
Abstract
Background
Macrophage-based cell therapy is promising in solid tumors, but the efficient acquisition of macrophages remains a challenge. Induced pluripotent stem cell (iPSC)-induced macrophages are a valuable source, but time-consuming and costly. The application of reprogramming technologies allows for the generation of macrophages from somatic cells, thereby facilitating the advancement of cell-based therapies for numerous malignant diseases.
Methods
The composition of CD45+ myeloid-like cell complex (MCC) and induced macrophage (iMac) were analyzed by flow cytometry and single-cell RNA sequencing. The engraftment capacity of CD45+ MCC was evaluated by two transplantation assays. Regulation of c-Myc on MafB was evaluated by ChIP-qPCR and promoter reporter and dual luciferase assays. The phenotype and phagocytosis of iMac were explored by flow cytometry and immunofluorescence. Leukemia, breast cancer, and patient-derived tumor xenograft models were used to explore the anti-tumor function of iMac.
Results
Here we report on the establishment of a novel methodology allowing for reprogramming fibroblasts into functional macrophages with phagocytic activity by c-Myc overexpression. Fibroblasts with ectopic expression of c-Myc in iPSC medium rapidly generated CD45+ MCC intermediates with engraftment capacity as well as the repopulation of distinct hematopoietic compartments. MCC intermediates were stably maintained in iPSC medium and continuously generated functional and highly pure iMac just by M-CSF cytokine stimulation. Single-cell transcriptomic analysis of MCC intermediates revealed that c-Myc up-regulated the expression of MafB, a major regulator of macrophage differentiation, to promote macrophage differentiation. Characterization of the iMac activity showed NF-κB signaling activation and a pro-inflammatory phenotype. iMac cells displayed significantly increased in vivo persistence and inhibition of tumor progression in leukemia, breast cancer, and patient-derived tumor xenograft models.
Conclusions
Our findings demonstrate that c-Myc alone is enough to reprogram fibroblasts into functional macrophages, supporting that c-Myc reprogramming strategy of fibroblasts can help circumvent long-standing obstacles to gaining “off-the-shelf” macrophages for anti-cancer immunotherapy.
Similar content being viewed by others


Introduction
Immune cell-based therapies have shown promise in potentially curing malignant cancers with chemo- and radio-resistance. Although chimeric antigen receptor (CAR)-T cell therapy has demonstrated potency against hematologic malignancies, the efficacy of CAR-T cells against solid tumors is limited [1, 2]. Macrophages are central effectors and regulators of the innate immune system, capable of phagocytosis, secretion of pro-inflammatory factors, and tumor infiltration [3]. Macrophages have now shown great potential as professional antigen-presenting cells for CAR-based therapies [4,5,6]. However, the collection and culture of macrophages in vitro is challenging.
The technological discovery of macrophage differentiation from induced pluripotent stem cells (iPSC-Mac) opened the door for the future application of autologous immune cell-based therapies in the clinic [6,7,8,9]. In this regard, the methodologies of lineage conversion have emerged as a complementary approach to iPSC-Mac technology [10, 11]. SOX2/miR-125b overexpression induced the conversion of fibroblasts into CD34+ immature hematopoietic progenitor-like cells, which could be specifically differentiated toward macrophage-like cells with phagocytic activity [10]. This indicates that overexpression of pluripotent stem cell-inducing factor(s) is possible to reprogram fibroblasts into macrophages.
c-Myc, together with Sox2, Oct4, and Klf4, reprograms fibroblasts into iPSCs [12, 13]. c-Myc controls macrophage proliferation [14, 15] and polarization [16]. In addition, v-raf oncogene converts Eµ-Myc transgenic B cells into macrophages [17]. Here we report a tractable approach in which the conditional expression of c-Myc in mouse embryonic fibroblasts (MEFs) cultured with iPSC medium induces reprogramming of fibroblasts into CD45+ myeloid-like cell complex (MCC) intermediates capable of engraftment and repopulation of distant hematopoietic compartments. MCC intermediates were stably maintained in iPSC medium and continuously generated functional and highly pure-induced macrophage (iMac) in macrophage differentiation medium. Profiling of MCC intermediates identified distinct subsets of macrophages, and characterization of the iMac’s activity mainly showed a pro-inflammatory phenotype. Moreover, transplantation of iMac significantly reduced leukemia and breast cancer progression in animal tumor xenograft models.
Materials and methods
Cells
E13.5 MEFs and murine induced pluripotent stem cells (iPSCs) were generated from indicated genotypes as we previously described [13]. HEK293T, 4T1, Raji, and Reh cells were obtained from American Type Culture Collection (ATCC, Manassas, Virginia, U.S.). Platinum-E retroviral packaging cells were obtained from Cell Biolabs, San Diego, CA. Sca-1 GFP MEFs were generated from Sca-1 GFP transgenic mice (The Jackson Laboratory, #012643) [18]. MCC intermediates and iMac cells were derived from MEFs in this study. Mouse peripheral blood (PB) and bone marrow mononuclear cells were generated from indicated genotypes in this study.
Cell culture
MEF, HEK293T, Platinum-E, and 4T1 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS). Raji and Reh cells were cultured in RPMI Medium 1640 basic (Gibco) supplemented with 10% FBS and 1% PS. MCC intermediate cells and iPSCs were cultured in the iPSC medium [Knockout DMEM, plus 20% FBS, 1% PS, mouse LIF (1000 U/ml), non-essential amino acids (NEAA, 100×), L-glutamine (100×), and β-mercaptoethanol (100×)]. iMac cells were cultured in macrophage differentiation medium [RPMI Medium 1640 basic (Gibco) supplemented with 10% FBS, 1% PS, and 100 ng/ml M-CSF (PeproTech)].
Generation of virus
The retroviral constructs pMXs-GFP, pMXs-Klf4, pMXs-Sox2, pMXs-Oct4, and pMXs-c-Myc were purchased from Addgene (Cambridge, MA). To produce retrovirus, the indicated plasmid was transfected into Platinum-E retroviral packaging cells using Hieff Trans® Liposomal Transfection Reagent (Yeasen) as previously described [13]. To produce lentivirus, the indicated plasmid with pMD2.G (Addgene #12259) and psPAX2 (Addgene #12260) was transfected into HEK293T cells. The virus supernatant was collected at 48 h and 72 h after transfection.
Reprogramming fibroblasts into macrophages
MEFs were seeded on a 6-well plate and transfected with the virus in the medium supplemented with 8 µg/ml polybrene. After transfection for 48 h, the culture medium was changed into iPSC medium. MEFs with overexpression of c-Myc were cultured in the iPSC medium for 7 days. Then the cells in suspension (named P0 MCC intermediates) were collected and resuspended in iPSC medium to get MCC intermediates that were passaged every 3 days. For macrophage differentiation, the iPSC medium was removed and replaced with macrophage differentiation medium on day 10. On day 15, cells (named induced macrophages, iMac) were harvested and analyzed.
