as a top candidate due to its high correlation coefficient and known responsiveness to ROS stimulation (Fig. 3A). Further analysis using two other public HCC databases consistently revealed a significant positive correlation between METTL16 and SENP3 expression (Fig. 3B). Importantly, the scRNA-seq dataset (GSE181515) further confirmed a concordant distribution pattern of Senp3 and Mettl16 at the single-cell level, particularly within HCC regions (Fig. S4A-B). Subsequent cell biological investigations indicated coherent alterations in the SENP3 and METTL16 expression profiles in HCC cells (Figs. 3C-D and S4C-D). MeRIP-seq data from GSE182607 demonstrated that METTL16 mediated the deposition of m6A within SENP3 mRNA (Fig. S4E). On the other hand, knocking down METTL3/14 expression had negligible effects on SENP3 expression, implicating the independent regulatory role of METTL16 (Fig. S4F). Furthermore, a dose-dependent decrease in SENP3 and METTL16 expression was observed following ferroptosis induction (Fig. S4G-H). Consistent trends were observed in the ferroptosis-related dataset GSE104462 (Fig. S4I) and ferroptosis-resistant dataset GSE262114 (Fig. S2F), hinting that METTL16 might regulate SENP3 mRNA stability in an m6A-dependent manner even under ferroptotic condition.

METTL16/IGF2BP2 facilitates the mRNA stability of SENP3 in an m6A-dependent manner. (A) The top 5 genes with the highest correlation coefficients linked to METTL16. (B) Correlation between METTL16 and SENP3 expression in public HCC databases. (C-D) The levels of METTL16 and SENP3 in the indicated cells. (E) Luciferase activities of the pGL3.0 plasmids containing SENP3 CDS and 3’UTR. (F) Schematic representation of the potential m6A sites and the mutation of m6A sites (T-A), as well as the primers used for MeRIP/RIP qPCR on SENP3 mRNA. (G) Luciferase activities of the pGL3.0 plasmids containing WT or MUT SENP3 CDS/3’UTR. (H) The relative m6A enrichment levels at the indicated site within the SENP3 mRNA via MeRIP-qPCR analysis. (I) RIP-qPCR was used to detect the binding of METTL16 and SENP3 at the indicated site. (J) The decay rate of SENP3 mRNA upon ActD (5 µg/mL) treatment at the indicated time points in PLC/PRF/5. (K-L) The SENP3 level was detected after multiply silencing different m6A readers in the indicated cells. (M) RIP-qPCR was used to detect the binding of IGF2BP2 and SENP3 at the indicated site upon METTL16 overexpression. (N) The decay rate of SENP3 mRNA in PLC/PRF/5. Statistical significance was analyzed using two-tailed Student’s t test (C, G, I, K and M, n = 3), one-way ANOVA with Tukey’s test (C, E and H, n = 3), two-way ANOVA with Tukey’s test (L, n = 3) and Pearson correlation analysis (B)
We conducted SENP3-CDS (coding sequence) and 3’UTR (untranslated region) luciferase reporter assays and the results showed increased luciferase activity in the wild-type METTL16 overexpression group (LV-METTL16) but not in the catalytically inactive mutant overexpression group (PP185/186AA and F187G [16], LV-METTL16 Mut) (Fig. 3E). Three potential m6A sites were found in both the CDS and 3’ UTR regions of SENP3 transcripts according to published literature [16, 24, 25] (Fig. 3F). As expected, relative luciferase activity increased in the wild-type group (SENP3 CDS/3’UTR WT) after METTL16 overexpression, but not in the mutant group (SENP3 CDS/3’UTR MUT) (Fig. 3G). Meanwhile, MeRIP-qPCR assays demonstrated increased m6A modification near potential sites (regions between forward primer and reverse primer of Primer 2# and Primer 3#) with METTL16 overexpression, confirmed by RIP-qPCR showing strong binding of METTL16 to m6A-modified regions of SENP3 transcripts (Figs. 3H-I and S5A). MAT2A, the well-established METTL16 mRNA target, was utilized as a positive control [24]. We further found that METTL16 overexpression increased SENP3 mRNA stability (Fig. 3J) using actinomycin D (ActD), while METTL16 knockdown reduced this stability (Fig. S5B), revealing that METTL16-mediated m6A modification enhances SENP3 mRNA stability, leading to its upregulation.
Next, we examined the effects of seven known stability-related m6A readers on SENP3 mRNA expression. Interestingly, only IGF2BP2 affected SENP3 expression in both cell lines (PLC/PRF/5 and Hep3B), whereas YTHDF1 and YTHDC2 showed inconsistency in the two cell lines (Figs. 3K and S5C). Meanwhile, we also observed that knockdown of IGF2BP2, but not of YTHDF1 or YTHDC2, attenuated SENP3’s upregulation induced by METTL16 overexpression (Figs. 3L and S5D). These data implied that IGF2BP2 might be the main m6A reader mediating METTL16’s effect on SENP3 expression. Furthermore, RIP-qPCR confirmed a direct interaction between IGF2BP2 and SENP3 mRNA (Figs. 3M and S5E) and IGF2BP2 knockdown decreased SENP3 mRNA stability even in METTL16 overexpression cells (Figs. 3N and S5F). In summary, our data uncovered that IGF2BP2 could bind to METTL16-methylated SENP3 mRNA and stabilize it in an m6A-dependent manner.
