
Abstract
Background
GMEB1 was originally identified via its interaction with GMEB2, which binds to the promoter region of the tyrosine aminotransferase (TAT) gene and modulates transactivation of the glucocorticoid receptor gene. In the cytosol, GMEB1 interacts with and inhibits CASP8, but the molecular mechanism is currently unknown.
Methods
Human non-small cell lung cancer cells and 293FT cells were used to investigate the function of GMEB1/USP40/CFLARL complex by WB, GST Pull-Down Assay, Immunoprecipitation, Immunofluorescence and Flow cytometry analysis. A549 cells overexpressing green fluorescent protein and GMEB1 shRNA were used for tumor xenograft using female athymic nu/nu 4-week-old mice.
Results
We found GMEB1 interacted with CFLARL (also known as c-FLIPL) in the cytosol and promoted its stability. USP40 targeted CFLARL for K48-linked de-ubiquitination. GMEB1 promoted the binding of USP40 to CFLARL. USP40 knockdown did not increase CFLARL protein level despite GMEB1 overexpression, suggesting GMEB1 promotes CFLARL stability via USP40. Additionally, GMEB1 inhibited the activation of pro-caspase 8 and apoptosis in non-small cell lung cancer (NSCLC) cell via CFLARL stabilization. Also, GMEB1 inhibited the formation of DISC upon TRAIL activation. CFLARL enhanced the binding of GMEB1 and CASP8. Downregulation of GMEB1 inhibited A549 xenograft tumor growth in vivo.
Conclusions
Our findings show the de-ubiquitinase USP40 regulates the ubiquitination and degradation of CFLARL; and GMEB1 acts as a bridge protein for USP40 and CFLARL. Mechanistically, we found GMEB1 inhibits the activation of CASP8 by modulating ubiquitination and degradation of CFLARL. These findings suggest a novel strategy to induce apoptosis through CFLARL targeting in human NSCLC cells.
Similar content being viewed by others


Background
Glucocorticoid modulatory element-binding protein 1 (GMEB1) was originally identified as a nuclear protein with a molecular weight of 88 kDa [1,2,3]. GMEB1 interacts with GMEB2 and binds to the promoter sequence glucocorticoid modulatory element (GME) of the tyrosine aminotransferase (TAT) gene to modulate glucocorticoid receptor transactivation [4]. GMEB1 also exists in the cytosol and functions at the protein level. It interacts with the heat shock protein HSP27, but the function is not well studied. GMEB1 also binds to pro-caspases and inhibits their activation and cell apoptosis. However, it is still unknown how GMEB1 does this [5,6,7,8].
CFLARL, also known as c-FLIPL, plays an important role in extrinsic ligand-induced apoptosis. In this pathway, FasL/tumor necrosis factor-alpha (TNF-α)/TRAIL binds to cell surface receptors, forms Death-Inducing Signaling Complex (DISC) and triggers the caspase-dependent apoptotic pathway. CFLARL interacts with CASP8 via DED domains and inhibits the activation of CASP8, and thus apoptosis [9,10,11,12]. ITCH is an E3 ligase of CFLARL, which enhances apoptosis by targeting CFLARL through ubiquitin-proteasome pathway [13, 14]. USP8 is a de-ubiquitination enzyme of CFLARL that enhances stability and inhibits apoptosis induced by extrinsic ligands [15]. Therefore, approaches to promote the ubiquitination and degradation of CFLARL are potential effective cancer therapies.
In the present study, we showed GMEB1 directly interacted with CFLARL and increased its stability at the protein level. GMEB1 inhibited the activation of pro-caspase 8 via CFLARL. We found the de-ubiquitination enzyme USP40 bound to CFLARL and GMEB, which enhanced the interaction between USP40 and CFLARL, resulting in reduced ubiquitination and degradation of CFLARL. These findings suggest GMEB1 inhibits the activation of CASP8 and apoptosis via CFLARL, which highlights potential implications for lung cancer therapy.