Compared to the iPSC medium, erythroid culture medium (EC-M) and granulocyte culture medium (GC-M) were used. EC-M: IMDM (50%) and Ham’s F12 (50%) plus ITS-X (100x), chemically defined lipid concentrate (100x), L-glutamine (100x), ascorbic acid (0.05 mg/ml), BSA (5 mg/ml), 1-thioglycerol (200 µM), SCF (100 ng/ml), IL-3 (10 ng/ml), erythropoietin (2 U/ml), IGF-1 (40 ng/ml), dexamethasone (1 µM), and holo transferrin (100 µg/ml). GC-M: IMDM (50%) and Ham’s F12 (50%), plus ITS-X (100x), chemically defined lipid concentrate (100x), L-glutamine (100x), ascorbic acid (0.05 mg/ml), BSA (5 mg/ml), 1-thioglycerol (200 µM), thrombopoietin (100 ng/ml), SCF (100 ng/ml), Flt3 ligand (100 ng/ml), granulocyte-colony stimulating factor (G-CSF) (100 ng/ml), and IL-3 (10 ng/ml).
Macrophage polarization and cytokine analysis
Mouse bone marrow cells were collected, lysed using red blood lysis buffer, and prepared into a single-cell suspension. Then, cells were cultured in RPIM1640, 10% FBS, 1% PS, and 20 ng/ml M-CSF to differentiate into macrophages. For polarization, iMac and bone marrow-derived macrophages were induced with LPS (100 ng/ml) and IFN-γ (20 ng/ml), or IL-4 (20 ng/ml) for 24–48 h. Cell supernatants were collected to detect cytokine levels by Luminex-MultiDTM, and cells were digested and collected for flow cytometry.
Flow cytometry
Cells were collected and separated into single cells using 100 μm cell strainers. Cells were collected, separated into single cells, and resuspended in 100 µl flow cytometry staining buffer. For cell surface markers, cells were incubated with antibodies at 4℃ for 30 min. For intracellular staining, cells were fixed and permeabilized with Intracellular Fixation and Permeabilization Buffer Set (eBioscience), then incubated with antibodies for 30 min. Samples were detected by BD Canto Plus or sorted by Beckman Coulter MoFlo XDP. FlowJo v 10.7.2 was used for data analysis. The following antibodies were used: anti-mouse CD16/32 (clone S17011E, Biolegend), APC-Cy7 anti-mouse CD45 (clone 30-F11, Biolegend), PE anti-mouse Sca-1 (clone E13-161.7, Biolegend), APC-eFluor™ 780 anti-mouse c-Kit (clone 2B8, eBioscience), PE-Cy7 anti-mouse/human B220 (clone RA3-6B2, Biolegend), PE anti-mouse Lineage Cocktail (Biolegend), PE-Cy7 anti-mouse CD3 (clone 17A2, eBioscience), APC anti-mouse CD11b (clone M1/70, eBioscience), Super Bright™ 436 anti-mouse F4/80 (clone BM8, Invitrogen), FITC anti-mouse CD11c (clone N418, eBioscience), Pacific Blue anti-mouse CD69 (clone H1.2F3, eBioscience), PE anti-mouse CD49b (clone DX5, eBioscience), PE anti-mouse CD206 (clone MR6F3, Invitrogen), Alexa Fluor™ 488 anti-mouse iNOS (clone CXNFT, eBioscience), PE-Cy7 anti-human/mouse Arginase1 (clone A1exF5, eBioscience), FITC anti-mouse CD107a (clone 1D4B, Biolegend), PE anti-human CD7 (clone CD7-6B7, Biolegend), APC-Cy7 anti-human CD45 (clone 2D1, Biolegend).
Cell transplantation
For cell transplantation, 6-8-week-old female C57BL/6J mice [Shanghai Laboratory Animal Center (SLAC), n = 5 per group] were used. MEFs were co-transfected with pMXs-GFP and pMXs-c-Myc to get MCC-GFP. In the first transplantation, a total of 1 × 106 MCC intermediate cells (labeled with GFP) or fetal liver mononuclear cells were first injected into lethally irradiated recipient mice via the tail vein. The peripheral blood of the mice was collected and analyzed by flow cytometry at indicated time points. Three months after the first transplantation, cells from different organs of one batch of mice were isolated and analyzed by flow cytometry. In the second transplantation, 1 × 107 bone marrow cells from recipient mice of the first transplantation were injected into lethally irradiated recipient mice via the tail vein. One month later, the peripheral blood of the mice was collected, and bone marrow was analyzed for lineage constitution.
Tumor xenograft animal models
In Reh leukemia animal model, 6-8-week-old female severe combined immunodeficiency (SCID) (SLAC, n = 5 per group) mice were used. SCID mice were irradiated with 2 Gy before transplantation. A total of 2 × 105 GFP-labeled Reh (Reh-GFP) cells with different quantities of MCC intermediates were co-injected into animals via the tail vein. On day 28 after injection, mice were euthanized when leukemia symptoms developed. The proportion of leukemia cells in BM and spleen was measured by flow cytometry.
In 4T1 breast cancer xenograft model, 6-8-week-old female BALB/c mice (SLAC, n = 5 per group) were used. A total of 2 × 105 Luciferase-labeled 4T1 (4T1-Luc) cells were injected into mice via the tail vein. On day 5, mice were randomized and injected with 1 × 106 iMac (from mice of the same strain) or vehicle (PBS) via the tail vein. Bioluminescence intensity (BLI) followed tumor growth using the IVIS Lumina imaging station (Caliper Life Sciences). Mouse lung tissues were analyzed by hematoxylin and eosin staining, immunohistochemistry and flow cytometry.
Patient-derived tumor xenograft (PDX) model
In the PDX model, 6-week-old male NCG mice (Gem Pharmatech Co., Ltd., Nanjing, China, n = 5 per group) were injected with 1 × 106 PDX cells by tail vein. On day 14 and 21, 2 × 106 iMac cells or vehicle (PBS) were injected into animals by tail vein. On day 35 after injection, mice were euthanized when leukemia symptoms developed. The proportion of leukemia cells in BM and spleen was measured by flow cytometry.
Western blotting assay
Cells were collected and lysed in RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 2 mM Na3VO4, 5 mM NaF, 1% Triton X-100) with a Protease inhibitor cocktail (Invitrogen) and eluted by SDS loading buffer. The proteins were resolved in SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Blots were incubated with primary antibodies overnight at 4℃ followed by HRP-conjugated species-specific antibodies. The membrane was visualized and analyzed by an imaging system (Bio-Rad Laboratories) after incubation with appropriate primary and secondary antibodies.
Wright-Giemsa stain
Cells were collected, then centrifuged and immobilized on the slides. Slides were stained with a Wright-Giemsa Stain kit (BaSO, Zhuhai) as previously described [19]. Images were acquired with an upright light microscope.
Hematopoietic colony-forming cell assay
Hematopoietic colony-forming cell (CFC) assay was performed in 35-mm low adherent plastic dishes using MethoCult GF M3434 (StemCell Technologies) according to the manufacturer’s instructions. Briefly, cells were pre-passed through a 100 µM cell strainer, and 3000 single cells were plated in a well. Colony-forming units (CFU) were identified after 14 days of incubation according to their colony morphology as granulocyte (CFU-G), macrophage (CFU-M), granulocyte-macrophage (CFU-GM), granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM), and erythroid (CFU-E).
shRNA knockdown
The sequences of all small hairpin RNA (shRNA) oligonucleotides were synthesized and cloned into an FH1TUTG vector. The murine MafB shRNA sequence (5’-CTACAAGGTCAAGTGCGAGAA-3’) was from Sigma. Virus packaging was performed as previously described. MEFs were infected with pMXs-c-Myc and FH1TUTG-shMafB and induced with doxycycline hyclate (2 µg/ml).