METTL16 promotes survival from ferroptosis by SENP3
Our investigations further focused on the role of SENP3 in METTL16-mediated anti-ferroptotic activity. Functional experiments revealed that SENP3 downregulation amplified the lipid peroxidation induced by RSL3-mediated ferroptosis and attenuated the inhibitory effects of METTL16 overexpression on ferroptosis (Fig. 4A). Similarly, SENP3 knockdown alleviated the inhibitory effects of METTL16 overexpression on the decreased levels of ROS, GSH depletion, iron and MDA (Fig. 4B-D). Conversely, SENP3 overexpression decreased ferroptotic activity and partially restricted the increase caused by METTL16 knockdown (Fig. S6A-D).

METTL16 exerts its anti-ferroptotic effect mediated by SENP3. (A-B) Intracellular lipid peroxidation (A) and ROS (B) content of Hep3B were observed (RSL3:1µM). (C) Intracellular lipid peroxidation and ROS content of Hep3B were quantified (RSL3:1µM). (D) Intracellular GSH levels, Iron content, and MDA levels of Hep3B (RSL3:1µM). (E) Representative images and statistical data of weights and volumes of xenografts from Hep3B (n = 5 mice per group). (F) Representative images of HE staining and METTL16, SENP3, MDA, 4-HNE IHC staining of xenografts from Hep3B. Scale bar, 100 μm. Statistical significance was analyzed using one-way ANOVA with Tukey’s test (C, D and E, n = 3 or 5), and two-way ANOVA with Tukey’s test (E, n = 5)
In vivo studies using murine xenograft tumor models demonstrated that METTL16 overexpression promoted tumor growth, which was suppressed by SENP3 knockdown (Fig. 4E). Conversely, METTL16 knockdown inhibited tumor growth, which was reversed by SENP3 overexpression (Fig. S6E). These results highlighted SENP3 as a downstream effector of METTL16 in HCC development. Consistent with the in vitro findings, SENP3 knockdown alleviated the decrease in MDA levels induced by METTL16 upregulation, whereas SENP3 overexpression reversed these effects in subcutaneous xenografts (Fig. S6F-G). IHC staining results for 4-HNE and MDA illustrated reduced lipid peroxidation products in xenografts overexpressing METTL16, and SENP3 knockdown reversed this effect (Fig. 4F). Taken together, SENP3 serves an essential role in enhancing the anti-ferroptotic activity of METTL16, consequently promoting tumorigenicity.
SENP3 de-SUMOylates lactotransferrin (LTF) and blocks its proteasome-dependent degradation
To explore the potential regulatory mechanism of SENP3 in ferroptosis, we performed immunoprecipitation (IP) coupled with Mass Spectrometry analysis in search of SENP3 interacting proteins. Among the candidates identified, Lactotransferrin (LTF) was selected for further investigation due to its reported roles in iron homeostasis regulation (Figs. 5A and S7A). Co-IP assays confirmed the interaction between SENP3 and LTF, both in ectopically expressed HEK293T cells and in endogenous HCC cell lysates (Fig. 5B-D). Moreover, we observed that LTF expression was changed with SENP3 overexpression or knockdown (Fig. 5E-F), implicating that SENP3 had a positive impact on LTF expression. Remarkably, SENP3-C532A, the catalytically inactive mutant form of SENP3 [26], failed to regulate LTF level (Fig. 5G), suggesting that the deSUMOylation of LTF protein by SENP3 might be involved in this regulation.

SENP3 de-SUMOylates LTF and blocks its proteasome-dependent degradation. (A) LTF was identified as an interacting protein of SENP3 through IP coupled with mass spectrometry, and bands containing LTF were observed after silver staining. (B-D) Co-immunoprecipitation assay analyzes the interaction of endogenous SENP3 and heterogenous expressed GFP-LTF in HEK293T cells (B), endogenous LTF and SENP3 in PLC/PRF/5 cells (C) and heterogenous expressed GFP-LTF with FLAG-SENP3 and MYC-SUMO3 in HEK293T cells (D). (E-G) The LTF and SENP3 expression were detected following GFP-LTF transfection. (H-I) The LTF expression was detected after simultaneous SENP3 knockdown (H) or SUMO3 overexpression (I) and MG132 treatment following GFP-LTF transfection. (J) The LTF expression was detected after simultaneous SUMO3 and SENP3 overexpression following GFP-LTF transfection. (K-L) The SUMOylation of GFP-LTF was determined in indicated cells using GFP-beads and western blotting using anti-MYC antibodies. (M) The LTF expression was detected after transfected with plasmids mutated with different predicted LTF SUMOylation modification sites (K-R). (N) The SUMOylation of GFP-LTF was determined after transfected with LTF K57R, K118R, K405R and 2KR (two lysine residues in 57 and 405 sites were simultaneously replaced by arginine) plasmids. (O) The SUMOylation of GFP-LTF was determined. (P-Q) The ubiquitination of GFP-LTF was determined using GFP-beads and western blotting using antibody against HA
It is well known that protein SUMOylation is involved in the ubiquitin-proteasome system (UPS) through SUMO-targeted ubiquitin ligases (STUbLs) [27, 28]. To exclude the involvement of the lysosomal degradation pathway, we examined LTF expression following the addition of MG132 or leupeptin into HCC cells. The results indicated that LTF degradation was primarily mediated through the ubiquitin-proteasome system (Fig. S7B). In the meantime, we found that SENP3 promoted LTF protein stability (Figs. 5H and S7C-D). Since SENP3 is a SUMO2/3-specific protease [26, 29,30,31], we then assessed whether SUMO3 had an impact on LTF expression. The western blot assay results indicated a dose-dependent decrease in LTF level upon SUMO3 overexpression, and this result could be blocked by MG132 (Fig. 5I). Interestingly, SENP3 overexpression attenuated the decrease in LTF levels induced by SUMO3 (Fig. 5J). More importantly, the Co-IP assay indicated an interaction between SUMO3 and LTF as shown in Fig. 5D. These results suggested that SUMOylation is involved in the regulation of SENP3 mediated LTF protein stability. Subsequently, the SUMOylation assay revealed that the SUMOylation level of LTF in HCC cells co-expressing LTF-GFP, MYC-SUMO3 and UBC9 was increased when SENP3 was knocked down (Fig. 5K). At the same time, SENP3 overexpression deconjugated SUMO3 from LTF, whereas the SENP3-C532A mutant did not affect the overall SUMOylation status of LTF (Fig. 5L). Additionally, we attempted to pinpoint potential SUMOylation sites within LTF by applying SUMOylation prediction tools (GPS-SUMO, SUMOplot™ and JASSA). As a result, three putative residues with the highest scores (K57, K405 and K118) were found (Fig. S7E). However, only mutations at K57 (K57R) or K405 (K405R) displayed a modest increase in LTF expression compared to the wild-type LTF, with SENP3 overexpression failing to elevate the levels of these mutants (Fig. 5M). As anticipated, the mutants K57R and K405R exhibited a conspicuous reduction in SUMOylation, with the double mutant variant (K57R/405R, 2KR) showing the same low level (Fig. 5N). Importantly, SENP3 overexpression did not affect the deSUMOylation of LTF 2KR (Fig. 5O).
SENP3 has been reported to stabilize specific proteins such as β-catenin by regulating SUMOylation and inhibiting proteasome-dependent degradation [26]. Through the ubiquitination assays, we found that the ubiquitination level of LTF was decreased with SENP3 overexpression, or increased with SENP3 knockdown (Fig. S7F and Fig. 5P). Intriguingly, overexpression of SENP3-C532A failed to modulate the level of LTF ubiquitination, suggesting that SENP3 indirectly influences LTF ubiquitination through its deSUMOylation activity (Fig. 5Q). These observations suggested a regulatory role for SENP3-mediated deSUMOylation at K57 and K405, countering proteasome-dependent degradation and suggesting a novel mechanism for modulating LTF protein stability.
METTL16/SENP3 inhibits ferroptosis via regulating the iron-chelation function of LTF
Considering the recognized neuroprotective potential of LTF as an iron chelator [32, 33], we conducted functional analyses to assess its role in HCC cells by manipulating Fe3+ concentrations using ferric ammonium citrate (FAC) and deferoxamine (DFO) to induce or inhibit ferroptosis. We took a specific focus on monitoring changes in the ferrous iron (Fe2+, active iron) and free iron in the labile iron pool (LIP) utilizing FerroOrange and Calcein-AM (The fluorescence intensity of Calcein-AM increases inversely with free iron content) probes. LTF overexpression decreased the FAC-induced intracellular accumulation of LIP and Fe2+ in PLC/PRF/5 cells, accompanied by decreases in lipid peroxidation and ROS levels. DFO reversed the effects of FAC and slightly enhanced this reduction caused by LTF through further diminishing intracellular iron levels (Figs. 6A and C and S8A-B). These findings implicated that LTF might function as a ferroptotic suppressor, potentially though reducing the LIP via iron chelation.

METTL16/SENP3 inhibits ferroptosis via regulating the iron-chelation function of LTF. (A, C) Intracellular lipid peroxidation content and LIP level of PLC/PRF/5 (FAC: 100 µg/mL, DFO: 100µM) with C11-BODIPY and Calcein-AM staining were observed by fluorescence microscope and quantified by flow cytometry. (B, D) Intracellular lipid peroxidation content and LIP level of PLC/PRF/5 (FAC: 100 µg/mL). (E) Schematic diagram of murine MYC/Trp53−/− HCC establishment with additional hydrodynamic tail vein injections of Mettl16-overpression plasmid or sgRNA against Senp3 or Ltf. (F) Representative images of livers (n = 7 mice per group) from the indicated mice. Scale bar, 1 cm. (G) The liver weight (Kruskal-Wallis with Dunn’s test), liver weight/body weight ratio (Kruskal-Wallis with Dunn’s test), tumor number per liver and max tumor diameter in the indicated mice were measured. (H) AST (Kruskal-Wallis with Dunn’s test) and ALT levels in mice serum. (I) The MDA concentration in mice livers. (J) Representative images of HE staining, Mettl16, Senp3, Ltf and 4-HNE IHC staining from mice livers. Scale bar, 2.5 mm (up) and 100 μm (down). Statistical significance was analyzed using one-way ANOVA with Tukey’s test (C, D, G, H and I, n = 3 or 7)
Further investigations into the regulatory interaction between LTF and METTL16/SENP3 have underscored that LTF effectively mitigated FAC-induced lipid peroxidation, iron overload (LIP and Fe2+), and MDA accumulation following SENP3 or METTL16 depletion. Moreover, LTF also reversed FAC-induced GSH depletion and growth inhibition in HCC cells with SENP3 knockdown. No significant alterations were observed in the LTF 2KR group, regardless of SENP3 alteration (Figs. 6B and D and S8C-K). These findings indicated that METTL16/SENP3 exerts an anti-ferroptosis effect that is intricately linked to the iron-chelating function of LTF in HCC cells.