Hematoxylin and eosin (H&E) staining
Frozen sections were fixed with acetone, treated with hematoxylin and 1% ethanol hydrochloride, and then washed. The sections were dehydrated and sealed after being treated with Eosin Y solution.
Immunohistochemistry
Tissue sections were dewaxed with xylene and ethanol. Endogenous peroxidase was inactivated with 3% H2O2. For antigen retrieval, tissue sections were treated with citrate buffer. Tissue sections were blocked for 1 h at room temperature. After that, tissue sections were incubated overnight at 4℃ with primary antibody. Tissue sections were incubated with HRP-conjugated secondary antibody and DAB staining solution.
Single-cell RNA sequencing and analysis
MCC intermediates and iMac cells were collected for single-cell RNA sequencing. DNBelab C4 System or Illumina NovaSeq 6000 was utilized for scRNA-seq library preparation according to the manufacturer’s protocol. Raw sequencing data were aligned and quantified using the CellRanger. The output was converted to a Seurat object using the R Seurat package for downstream analysis. To remove low-quality cells, we used the following criteria: less than 1000 unique genes expressed, or more than 20% of reads mapping to mitochondria. The gene expression matrix of the remaining cells was normalized through the function of ScaleData. Cell clustering was performed using the FindClusters, and the clusters were annotated by the expression of canonical marker genes. FindAllMarkers was used to define marker genes for each cluster. Functional enrichment analysis was done by clusterProfiler. Monecle2 was used to infer the potential developmental trajectory. Enriched motif for each cell type was done using pySCENIC. Pathway activation score was calculated by GSVA on average expression on each subtype.
Bulk RNA sequencing and analysis
Total RNA (iMac, MCC, MEF, iPSC, BM, and PB CD45+ cells) was extracted using TRIzol (Thermo Fisher Scientific) and sequenced by Mingma Technologies Co., Ltd. at Shanghai according to the manufacturer’s instructions. Libraries were sequenced on Illumina NovaSeq 6000 or Illumina HiSeqX Ten platform. Raw reads were mapped to the mouse genome (mm10) and GENECODE vM25 using STAR. Quantification for each expressed gene was performed using featureCounts from Rsubread. Differentially expressed genes were calculated by DESeq2 with cutoffs as follows: Padj < 0.05 and absolute log2 fold change > 1. Functional annotations were done using enrichKEGG from clusterProfiler package.
Quantitative RT-PCR (qRT-PCR)
Cells were collected, and RNA was extracted with TRIzol. The eluted RNA was reverse-transcribed using Hifair II 1st Strand cDNA Synthesis SuperMix for qPCR (11123ES10, YEASEN) according to the manufacturer’s instructions. Quantitative RT-PCR was performed with qPCR SYBR Green Master Mix (11201ES03, YEASEN), and results were analyzed using BioRad CFX Maestro Software. Primers were as follows: 5’-CCAAGTGCTGCCGTCATTTTC-3’ and 5’-GGCTCGCAGGGATGATTTCAA-3’ for Cxcl10, 5’-GAGCCTGGCTAGGAAGGTG-3’ and 5’-TGCTGAAACCAATAAGGAACCTG-3’ for Fpr2, 5’-ATGCCAATCACTCGAATGCG-3’ and 5’-TTGTATCGGCCTGTGTGAATG-3’ for Irf1.
Chromatin immunoprecipitations (ChIP)-qPCR
For each ChIP assay, 1 × 106 cells were harvested and followed the manufacturer’s protocol of Simple ChIP Plus Enzymatic Chromatin IP Kit (Magnetic Beads, Cell Signaling Technology, 9005S). ChIP DNA was purified and subsequently quantified by qPCR. Primers: 5’-ggggccacaagagtcctcttca-3’ and 5’-gcgcttcccgagtacgcg-3’.
Promoter reporter and dual luciferase assays
The MafB promoter was amplified by PCR and then inserted into a pGL3.0-Basic vector. pGL3.0 basic-MafB promoter was co-transfected with or without c-Myc WT or mutants according to the manufacturer’s protocol. For normalization of transfection efficiency, the pRL-TK (Renilla luciferase) reporter plasmid was added to each transfection. Luciferase activity was quantified with a Dual-Specific Luciferase Assay Kit (E1910; Promega).
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 20 min at room temperature. Then, cells were penetrated with 0.3% Triton X-100 in PBS for 20 min and blocked with 10% goat serum for 60 min. For the immunofluorescence assay, cells were incubated with primary antibody overnight at 4℃ followed by secondary antibody for 1 h at room temperature. Nuclei were counterstained with DAPI. Images were captured by Leica SP8 confocal microscope.
Fluorescent beads phagocytosis assay
Fluroresbrite Polychromatic Red 1.0-µm Latex beads (Polyscience Inc., 18660) were added to the cell culture medium and incubated with cells for 24 h. Then, cells were washed several times and analyzed by Leica SP8 confocal microscope or flow cytometry.
Tumor cell phagocytosis assay
Raji and Reh cell lines were washed with PBS, resuspended in 1 ml CFSE staining solution, and incubated for 20 min at 37℃. To remove free dye remaining in the solution, 5 ml complete culture medium was added to the cells and incubated for 5 min. Cells were centrifuged and resuspended in culture medium. MEFs were pre-stained with DiD dye, then iMac and MEF cells were co-incubated with tumor cells at a ratio of 1:4 at 37℃ for 2 h. For flow cytometry, cells in iMac group were harvested and stained with CD11b antibody, and the percentage of phagocytosis was the ratio of CFSE-positive cells in the CD11b+ population. In MEF group, cells were harvested, and the percentage of phagocytosis was the ratio of CFSE-positive cells in the DiD+ population.
Statistical analysis
All grouped data are presented as mean ± SEM. All in vitro experiments were repeated at least three times. Two-tailed unpaired Student’s t test was used to analyze the data from two groups, and one-way ANOVA was used to analyze multiple groups. For the survival analysis, Kaplan-Meier survival curves were analyzed by using log-rank test comparing the different mouse groups. Data were considered statistically significant when P values were less than or equal to 0.05 in this study (*P < 0.05, ** P < 0.01, *** P < 0.001). Statistical analyses were performed with GraphPad Prism 8.2.1.