Additionally, we utilized the MYC/Trp53-/- HCC model to investigate the in vivo effects of the METTL16-SENP3-LTF regulatory axis. By employing HDTVi, we delivered sgRNAs targeting Senp3 or Ltf, along with Mettl16 overexpression plasmids, to mouse livers (Fig. 6E). Mettl16 overexpression (Mettl16OE + sgNC) in the liver resulted in more severe tumors, which was attenuated by Senp3 knockout (Mettl16OE + sgSenp3) or Ltf depletion (Mettl16OE + sgLtf) (Fig. 6F-G), and liver damage was milder after Senp3 or Ltf depletion (Fig. 6H). The absence of Senp3 or Ltf (Mettl16OE + sgSenp3/sgLtf) slightly elevated the hepatic MDA levels (Fig. 6I) and increased the 4-HNE positive cells proportion as revealed by IHC staining (Fig. 6J) compared to Mettl16OE + sgNC group, supporting the hypothesis that METTL16/SENP3 attenuates ferroptotic activity via LTF to promote hepatocarcinogenesis.
The METTL16/SENP3 are significantly elevated in HCC patients
By analyzing two extensive cell survival- and fitness-associated genome-wide CRISPR-Cas9 knockout screening datasets [34, 35], we revealed the essential role of METTL16 in cancer cell survival and proliferation, particularly in HCC, in the human METTL family (Fig. S9A-B). In our HCC cohort (n = 37), elevated levels of METTL16 and SENP3 were observed in cancer tissues compared to paired normal tissues, with a highly positive correlation between their expression (Fig. 7A-B). Consistent results were also manifested in the CHCC cohort from Zhongshan Hospital (Fig. S9C-D). METTL16 and SENP3 were observed to be upregulated in advanced-stage HCC, particularly in stage III, compared to normal samples in the TCGA-LIHC cohort (Fig. S9E). Furthermore, high expression of METTL16 or SENP3 was correlated with lower overall survival rates in both TCGA-LIHC and CHCC cohorts (Fig. S9F-G). Interestingly, simultaneous high expression of METTL16 and SENP3 in the CHCC cohort was associated with the poorest overall survival (Fig. S9H).

METTL16 expression is positively correlated with SENP3 in HCC patients and increased METTL16 level induces ferroptosis resistance in PDOs. (A) The METTL16 (left) and SENP3 (right) mRNA expression in our cohort HCC samples. Data are presented as fold changes of METTL16 and SENP3 mRNA expression in liver tissue (C: hepatocarcinoma tissues; N, paired adjacent normal tissues). Paired line scatter plot showing METTL16 and SENP3 mRNA expression in cancer (orange dot) and adjacent normal tissues (green dot). (B) The relativity analysis of METTL16 and SENP3 in liver cancer tissues. (C) Representative images of IHC staining of METTL16 and SENP3 expression in human normal liver tissues and HCC specimens (n = 90 per group) from Tissue Microarray. Scar bar: 50 μm. (D) Quantification study of METTL16 and SENP3 expression levels in liver tissues from Tissue Microarray. (E) Representative images of IHC staining of METTL16, SENP3 and LTF expression in HCC specimens. Scar bar: 50 μm. (F) Correlational analysis of METTL16 and SENP3, SENP3 and LTF at protein levels obtained from IHC staining results. (G) Representative images of PDOs treated with Vehicle, RSL3 and Sorafenib. (H) Quantification of numbers of PDOs. Scale bar, 100 μm. Statistical significance was analyzed using paired two-tailed Student’s t test (A), Pearson correlation analysis (B and F), Chi-square test (D) and one-way ANOVA with Tukey’s test (H, n = 5)
Subsequently, IHC analysis of Tissue Microarray (TMA) confirmed higher METTL16 and SENP3 levels in hepatic cancer tissues, with no significant difference observed for LTF (Fig. 7C-D and S10A-B). A positive correlation was discovered between SENP3 and METTL16 expression, as well as between SENP3 and LTF expression (Fig. 7E-F). Upregulation of METTL16 and SENP3 protein expression was displayed within the CHCC cohort, and the positive correlations between SENP3 with METTL16 and SENP3 with LTF in HCC tissues were observed (Fig. S10C-E). Furthermore, we discovered a positive correlation between METTL16/SENP3 mRNA levels and preoperative alpha-fetoprotein (AFP) levels in HCC samples (Fig. S10F).