Results
Ectopic expression of c-Myc reprograms fibroblasts into macrophages
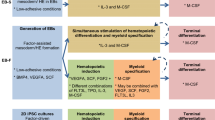
To establish a novel lineage conversion strategy for the generation of functional macrophages, we investigated whether overexpression of pluripotency-related genes could reprogram fibroblasts into macrophages. As shown in Fig. 1A, we overexpressed c-Myc, Klf4, Sox2, and Oct4 in mouse embryonic fibroblasts (MEFs), and MEFs were cultured in iPSC culture medium. After 7 days, suspension cells were generated from MEFs. Flow cytometry identified more than 90% of these cells as CD45+ cells (Fig. 1B). We plated ~ 1 × 105 MEFs in a well and obtained ~ 32 × 105 CD45+ P0 cells on day 7 (Fig. 1C). Compared to c-Myc, a few CD45+ cells were induced by ectopic expression of Oct4, Sox2, or Klf4 (Fig. 1C and S1A). In addition, compared to MEF medium (MEF-M), erythroid culture medium (EC-M), and granulocyte culture medium (GC-M), iPSC medium was the most effective to induce the conversion of fibroblasts (Fig. 1D). To exclude the contamination of hematopoietic cells in MEFs, MEFs were depleted of CD45+ cells, and the number and proportion of CD45+ cells were similar to those in the undepleted group (Figure S1B and S1C). To identify the occurrence of transdifferentiation, we overexpressed c-Myc in MEFs from Sca-1-GFP transgenic mice. Sca-1-GFP was not expressed in MEFs but expressed in generated suspension cells, demonstrating the presence of conversion (Figure S1D). In addition, we assessed the expression of pluripotency markers Sox2 and Oct4 and revealed that they were not expressed on day 2, 4, and 7 (Figure S1E), suggesting that overexpression of c-Myc alone did not lead to iPSCs production. Then, P0 cells were collected and cultured for another 3 days to get progeny 1 (P1) cell complex. The P1 cells were further cultured in the macrophage differentiation medium for 5 days to differentiate into high-pure induced macrophages (iMac) (Fig. 1A). Compared with P1 cells, ~ 92% of iMac were double positive for both macrophage markers CD11b and F4/80 (Fig. 1E and F). These data demonstrate that overexpression of c-Myc reprograms MEFs into macrophage-biased hematopoietic-like cells, which can be further differentiated into iMac.
To investigate the components of P1 cells, flow cytometry was performed and identified that there are ~ 1% LSK (Lin− Sca-1+ c-Kit+) cells, which are enriched for hematopoietic progenitor-like cells (HPCs) [20] (Fig. 1G, S1F-S1G), and ~ 30% myeloid progenitor (MP, Lin− Sca-1− c-Kit+) cells in Lin− cells (Fig. 1G). Interestingly, we found that c-Kit+Sca-1+ cells accounted for a large part of P1 cells, almost all of which expressed CD11b (Figure S1H and S1I). P1 cells expressed the markers of lymphocytic and myeloid lineages of blood cell types (Fig. 1H and S1J) with corresponding cell morphology (Fig. 1I), illustrating that CD45+ P1 cells may possess the functional hematopoietic potential of multiple blood cell types. We further performed hematopoietic colony-forming cell assays and found that similar to murine bone marrow (BM)-derived progenitors, P1 cells gave rise to all types of well-defined myeloid colonies, including CFU-granulocyte macrophage (CFU-GM), CFU-granulocyte (CFU-G), and CFU-macrophage (CFU-M) (Fig. 1J and K), indicating the developmental potential of myeloid lineages of P1 cells. Besides, compared to BM-derived progenitors, CD45+ cells gave rise to more CFU-GM, CFU-M, and CFU-E colonies. Therefore, P0 and P1 cells generated from fibroblasts were named myeloid-like cell complex (MCC) intermediates.
To further evaluate the ability of MCC intermediates, we performed engraftment and differentiation in vivo. GFP-tagged MEF-derived MCC intermediate cells were isolated and transplanted into C57BL/6J mice pre-treated with 8 Gy irradiation as shown in Fig. 1L. MCC intermediates engrafted and continued to proliferate in animal recipients in the first three months after transplantation, as indicated by GFP-tagged cells (Fig. 1M and S1K). MCC intermediates were also able to engraft into other organs, such as bone marrow, spleen, liver, and lymph gland (Fig. 1N). After three months, a second transplantation was conducted to further investigate the potential and safety of MCC intermediates. Engraftment of cells in the second transplantation was also detected, and there was no significant difference in peripheral blood (PB) reconstitution compared to the first one (Fig. 1O). We tested the proportion of multiple blood cell lineages in the GFP-tagged MCC intermediates after the second transplantation and found that the MCC intermediates could also be differentiated into lymphocytic and myeloid lineages in vivo, which were composed of ~ 30% B220+ B cells, 20% CD3+ T cells, and 50% CD11b+ myeloid cells (Fig. 1P). To further prove the self-renewal potential of MCC intermediate cells, we used fetal liver mononuclear cells as a control in the transplantation experiment, which have been reported to contain highly proliferative CD11b+ hematopoietic stem cells [21]. The ratio of PB reconstitution in MCC intermediates was equivalent to fetal liver (Fig. 1O). In addition, MCC intermediates reconstituted a lower proportion of T cells and a higher proportion of myeloid cells compared with fetal liver (Fig. 1P). These results demonstrate that only ectopic expression of c-Myc can reprogram murine fibroblasts into MCC intermediates, which have engraftment capacity as well as the repopulation of distant hematopoietic compartments.

c-Myc reprograms fibroblasts into macrophages.
A. Schematic diagram of the protocol of c-Myc reprogramming fibroblasts into macrophages. Representative bright-field images of murine MEF/c-Myc, P0 MCC intermediates, P1 MCC intermediates, and iMac. Scale bars, 100 μm.
B. Flow cytometry of the percentage of CD45+ cells in MEF and P0 MCC intermediates.
C. Effects of different iPSC factors on the yield of CD45+ cells derived from MEF. MEFs with overexpression of c-Myc, Klf4, Sox2, or Oct4 were cultured in an iPSC medium.
D. Effects of different culture mediums on the yield of CD45+ cells derived from MEF/c-Myc. MEF-M: MEF medium. EC-M: Erythroid culture medium. GC-M: granulocyte culture medium. iPSC-M: iPS cell medium.
E. Flow cytometry analysis of CD11b and F4/80 expression in MCC intermediates and iMac cells.
F. Quantification of CD11b+F4/80+ macrophages in MCC intermediates and iMac cells.
G. Flow cytometry of the percentage of LSK (Lin-Sca-1+c-Kit+) and MP (Lin-Sca-1-c-Kit+) cells in Lin- cells of MCC intermediates.
H. The percentage of LSK, CD45+B220+, CD45+CD3+, and CD45+CD11b+ cells in MCC intermediates.
I. Representative images of Wright-Giemsa staining of B220+, CD3+, and CD11b+ cells in MCC intermediates. Scale bar, 20 μm.
J. Comparison of hematopoietic colony formation between MCC intermediates and murine bone marrow cells. Hematopoietic colony formation in methylcellulose was performed using MCC intermediates and murine bone marrow-derived progenitors. CFU, colony-forming units. CFU-G, granulocyte. CFU-GM, granulocyte-macrophage. CFU-M, macrophage. CFU-GEMM, granulocyte, erythroid, macrophage, megakaryocyte. CFU-E, erythroid.
K. Representative images of hematopoietic colonies ex-vivo. Scale bar, 100 μm.
L. Schematic diagram for the evaluation of the engraftment ability of MCC intermediates. C57BL/6J mice were pre-treated with 8 Gy irradiation and then GFP-tagged MCC intermediates were transplanted via tail veins. Three months later, a second transplantation was conducted to further investigate the potential and safety of MCC intermediates. n = 5.
M. Quantification of PB reconstitution in recipient animals at different time points after the first transplantation of GFP-tagged MCC intermediates.
N. Quantification of reconstitution in different tissues of recipient mice at 3 months after the first transplantation.
O. PB reconstitution of fetal liver and MCC first and second transplants at 1 month after transplantation.
P. Comparison of the lineage reconstitution of MCC (second transplantation) and fetal liver transplants in the recipient mice BM.
Data are presented as mean ± SEM. *P < 0.05, ***P < 0.001, by two-tailed t-test or one-way ANOVA.
c-Myc upregulates MafB in fibroblast conversion to macrophages
To investigate the mechanism by which c-Myc induces fibroblast conversion to macrophages, we performed single-cell RNA sequencing of MCC intermediate cells. A total of 4591 MCC cells were analyzed. Unsupervised t-SNE (t-distributed stochastic neighbor embedding) analysis of the total cell populations identified 6 sub-clusters (C0-C5, Fig. 2A), including Anp32b and Birc5-positive erythroid progenitor cells (C0), Cd74-positive macrophages (C1), Cytip-positive lymphocytes (C2), Ms4a7-positive macrophages (C3), Ddx39-positive hematopoietic stem cells (C4), and Col3a1 and Col1a2-positive MEFs (C5) (Fig. 2B). The hematopoietic or erythroid progenitor cells represent a heterogeneous and poorly characterized population of cells with modifiable self-renewal, proliferation, and differentiation capabilities [22]. In addition to the erythroid progenitor cell subset (C0), macrophages (C1 and C3) accounted for a large proportion (Fig. 2C). We also performed RNA-seq and found that the profiles of MCC intermediates were markedly different from those of CD45+ PB cells, BM cells, murine iPSCs, and MEFs (Figure S2A). MCC intermediates, unlike MEFs, showed strong enrichment of up-regulated genes of the “Inflammatory cytokines pathway” (Figure S2B-S2D), which was mainly enriched in the macrophage subsets (Fig. 2C).
Next, we conducted a trajectory analysis of the six subsets and found that macrophage subsets were clustered in one direction branch, while erythroid progenitor cells and lymphocytes were clustered in the other direction branch (Fig. 2D), suggesting the process from fibroblast reprogramming into hematopoietic stem cells, which further differentiate into macrophages. By gene expression regulation assessment, five transcriptional factors, Nr6a1, Ddit3, Cebpa, MafB, and Vezf1, were ranked on the top (Fig. 2E). Among them, Cebpa and MafB are important transcriptional regulators of macrophage development and differentiation. A previous study reported that PU.1 and C/EBPα/β converted fibroblasts into macrophages, but this process was PU.1 dependent [11]. MafB is essential for the differentiation from monocytes to macrophages [15, 23]. MafB and c-Maf belong to the Maf transcription factor family, which can regulate macrophage self-renewal cooperatively [15]. In contrast to c-Maf, which was only expressed in a small subset of macrophages, MafB was highly expressed in C1 and C3 macrophage subsets (Fig. 2F). Thus, we focused on MafB in the following study. We downloaded the ChIP-seq data of c-Myc in MEFs (GSE90893) and macrophages (GSE84520) and found that c-Myc was present at the MafB promoter in macrophages but not in MEFs (Fig. 2G and S2E). We performed ChIP-qPCR analysis and verified this in MCC intermediates (Fig. 2H). Overexpression of C-Myc activated MafB promoter, while the luciferase activity of MafB promoter was inhibited after mutating the binding sites of c-Myc (Fig. 2I and J). This data indicates that c-Myc may upregulate MafB in fibroblast conversion to macrophages.
To further evaluate the function of MafB in c-Myc reprogramming fibroblasts into macrophages, MafB was knocked down in MEF/c-Myc with an inducible shRNA (tetO-shMafB). The expression of MafB in MCC intermediates increased with induction (Fig. 2K), and the proportion of CD45+CD11b+ cells decreased after MafB knockdown (Fig. 2L and M), but not CD45+CD3+, CD45+B220+ (Fig. 2L and M), and LSK cells (Figure S2F), compared to the control (Fig. 2L and M, and S2F). These results demonstrate that c-Myc upregulates MafB in fibroblast conversion to macrophages.

c-Myc upregulates MafB and promotes fibroblasts conversion to macrophages.
A. A t-SNE plot of total cells from MCC intermediates, with each cell color-coded for cell cluster and cell type.
B. Heatmap shows expression of marker genes in each subcluster.
C. The cell proportion and inflammatory cytokines pathway activation rate of the six clusters in A.
D. Pseudotime analysis of six MCC intermediates clusters indicates two developmental directions. Arrow, the process of fibroblast reprogramming.
E. TF regulatory networks (regulon, x-axis) derived by SCENIC, plotted against their normalized specificity score (y-axis).
F. The t-SNE plots of expression levels of MafB and c-Maf.
G. ChIP-seq peaks of c-Myc on MafB gene in MEFs (GSE90893) and macrophages (GSE84520) using IGV (Integrative Genomics Viewer).
H. ChIP-qPCR of c-Myc binding to MafB promoter in MEFs and MCC intermediates.
I. c-Myc overexpression activated the expression of MafB. HEK293T was transfected with c-Myc plus MafB promoter-reporter wild type (WT), Site 1 mutation (Mut1), Site 2 mutation (Mut2), or an empty vector (EV).
J. A schematic diagram of WT and two mutation motifs of the c-Myc binding site in MafB promoter.
K. Western blotting of MafB expression during MEF/c-Myc conversion to MCC intermediates. MEF/c-Myc/tetO-shMafB was treated with or without Dox.
L. Flow cytometry of CD45+B220+, CD45+CD3+, and CD45+CD11b+ cells in MCC intermediates treated with or without Dox at day 12.
M. Quantification of CD45+B220+, CD45+CD3+, and CD45+CD11b+ cells in L.
Data are presented as mean ± SEM. ***P < 0.001, by two-tailed t-test or one-way ANOVA.
c-Myc-induced macrophages present a pro-inflammatory phenotype
We further performed a single-cell RNA sequencing of iMac cells and stratified them into five subsets (C0-C4) based on gene signatures (Fig. 3A and B), Ctsb and Apoe-positive macrophages (C0), Cdkn1a-positive macrophages (C1), Nhp2-positive macrophages (C2), Runx1-positive macrophages (C3), and Arpc1b-positive macrophages (C4). NF-κB pathway was activated, and the expression of NF-κB pathway-related genes was upregulated in the C0 subset (Fig. 3C and D). In addition, pro-inflammatory genes were highly expressed in the C0, C1, and C3 subsets (Fig. 3E). This data indicates that the iMac cells display characteristics of a pro-inflammatory state with NF-κB pathway activation.