Given sorafenib’s dual roles as both a tumor suppressor and ferroptosis inducer in HCC, we conducted additional investigations into the impact of METTL16 with sorafenib treatment. As illustrated in Fig. S11A-D, METTL16 overexpression attenuated sorafenib-induced clonogenicity and growth inhibition, whereas METTL16 knockdown exerted the opposite effects. Elevated METTL16 expression levels were also observed in sorafenib-resistant primary liver cancer organoids and Hep3B cells, consistent with observations in samples resistant to RSL3 or erastin (Figs. S11E-F, S2F). Additionally, analysis of data from GSE109211 revealed the positive correlation between METTL16 expression levels and sorafenib resistance (Fig. S11G), and patients with high METTL16 expression were more likely to exhibit sorafenib resistance (Fig. S11H). Conversely, no significant variances were observed in response to TACE (Transcatheter Arterial Chemoembolization) (GSE104580), another classic therapeutic modality for HCC (Fig. S11H). Interestingly, a highly consistent expression pattern was also observed between SENP3 and METTL16 (Fig. S11E-F). Patients with high SENP3 expression were more prone to sorafenib resistance (Fig. S11I).
Finally, we evaluated the impact of METTL16 on ferroptosis resistance utilizing the preclinical model. Patients-derived organoids (PDOs) from primary liver cancer patients were categorized into high- and low-expression groups based on METTL16 levels in the cancerous tissue (Fig. S11J). Treatments with sorafenib and RSL3 efficiently restricted the size of METTL16Low HCC organoids (Fig. 7G-H). Interestingly, PDOs from the METTL16High group displayed resistance to the tumor-suppressive function of ferroptosis inducers, indicating varying responses to antitumor interventions. In summary, our investigations elucidated that heightened METTL16 and SENP3 expression in HCC tissues correlates with unfavorable prognostic outcomes and confers HCC cell ferroptosis resistance.
Discussion
Primary liver cancer poses a major threat to global health. It is projected that over three-quarters of patients will succumb to this disease by 2022, positioning liver cancer as the third leading cause of cancer-related mortality [1]. The efficacy of therapies against HCC varies due to the multifaceted molecular abnormalities within tumors. Emerging research suggests that ferroptosis holds promise for inhibiting tumor growth and overcoming drug resistance. However, the clinical application of targeted ferroptosis remains limited, highlighting the need for more effective strategies to induce ferroptosis and inhibit tumor development.
Current investigations into ferroptosis predominantly center on the complex interplay between lipid metabolism and oxidation-reduction reactions. For example, CPT1A boosted cellular antioxidant capacity and inhibited ferroptosis by reducing polyunsaturated fatty acids (PUFA) [36]. LGR4/Wnt/TCF7 signaling upregulated SLC7A11 to confer drug resistance [37]. However, studies on iron homeostatic regulation are still limited despite the critical role of iron metabolism in ferroptosis. Free iron in LIP triggers non-enzymatic Fenton reactions or acts as a cofactor for lipoxygenase and cytochrome P450 oxidoreductase to catalyze PUFA phospholipid peroxidation. Therefore, a comprehensive understanding of the iron imbalance in ferroptosis is essential for elucidating its underlying mechanisms. Our study elucidates a novel mechanism whereby METTL16, which is screened and identified experimentally, modulates the progression of ferroptosis by regulating intracellular LIP concentration, addressing the deficiency in research on ferroptosis-associated iron metabolism.
Recent research has stressed the significant function of m6A modification in different cellular processes, yet its connection to tumor ferroptosis remains elusive. Specifically, PRMT3 inhibition exacerbated lipid peroxidation by destabilizing GPX4 mRNA through a METTL14-YTHDF2 dependent mechanism [38]. Moreover, YTHDC1 epigenetically suppressed ferroptosis via controlling FSP1 [39]. METTL3/IGF2BP1 enhanced resistance to ferroptosis by stabilizing SLC7A11 mRNA [40]. METTL16, a methyltransferase, modulates mRNA stability and splicing processes distinct from METTL3/14 [16, 24, 25], with mRNA targets like PRDM15 [41], BCAT1/2 [42], SOGA1 [43] and FDX1 [44]. However, its impact on regulating ferroptosis in HCC remains unclear. In our study, elevated METTL16 expression in HCC tissues correlates strongly with ferroptosis resistance and adverse prognostic outcomes. Moreover, METTL16 potentiates HCC progression and regulates ferroptosis in an m6A-mediated manner.
SUMOylation, a crucial post-translational modification process similar to ubiquitylation, regulates various biological functions including protein stability, localization and interactions. SENP3 has been implicated in tumorigenesis and progression by deconjugating the SUMO2/3 precursors. The inhibitory effect of ROS on SENP3 degradation under basal conditions suggests that SENP3 is an oxidative stress sensor [45]. Moreover, evidence suggested the involvement of SENP3 in tumor immune evasion [46] and vascular remodeling [26] under the condition of oxidative stress. Nevertheless, its precise influence on HCC and ferroptosis remains unclear. In this investigation, we identified SENP3 as a target gene of m6A modification mediated by METTL16/IGF2BP2. We also revealed a dose-dependent reduction in SENP3 expression levels under ferroptosis conditions. Crucially, SENP3-C532A lost the ability to impede LTF degradation through deSUMOylation modification. Notably, SENP3 expression correlated strongly with METTL16 in both HCC and ferroptotic samples, along with significant associations with sorafenib sensitivity and adverse patient prognosis. These discoveries unveil the undisclosed roles of SENP3 in attenuating ferroptosis and fostering HCC progression.