Next, to characterize the property of iMac, we first revealed that the proportion of M1-like macrophages (CD11c+CD206−) was higher than that of M2-like macrophages (CD11c−CD206+) in iMac (Fig. 3F). The high expression levels of pro-inflammatory genes Cxcl10, Fpr2, and Irf1 in iMac were verified by qRT-PCR (Figure S3A). Second, we treated MEF and iMac cells with LPS and found that the phosphorylation of IKKα/β (p-IKKα/β) markedly increased in iMac compared to MEF, indicating the activation of NF-κB signaling in iMac (Fig. 3G). Then, the phenotypic plasticity of iMac was challenged with LPS/IFN-γ or IL-4 to analyze the expression of macrophage polarization markers (Fig. 3H). Similar to murine bone marrow-derived macrophage (BMDM), the expression of CD11c and iNOS increased with LPS/IFN-γ treatment, while the expression of CD206 and Arg1 increased with IL-4 treatment (Fig. 3H). Interestingly, unlike BMDM, expression of CD11c and iNOS didn’t decrease or even increased after IL-4 treatment (Fig. 3H). Quantitative analysis of cytokines and chemokines displayed higher levels of pro-inflammatory cytokines and chemokines, including IL-6, CXCL10, and CXCL1 in LPS/IFN-γ stimulated iMac than M-CSF control, which was similar to the profiles in murine BMDMs (Fig. 3I). We also found that IL-4 stimulation induced higher levels of IL-6, CXCL10, CXCL1, IL-1β, and TNF-α in iMac, while no significant changes were seen in BMDMs (Fig. 3I).
Next, we examined the phagocytic function of iMac. Immunofluorescence verified the phagocytosis of iMac toward tumor cells and fluorescent beads, which was also confirmed by flow cytometry (Fig. 3J and K and S3B-S3C). We further performed the bulk RNA-seq of MEF, MCC, and iMac cells (Figure S4A). Compared to MEFs, stemness-related and macrophage-related genes were upregulated in MCC cells (Figure S4B-S4C). Macrophage-related genes were upregulated in iMac cells compared to MEFs, but M2-like macrophage-related genes were downregulated compared to MCC cells (Figure S4B). Pathway analysis further revealed that NF-κB and chemokine signaling pathways were enriched in MCC and iMac, and the pathway of intestinal immune network for IgA production was specifically enriched in iMac (Figure S4D). These results demonstrate that iMac has phagocytic ability in vitro and is more inclined to a pro-inflammatory phenotype overall, which is resistant to anti-inflammatory challenge.

c-Myc-induced macrophages present a pro-inflammatory phenotype.
A. The t-SNE plot displays five sub-clusters (C0-C4) of iMac.
B. Heatmap shows expression of marker genes in each subcluster.
C. Relative percent of cell resource and NF-κB pathway activation rate in the five sub-clusters.
D. The heatmap of NF-κB pathway gene expression in different clusters in iMac.
E. The heatmap of pro-inflammatory gene expression and the pro-inflammatory score in different clusters.
F. Flow cytometry analysis of CD11c and CD206 expression in iMac and quantification of CD11c+CD206- and CD11c-CD206+ macrophages.
G. Western blotting of phospho-IKKα/β (phospho-IKKα/β), IKKα, IKKβ, and β-actin in MEF and iMac with or without LPS stimulation.
H. Flow cytometry analysis of CD11c, iNOS, CD206, and Arg1 expression in iMac with or without LPS/IFN-γ or IL-4 stimulation. BMDMs were used as a control.
I. The expression levels of pro-inflammatory cytokines and chemokines in BMDM and iMac with or without LPS/IFN-γ or IL-4 stimulation.
J. Representative images of immunofluorescence of the phagocytosis of Reh and Raji (green) by MEF or iMac (red). Scale bars, 50 μm.
K. Flow cytometry and quantification of the phagocytosis of Reh and Raji cells by MEF and iMac cells.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, by two tailed t-test.
c-Myc-induced macrophages inhibit the progression of leukemia and breast cancer
Tumor-associated macrophages (TAMs) commonly play a tumor-promoting role in tumor microenvironment, and many re-educating TAM strategies play beneficial effects in tumor therapy, such as inhibition of PD-1/PDL-1 [24], PI3Kγ [25], and CD40 agonists [26]. Based on this, we hypothesized pro-inflammatory iMac may traffic to the tumor and induce endogenous immune response. To evaluate the role of iMac in tumor therapy, we first employed the Reh leukemia animal model. GFP-labeled Reh (Reh-GFP) cells with different doses of iMac were injected into SCID mice via tail veins (Fig. 4A). After one month, the percentages of Reh-GFP leukemia cells in bone marrow significantly decreased in the groups treated with iMac (Fig. 4B and S5A). Moreover, the percentage of the leukemia cells decreased with the increased iMac numbers (Fig. 4B and S5A). The percentage of the leukemia cells in the spleen also decreased, but there was no significant difference among the different doses of iMac (Fig. 4C, S5A, and S5B). In addition, we employed a patient-derived T-cell acute lymphoblastic leukemia (T-ALL) xenograft model to evaluate the therapeutic function of iMac (Fig. 4D). iMac markedly decreased human CD45+CD7+ PDX cell proliferation in animal BM but not in spleen, compared to the control (Fig. 4E and F, and S5C).
To further demonstrate the antitumor efficacy of iMac, we established the 4T1 breast cancer metastasis model (Fig. 4G). Mice were injected with 4T1 on day 1, then randomly grouped and treated with iMac or vehicle (PBS) on day 5. Bioluminescence imaging (BLI) signal showed a lower tumor burden after iMac treatment (Fig. 4H and I). Kaplan-Meier survival analysis showed a significant improvement in animal survival of the iMac-treated cohort compared with the vehicle cohort (P < 0.05) (Fig. 4J). Both the number of lung nodules and lung weight decreased in the iMac treatment group (Fig. 4K and M). Compared with the control group, the protein levels of proliferation marker Ki-67 in lung tissue were lower in the iMac treatment group (Fig. 4N and O), whereas the protein levels of macrophage marker CD68 were higher (Fig. 4N and P). To investigate the function of infuse iMac, we evaluated the ratio of macrophages in the lungs with 4T1 breast cancer metastatic tumors. Compared to the control, iMac treatment increased the ratio of CD11c+CD206− M1-like macrophages (Fig. 4Q and S5D) but decreased the percentage of CD11c−CD206+ M2-like macrophages (Fig. 4R and S5D), suggesting that iMac cells were highly infiltrated in tumor. In addition, we investigated the impact of iMac on various immune cells within the tumor microenvironment in the lungs. Compared to the control, iMac treatment increased the percentages of CD69+ T cells (Fig. 4S and S5D), CD107a+ natural killer (NK) cells (Fig. 4T and S5D), and dendritic cells (DC) (Fig. 4U and S5D), suggesting that the application of iMac cells improves the immunosuppressive nature of the tumor microenvironment. Taken together, these data demonstrate that iMac cells are functional macrophages, providing a new therapeutic possibility for tumor treatment.

c-Myc-induced macrophages reduce leukemia and breast cancer progression.
A. Treatment scheme for the evaluation of in vivo efficacy of iMac in Reh leukemia xenografts. SCID mice were pre-treated with 2 Gy irradiation and then injected with 2 × 105 Reh-GFP cells with vehicle (PBS) or indicated iMac by tail vein.
B and C. The percentage of leukemia cells in bone marrow (B) and spleen (C) of mice at day 28 after iMac treatment. n = 5.
D. Treatment scheme for the evaluation of in vivo efficacy of iMac in ALL PDX model. NCG mice were injected with 1 × 106 PDX cells on day 1 by tail vein. Vehicle (PBS) or iMac (2 × 106) was injected on day 14 and 21, respectively.
E and F. The percentage of PDX cells in bone marrow (E) and spleen (F) of mice on day 35. n = 5.
G. Treatment scheme for the evaluation of in vivo efficacy of iMac in 4T1 breast cancer xenografts. BALB/c mice were injected with 2 × 105 luciferase-labeled 4T1 cells by tail vein on day 1 and treated with 1 × 106 iMac or vehicle (PBS) on day 5. n = 5.
H. Representative images of BLI at day 16.
I. Quantification of the BLI activity in H.
J. Kaplan-Meier survival curves of animals bearing 4T1 tumors. Data were analyzed using the log-rank Mantel-Cox test. Median survival: Vehicle (17.0 days) and iMac (20.0 days).
K. Representative images of bright field (upper panel) and H&E-stained sections (lower panel) of the lung tissue with or without iMac treatment. Scale bar, 100 μm.
L-M. Quantification of the number of lung metastases (L) and the weight of lungs (M) in K.
N. Representative IHC staining of Ki-67 and CD68 in lung sections. Scale bar, 50 μm.
O and P. Quantification of Ki-67 (O) and CD68 (P) positive cells in N.
Q-U. Quantification of CD11c+CD206− M1-like macrophages (% of CD11c+CD206− in CD11b+F4/80+ cells) (Q), CD11c−CD206+ M2-like macrophages (% of CD11c−CD206+ in CD11b+F4/80+ cells) (R), CD69+ T cells (% of CD69+ in CD3+ cells) (S), CD107a+ NK cells (% of CD49b+CD107a+ in CD3− cells) (T), and DC (% of CD11c+ in CD11b+F4/80− cells) (U) in lungs by flow cytometry.
V. A model of c-Myc reprogramming of fibroblasts into macrophages.
Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, by two-tailed t-test or one-way ANOVA.
Discussion
In this study, we present that overexpression of c-Myc can reprogram fibroblasts into functional macrophages with a pro-inflammatory phenotype through upregulation of MafB (Fig. 4V). The iPSC culture medium is critical for c-Myc reprogramming fibroblasts into functional macrophages. MCC intermediates can be stably maintained in iPSC medium and continuously generate functional and highly pure iMac. More importantly, iMac is functional, which inhibits leukemia and breast cancer progression in animal tumor xenograft models.
Compared to iPSC-Mac, our MEF-Mac technology is easy and efficient. IPSC-Mac technology has been reported by several groups [5, 8], including ourselves [27]. Macrophage differentiation of iPSCs needs a variety of cytokines [5, 8, 27], whereas our MEF-Mac technology is simple and only needs the iPSC medium and macrophage differentiation medium. iPSC-Mac technology usually takes 20 to 30 days [5, 8, 27], yet our iMac can be generated in two weeks.
Our study demonstrates that compared to other pluripotency-related factors, only c-Myc can efficiently reprogram fibroblasts into macrophages. Transcription factor c-Myc plays important functions in development and diseases, such as cell proliferation, apoptosis, cell survival, tissue remodeling, angiogenesis, cell metabolism, production of inflammatory and anti-inflammatory cytokines, as well as cell transformation [28, 29]. Accumulated data also have demonstrated that c-Myc controls macrophage proliferation and polarization [14,15,16]. In this study, we demonstrate that c-Myc has a new function in reprogramming fibroblasts into macrophages. In addition, we also show that iPSC medium is required for c-Myc-induced conversion of fibroblasts into macrophages, suggesting that c-Myc-reprogramming macrophages depend on a specific microenvironment.
M1/M2 classification is somewhat of a simplification, and macrophages can display overlapped phenotypes depending on their tissue location and type of signaling [30]. Our data indicated the high expression of M1-related markers (Cd86 and Cd74) and M2-related markers (Cd163 and Arg1) in MCC intermediate cells, indicating a continuum of classically and alternatively activated phenotypes among differentiated macrophages. Our results also showed that NF-κB and chemokine signaling pathways were activated in MCC intermediate cells, which may promote the high ratio of M1-like macrophages produced in the iPSC medium.
Importantly, we identify that c-Myc-reprogrammed macrophages are functional. Immortalized macrophage cell lines are not functional, and thus they are not applicable to clinical settings, and bone marrow or PBMC-derived primary macrophages are not easily efficiently engineered [5, 27, 31]. The functions of iPSC-Mac were also limited and most clusters of iPSC-Mac were in the M2 state (anti-inflammatory phenotype) [5]. c-Myc has been detected in human M2 macrophages [16, 32] and was identified as a murine M2 macrophage marker in vitro [33]. In addition, c-Myc is related to TAMs, which have an M2-like activity [34]. In this study, our scRNA-seq data indicated that the major part of c-Myc-reprogrammed macrophages displayed characteristics of a pro-inflammatory state with NF-κB pathway activation. Phenotype analysis also demonstrated the bias of M1-like polarity state of iMac. When exposed to IL-4, iMac cells maintained high levels of CD11c and iNOS expression, along with elevated pro-inflammatory cytokines and chemokines, demonstrating a distinct response compared to BMDM. c-Myc-reprogrammed macrophages indicated strong phagocytosis activity in vitro and in vivo. Thus, our data indicate that c-Myc is also important for M1 macrophages.
We indicate c-Myc-reprogrammed MCC intermediates with engraftment capacity as well as the repopulation of distinct hematopoietic compartments. Accumulated data have indicated that c-Myc plays a critical role in controlling self-renewal versus differentiation in hematopoietic stem cells [35] and cell fate in megakaryocyte-erythrocyte [36]. Overexpression of hematopoietic transcription factors also converted fibroblasts into ‘induced hematopoietic progenitors’ (iHPs) with engraftment of immunodeficient mice, whereas iHPs only had a phagocytic function in vitro [37]. SOX2/miR-125b overexpression also induced the conversion of fibroblasts into CD34+ immature hematopoietic progenitor-like cells with an immature phenotype [10]. Here, we show that MCC intermediate cells can engraft immunocompetent animals, and iMac have phagocytic functions in vitro and in vivo. MCC intermediate cells not only express the markers of hematopoietic stem cells and myeloid progenitor cells but also express the markers of B, T, and macrophage cells. In addition, the intermediates are stably maintained in iPSC medium and continuously generate functional iMac.
Our study also reveals that c-Myc upregulates MafB to promote the conversion of fibroblasts into macrophages. MafB is a specific marker of macrophages that can be regulated by various signals, including lipid metabolism, immunological regulators, and microRNAs [38]. MafB is essential for the differentiation from monocytes to macrophages [15, 23]. Here, we demonstrate that c-Myc regulates the expression of MafB in fibroblasts’ conversion to macrophages. MafB expression is required for c-Myc-reprogramming macrophages, and depletion of MafB specifically reduced fibroblasts’ conversion to macrophages but not B and T cells. Our data reveal that MafB has a novel function in fibroblast reprogramming.
Conclusion
Taken together, the technology of c-Myc reprogramming fibroblasts into functional macrophages is highly relevant to various fields ranging from developmental biology, immunology, and cell therapy to industrial applications for drug discovery. The subsequent direct reprogramming of human fibroblasts into functional macrophages is a target for further research.
Data availability
All data generated or analyzed during this study are present in this article and its supplementary data. The Bulk RNA-seq and scRNA-seq data of this article are available in the gene expression omnibus (GEO): GSE237621, GSE275456, and GSE275457.
References
Flugel CL, Majzner RG, Krenciute G, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours [J]. Nat Rev Clin Oncol. 2023;20(1):49–62.