LTF, an iron-binding glycoprotein belonging to the transferrin family, plays a crucial part in modulating iron metabolism [47]. LTF strengthened host immune defenses by sequestering the iron essential for bacterial growth [48]. Moreover, its iron-binding properties were promising as a safe therapeutic agent for Parkinson’s disease [32]. Furthermore, its absence in neurons exacerbated ferroptosis and neuronal dysfunction [33]. Remarkably, apo-LTF, unlike holo-LTF (another form of LTF with iron-binding and transporting properties), diminished ROS generation by capturing free iron and increasing SLC7A11 expression, thereby alleviating ferroptosis [49]. Consistently, we overexpressed LTF utilizing a plasmid directly in HCC cells, mimicking the functions of apo-LTF. Notably, LTF was effective in reducing hepatic iron accumulation and boosting hepatic iron output through ferroportin (FPN), inhibiting hepatocyte death [50]. Nevertheless, our study unveiled an interesting phenomenon where “devious” liver cancer cells exploit “virtuous” LTF to evade ferroptosis by sequestering free iron, similar to classic iron-chelator DFO. Our investigation revealed that SENP3-mediated deSUMOylation at specific sites effectively antagonized ubiquitin-mediated degradation of LTF. Mutation of these sites results in LTF being able to function independently of SENP3 regulation, enabling it to autonomously execute its antioxidant function. Remarkably, intracellular iron-regulatory protein 1 (IRP1) mRNA levels exhibited a notable decrease following LTF overexpression (data not shown). And IRP1 is crucial for regulating iron metabolism. Previous reports have indicated that LTF interacted with specific DNA sequences, facilitating transcriptional activation [51], but we did not find DNA sequences in the IRP1 promoter region that fully meet these requirements, and further investigations are necessary to elucidate the regulatory mechanisms involved in these processes.
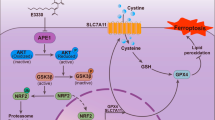
This investigation employed a multifaceted approach encompassing cancer cell lines, organoids, murine de novo MYC/Trp53-/- HCC and xenograft liver tumor models, to corroborate the crucial role of the METTL16-SENP3-LTF signaling axis in ferroptosis inhibition and HCC development. Our study delineated a regulatory cascade wherein METTL16 mediates SENP3 mRNA stability through precise m6A modifications, subsequently influencing SENP3-mediated de-SUMOylation modification of LTF, serving to fine-tune iron metabolism within HCC cells, ultimately inhibiting ferroptosis. Notedly, the augmented abundance of LTF protein enhances its capability to sequester free iron, thereby conferring a survival benefit to HCC cells (Fig. 8). Eventually, by underscoring the intricate interplay between RNA modification, deSUMOylation and ubiquitination, our research not only expands our understanding of the molecular basis of HCC but also presents promising avenues for therapeutic intervention. Given the potent antitumor effects of ferroptosis inducers, such as sorafenib, in HCC primary organoids with low METTL16 expression, the discernment of METTL16 expression profiles in patients with HCC holds promise for stratifying individuals based on their molecular signatures. This facilitates the application of personalized therapeutics aimed at impeding HCC progression. Consequently, our identification of METTL16 as a potential therapeutic target opens new avenues for the development of precision medicine approaches to optimize patient outcomes and enhance the overall effectiveness of HCC treatment.

METTL16-SENP3-LTF axis conferring ferroptosis resistance and facilitating tumorigenesis in liver cancer. Through enhancing SENP3 expression in an m6A-dependent manner, METTL16 emerges as a linchpin in the regulatory cascade involving SENP3-mediated LTF de-SUMOylation, shaping the dynamics of iron metabolism in HCC cells
Conclusion
In summary, our investigations elucidated that heightened METTL16 and SENP3 expression in HCC tissues correlated with unfavorable prognostic outcomes. Through comprehensive assays, we uncovered novel mechanisms where METTL16 increased SENP3 expression via m6A modification and SENP3 stabilized LTF protein through de-SUMOylation. These findings illustrated the role of the METTL16-SENP3-LTF axis in impacting ferroptosis and HCC progression, highlighting therapeutic potential by targeting this axis to disrupt iron homeostasis and ferroptosis susceptibility.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- CDS:
-
Coding sequences
- DFO:
-
Deferoxamine
- FAC:
-
Ferric ammonium citrate
- Fer-1:
-
Ferrostatin-1
- 4-HNE:
-
4-hydroxynonenal
- GSH:
-
Glutathione
- HCC:
-
Hepatocellular carcinoma
- HDTVi:
-
Hydrodynamic tail vein injection
- IHC:
-
Immunohistochemistry
- IP:
-
Immunoprecipitation
- LIP:
-
Labile iron pool
- LTF:
-
Lactotransferrin
- MDA:
-
Malondialdehyde
- m6A:
-
N6-methyladenosine
- PDOs:
-
Patient-derived organoids
- ROS:
-
Reactive Oxygen Species
- SENPs:
-
The SUMO proteases
- TKIs:
-
Tyrosine Kinase Inhibitors
- UTR:
-
The untranslated region
References
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024.
Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–62.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12(1):34.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014, 10(1).
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–96.
Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, Sun Y, Zeng F, Chen X, Deng G. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9(1):55.
Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–55.
Sun S, Shen J, Jiang J, Wang F, Min J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct Target Ther. 2023;8(1):372.
Tang D, Kroemer G, Kang R. Ferroptosis in hepatocellular carcinoma: from bench to bedside. Hepatology 2023.
He F, Zhang P, Liu J, Wang R, Kaufman RJ, Yaden BC, Karin M. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J Hepatol. 2023;79(2):362–77.