Cappell KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far [J]. Nat Rev Clin Oncol. 2023;20(6):359–71.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy [J]. Nat Rev Drug Discov. 2022;21(11):799–820.
Klichinsky M, Ruella M, Shestova O, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy [J]. Nat Biotechnol. 2020;38(8):947–53.
Zhang L, Tian L, Dai X, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions [J]. J Hematol Oncol. 2020;13(1):153.
Lei A, Yu H, Lu S, et al. A second-generation M1-polarized CAR macrophage with antitumor efficacy [J]. Nat Immunol. 2024;25(1):102–16.
Wang X, Su S, Zhu Y, et al. Metabolic reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors [J]. Nat Commun. 2023;14(1):5778.
Ackermann M, Rafiei Hashtchin A, Manstein F, et al. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors [J]. Nat Protoc. 2022;17(2):513–39.
Shen J, Lyu S, Xu Y, et al. Activating innate immune responses repolarizes hPSC-derived CAR macrophages to improve anti-tumor activity [J]. Cell Stem Cell. 2024;31(7):1003–19.
Pulecio J, Nivet E, Sancho-Martinez I, et al. Conversion of human fibroblasts into monocyte-like progenitor cells [J]. Stem Cells. 2014;32(11):2923–38.
Feng R, Desbordes SC, Xie H, et al. PU.1 and C/EBPalpha/beta convert fibroblasts into macrophage-like cells [J]. Proc Natl Acad Sci U S A. 2008;105(16):6057–62.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors [J]. Cell. 2006;126(4):663–76.
Li Y, Feng H, Gu H, et al. The p53-PUMA axis suppresses iPSC generation [J]. Nat Commun. 2013;4:2174.
Gerlach BD, Ampomah PB, Yurdagul A Jr., et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury [J]. Cell Metab. 2021;33(12):2445–63.
Aziz A, Soucie E, Sarrazin S, Sieweke MH. MafB/c-Maf deficiency enables self-renewal of differentiated functional macrophages [J]. Science. 2009;326(5954):867–71.
Pello OM, De Pizzol M, Mirolo M, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology [J]. Blood. 2012;119(2):411–21.
Klinken SP, Alexander WS, Adams JM. Hemopoietic lineage switch: v-raf oncogene converts Emu-myc transgenic B cells into macrophages [J]. Cell. 1988;53(6):857–67.
de Bruijn MF, Ma X, Robin C, et al. Hematopoietic stem cells localize to the endothelial cell layer in the midgestation mouse aorta [J]. Immunity. 2002;16(5):673–83.
Wang W, Li X, Qin X, et al. Germline neurofibromin 1 mutation enhances the anti-tumour immune response and decreases juvenile myelomonocytic leukaemia tumourigenicity [J]. Br J Haematol. 2023;202(2):328–43.
Spevak CC, Elias HK, Kannan L, et al. Hematopoietic stem and progenitor cells exhibit stage-specific translational programs via mTOR- and CDK1-Dependent mechanisms [J]. Cell Stem Cell. 2020;26(5):755–65.
Morrison SJ, Hemmati HD, Wandycz AM, Weissman IL. The purification and characterization of fetal liver hematopoietic stem cells [J]. Proc Natl Acad Sci U S A. 1995;92(22):10302–6.
Dulmovits BM, Hom J, Narla A, et al. Characterization, regulation, and targeting of erythroid progenitors in normal and disordered human erythropoiesis [J]. Curr Opin Hematol. 2017;24(3):159–66.
Goudot C, Coillard A, Villani AC, et al. Aryl Hydrocarbon receptor controls monocyte differentiation into dendritic cells versus macrophages [J]. Immunity. 2017;47(3):582–96.
Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity [J]. Nature. 2017;545(7655):495–9.
Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression [J]. Nature. 2016;539(7629):437–42.
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans [J]. Science. 2011;331(6024):1612–6.
Li S, Song L, Zhang Y et al. Optimizing the Method for Differentiation of Macrophages from Human Induced Pluripotent Stem Cells [J]. Stem Cells International, 2022, 2022: 6593403.
Dhanasekaran R, Deutzmann A, Mahauad-Fernandez WD, et al. The MYC oncogene - the grand orchestrator of cancer growth and immune evasion [J]. Nat Rev Clin Oncol. 2022;19(1):23–36.
Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming [J]. Nat Rev Genet. 2013;14(6):427–39.
Strizova Z, Benesova I, Bartolini R, et al. M1/M2 macrophages and their overlaps - myth or reality? [J]. Clin Sci (Lond). 2023;137(15):1067–93.
Lee CZW, Kozaki T, Ginhoux F. Studying tissue macrophages in vitro: are iPSC-derived cells the answer? [J]. Nat Rev Immunol. 2018;18(11):716–25.
Martinez FO, Helming L, Milde R, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences [J]. Blood. 2013;121(9):e57–69.
Jablonski KA, Amici SA, Webb LM, et al. Novel markers to delineate murine M1 and M2 macrophages [J]. PLoS ONE. 2015;10(12):e0145342.
Maddipati R, Norgard RJ, Baslan T, et al. MYC levels regulate metastatic heterogeneity in pancreatic adenocarcinoma [J]. Cancer Discov. 2022;12(2):542–61.
Satoh Y, Matsumura I, Tanaka H, et al. Roles for c-Myc in self-renewal of hematopoietic stem cells [J]. J Biol Chem. 2004;279(24):24986–93.
Guo Y, Niu C, Breslin P, et al. c-Myc-mediated control of cell fate in megakaryocyte-erythrocyte progenitors [J]. Blood. 2009;114(10):2097–106.
Cheng H, Ang HY, C A E F, et al. Reprogramming mouse fibroblasts into engraftable myeloerythroid and lymphoid progenitors [J]. Nat Commun. 2016;7:13396.
Hamada M, Tsunakawa Y, Jeon H, et al. Role of MafB in macrophages [J]. Exp Anim. 2020;69(1):1–10.
Funding
This work was supported in part by National Natural Science Foundation of China (81972341, 32271007 to Y.L.; 32371004, 82072896 to H.F.); the National Key Research and Development Program of China (2022YFA1104001) to H.F.; Shanghai Natural Science Foundation (23ZR1441000), Shanghai Municipal Science and Technology Commission (201409002700) to Y.L.; Program of Shanghai Academic/Technology Research Leader (21XD1403100) to H.F.
Author information
Authors and Affiliations
Contributions
YL and HF designed and supervised the project. SL, GC, XH, and YZ performed experiments and analyzed the data. YL, HF, and SS interpreted and/or reviewed the data. SL, YL, and HF wrote or edited the manuscript. All the coauthors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures in mice were performed in accordance with the relevant laws and guidelines of the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiao Tong University (SCMC-LAWEC-2023-061).
The studies involving human participants were reviewed and approved by the Ethics Committee of the Children’s Medical Center affiliated with Shanghai Jiao Tong University. Written informed consent was provided by the participants’ legal guardians/next of kin (SCMCIRB-K2023093-1).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, S., Chen, G., Huang, X. et al. c-Myc alone is enough to reprogram fibroblasts into functional macrophages. J Hematol Oncol 17, 83 (2024). https://doi.org/10.1186/s13045-024-01605-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-024-01605-x