Tang J, Long G, Hu K, Xiao D, Liu S, Xiao L, Zhou L, Tao Y. Targeting USP8 inhibits O-GlcNAcylation of SLC7A11 to promote Ferroptosis of Hepatocellular Carcinoma via stabilization of OGT. Adv Sci (Weinh). 2023;10(33):e2302953.
Ding Z, Pan Y, Shang T, Jiang T, Lin Y, Yang C, Pang S, Cui X, Wang Y, Feng XF, et al. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat Commun. 2023;14(1):6269.
Qi Y-N, Liu Z, Hong L-L, Li P, Ling Z-Q. Methyltransferase-like proteins in cancer biology and potential therapeutic targeting. J Hematol Oncol. 2023;16(1):89.
Wang Y, Yang Y, Yang Y, Dang Y, Guo Z, Zhuang Q, Zheng X, Wang F, Cheng N, Liu X, et al. Hypoxia induces hepatocellular carcinoma metastasis via the HIF-1α/METTL16/lnc-CSMD1-7/RBFOX2 axis. iScience. 2023;26(12):108495.
Dai Y-Z, Liu Y-d, Li J, Chen M-T, Huang M, Wang F, Yang Q-S, Yuan J-H, Sun S-H. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m6A-dependent manner. Cell Mol Biol Lett. 2022;27(1):41.
Su R, Dong L, Li Y, Gao M, He PC, Liu W, Wei J, Zhao Z, Gao L, Han L, et al. METTL16 exerts an m6A-independent function to facilitate translation and tumorigenesis. Nat Cell Biol. 2022;24(2):205–16.
Liu P, Ge M, Hu J, Li X, Che L, Sun K, Cheng L, Huang Y, Pilo MG, Cigliano A, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology. 2017;66(1):167–81.
Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514(7522):380–4.
Zhang Z, Xie Z, Lin J, Sun Z, Li Z, Yu W, Zeng Y, Ye G, Li J, Ye F et al. The m6A methyltransferase METTL16 negatively regulates MCP1 expression in mesenchymal stem cells during monocyte recruitment. JCI Insight 2023, 8(6).
Gao Q, Zhu H, Dong L, Shi W, Chen R, Song Z, Huang C, Li J, Dong X, Zhou Y et al. Integrated Proteogenomic characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179(2).
Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, Wu J, Wang Y, Lin Z, Chen L, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol. 2022;24(7):1113–25.
Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, Lindblad KE, Maier B, Sia D, Puigvehi M, Miguela V, Casanova-Acebes M, Dhainaut M, et al. β-Catenin activation promotes Immune escape and resistance to Anti-PD-1 therapy in Hepatocellular Carcinoma. Cancer Discov. 2019;9(8):1124–41.
Yuen VW-H, Chiu DK-C, Law C-T, Cheu JW-S, Chan CY-K, Wong BP-Y, Goh C-C, Zhang MS, Xue HD-G, Tse AP-W, et al. Using mouse liver cancer models based on somatic genome editing to predict immune checkpoint inhibitor responses. J Hepatol. 2023;78(2):376–89.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase Intron Retention. Cell 2017, 169(5).
Satterwhite ER, Mansfield KD. RNA methyltransferase METTL16: targets and function. Wiley Interdiscip Rev RNA. 2022;13(2):e1681.
Cai Z, Wang Z, Yuan R, Cui M, Lao Y, Wang Y, Nie P, Shen L, Yi J, He B. Redox-sensitive enzyme SENP3 mediates vascular remodeling via de-SUMOylation of β-catenin and regulation of its stability. EBioMedicine. 2021;67:103386.
Schimmel J, Larsen KM, Matic I, van Hagen M, Cox J, Mann M, Andersen JS, Vertegaal ACO. The ubiquitin-proteasome system is a key component of the SUMO-2/3 cycle. Mol Cell Proteom. 2008;7(11):2107–22.
Geoffroy M-C, Hay RT. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat Rev Mol Cell Biol. 2009;10(8):564–8.
Liu K, Guo C, Lao Y, Yang J, Chen F, Zhao Y, Yang Y, Yang J, Yi J. A fine-tuning mechanism underlying self-control for autophagy: deSUMOylation of BECN1 by SENP3. Autophagy. 2020;16(6):975–90.
Zhang Y, Yang K, Yang J, Lao Y, Deng L, Deng G, Yi J, Sun X, Wang Q. SENP3 suppresses osteoclastogenesis by de-conjugating SUMO2/3 from IRF8 in bone marrow-derived monocytes. Cell Rep 2020, 30(6).
Jin S, He X, Ma L, Zhuang Z, Wang Y, Lin M, Cai S, Wei L, Wang Z, Zhao Z, et al. Suppression of ACE2 SUMOylation protects against SARS-CoV-2 infection through TOLLIP-mediated selective autophagy. Nat Commun. 2022;13(1):5204.
Xu S-F, Zhang Y-H, Wang S, Pang Z-Q, Fan Y-G, Li J-Y, Wang Z-Y, Guo C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019;21:101090.
Xiao Z, Shen D, Lan T, Wei C, Wu W, Sun Q, Luo Z, Chen W, Zhang Y, Hu L, et al. Reduction of lactoferrin aggravates neuronal ferroptosis after intracerebral hemorrhagic stroke in hyperglycemic mice. Redox Biol. 2022;50:102256.
Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–84.
Behan FM, Iorio F, Picco G, Gonçalves E, Beaver CM, Migliardi G, Santos R, Rao Y, Sassi F, Pinnelli M, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature. 2019;568(7753):511–6.
Ma L, Chen C, Zhao C, Li T, Ma L, Jiang J, Duan Z, Si Q, Chuang T-H, Xiang R, et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct Target Ther. 2024;9(1):64.
Zheng H, Liu J, Cheng Q, Zhang Q, Zhang Y, Jiang L, Huang Y, Li W, Zhao Y, Chen G et al. Targeted activation of ferroptosis in colorectal cancer via LGR4 targeting overcomes acquired drug resistance. Nat Cancer 2024.
Wang Y, Wang C, Guan X, Ma Y, Zhang S, Li F, Yin Y, Sun Z, Chen X, Yin H. PRMT3-Mediated arginine methylation of METTL14 promotes malignant progression and Treatment Resistance in Endometrial Carcinoma. Adv Sci (Weinh). 2023;10(36):e2303812.
Yuan S, Xi S, Weng H, Guo M-M, Zhang J-H, Yu Z-P, Zhang H, Yu Z, Xing Z, Liu M-Y, et al. YTHDC1 as a tumor progression suppressor through modulating FSP1-dependent ferroptosis suppression in lung cancer. Cell Death Differ. 2023;30(12):2477–90.
Liu L, He J, Sun G, Huang N, Bian Z, Xu C, Zhang Y, Cui Z, Xu W, Sun F, et al. The N6-methyladenosine modification enhances ferroptosis resistance through inhibiting SLC7A11 mRNA deadenylation in hepatoblastoma. Clin Transl Med. 2022;12(5):e778.
Liu N, Zhang J, Chen W, Ma W, Wu T. The RNA methyltransferase METTL16 enhances cholangiocarcinoma growth through PRDM15-mediated FGFR4 expression. J Exp Clin Cancer Res. 2023;42(1):263.
Han L, Dong L, Leung K, Zhao Z, Li Y, Gao L, Chen Z, Xue J, Qing Y, Li W et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell 2023, 30(1).
Wei W, Zhang Z-Y, Shi B, Cai Y, Zhang H-S, Sun C-L, Fei Y-F, Zhong W, Zhang S, Wang C, et al. METTL16 promotes glycolytic metabolism reprogramming and colorectal cancer progression. J Exp Clin Cancer Res. 2023;42(1):151.
Sun L, Zhang Y, Yang B, Sun S, Zhang P, Luo Z, Feng T, Cui Z, Zhu T, Li Y, et al. Lactylation of METTL16 promotes cuproptosis via m6A-modification on FDX1 mRNA in gastric cancer. Nat Commun. 2023;14(1):6523.
Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ETH, Chen Y, Cang H, Li H, Shi G, et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO J. 2009;28(18):2748–62.
Yu X, Lao Y, Teng X-L, Li S, Zhou Y, Wang F, Guo X, Deng S, Chang Y, Wu X, et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat Commun. 2018;9(1):3157.
Sienkiewicz M, Jaśkiewicz A, Tarasiuk A, Fichna J. Lactoferrin: an overview of its main functions, immunomodulatory and antimicrobial role, and clinical significance. Crit Rev Food Sci Nutr. 2022;62(22):6016–33.
Singh A, Ahmad N, Varadarajan A, Vikram N, Singh TP, Sharma S, Sharma P. Lactoferrin, a potential iron-chelator as an adjunct treatment for mucormycosis - A comprehensive review. Int J Biol Macromol. 2021;187:988–98.
Zhang Z, Lu M, Chen C, Tong X, Li Y, Yang K, Lv H, Xu J, Qin L. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. 2021;11(7):3167–82.
Guo C, Xue H, Guo T, Zhang W, Xuan W-Q, Ren Y-T, Wang D, Chen Y-H, Meng Y-H, Gao H-L, et al. Recombinant human lactoferrin attenuates the progression of hepatosteatosis and hepatocellular death by regulating iron and lipid homeostasis in ob/ob mice. Food Funct. 2020;11(8):7183–96.
He J, Furmanski P. Sequence specificity and transcriptional activation in the binding of lactoferrin to DNA. Nature. 1995;373(6516):721–4.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 82372706 and No. 82273015) and the Health and Family Planning Scientific Research Project of Pudong New Area Health Committee (No. PW2022E-02). Graphical abstract was created with BioRender (https://biorender.com).
Author information
Authors and Affiliations
Contributions
WJL performed the experiments, analyzed the data and drafted the manuscript; WJ and XMX assisted in the animal experiments; XMX supported the hepatocellular carcinoma organoid experiments; LYD and GY commented on the study, supervised the study and revised the manuscript. All authors have commented on the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was reviewed and approved by the Ethical Committee of Shanghai East Hospital, School of Medicine, Tongji University, China (No. 2023 − 212) and all animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health (NIH) and the study was reviewed and approved by the Institutional Animal Care and Use Committee at Tongji University with license number TJ-HB-LAC-2023-19.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Xiu, M., Wang, J. et al. METTL16-SENP3-LTF axis confers ferroptosis resistance and facilitates tumorigenesis in hepatocellular carcinoma. J Hematol Oncol 17, 78 (2024). https://doi.org/10.1186/s13045-024-01599-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01599-6