Methods
Reagents and antibodies
SAHA was purchased from Sigma (US). CHX, MG132 and E64D were purchased from Selleck (US). PARP (#9542), Caspase 8 (9746 L) antibodies were purchased from Cell Signaling Technology (US). Caspase 3 (NB100–56708) antibody was purchased from Imgenex (US). GMEB1 (sc-376,775), USP40 (sc-514,248) and FLIPL (sc-8346) antibodies were purchased from Santa Cruz (US). CFLARL (ALX-804-961-0100) antibody was purchased from Enzo (US). HA (D110004) tag antibody was purchased from Sangon Biotech (China). FLAG (F7425) tag antibody was purchased from Sigma (US). His (D291–3) tag antibody was purchased from MBL (Japan). USP8 (27791–1-AP) and FADD (14906–1-AP) antibodies were purchased from proteintech (US).
Cell lines and cell culture
The human NSCLC cell lines A549, H1299, H1792. H157, H460, Calu-1 and HEK293FT cell lines were originally obtained from the American Type Culture Collection (ATCC). A549 and H1792 cell lines have been authenticated in Microread Gene Technology by STR analysis. The NSCLC cells were grown in monolayer culture in RPMI 1640 with 5% FBS (Gibco, US) at 37 °C in a humidified atmosphere consisting of 5% CO2 and 95% air. HEK293FT cells were grown in DMEM with 5% FBS (Gibco, US) at 37 °C.
RNA interference and plasmid transfection
The GMEB1 siRNA targets the sequence: 5′-GCACCAAAUUUGAUCUUCU-3′ and 5′-GCACACACAUUUGGCCUAA -3′; USP40 siRNA targets the sequence 5′-GCAGCAAAGUCGGCCAAAU-3′ and 5′-GGAUGCAGCUAACAUUGAA-3′. The siRNAs were synthesized by GenePharma and used as the manufacture’s protocol.
GMEB1 and USP40 coding regions were amplified by PCR from A549 genomic DNA using following primers:
GMEB1 sense: CGGATCCGCCGCCACCATGGCTAATGCAGAAGTGAG
GMEB1 antisense: CCTCGAGTTAATCCTCTAAGACCACAATC
USP40 sense: CTAGCTAGCGCCGCCACCATGTCACTTTTTTTAAGGGTAG
USP40 antisense: CGCGGATCCTTATCTGAAGCTCCCCACG
HA tag was cloned to the N-terminal of GMEB1, His tag was cloned to the C-terminal of USP40. His-tagged, FLAG-tagged and GST-tagged CFLARL were cloned previously by our team.
Western blot analysis
Cells were harvested and rinsed with pre-chilled PBS on ice. They were lysed in lysis buffer on ice for 30 min and then purified via centrifugation for 15 min at 4 °C. Samples of the whole-cell protein lysates (35 μg) were prepared for SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane by electro blotting. The proteins were probed with the appropriate primary and secondary antibodies. Antibody binding was detected by an HRP system according to the manufacturer’s protocol [16].
Immunoprecipitation
Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM Na2EDTA; 1 mM EGTA; 2.5 mM sodium pyrophosphate; 1 mM β-glycerophosphate; 1 mM Na3VO4; 0.5% Triton) on ice for 30 min then purified via centrifugation for 15 min at 4 °C. The supernatants were incubated with antibody at 4 °C for 1 h. Then the mixture was incubated with protein A beads (ThermoFisher) at 4 °C for 2 h. The beads were washed twice with 1 ml of lysis buffer. 20 μl 2 × SDS buffer were added for elution (100 °C, 10 min). Samples were centrifuged for western blot analysis.
GST pull-down assay
Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM Na2EDTA; 1 mM EGTA; 2.5 mM sodium pyrophosphate; 1 mM β-glycerophosphate; 1 mM Na3VO4; 0.5% Triton) on ice for 30 min, then purified via centrifugation for 15 min at 4 °C. The supernatants were incubated with rotation in 20 μl of Glutathione Sepharose beads (GE) at 4 °C for 2 h. Beads were washed twice with 1 ml of lysis buffer. 20 μl 2 × SDS buffer was added to beads for elution (100 °C, 10 min). Samples were centrifuged for western blot analysis.
Immunofluorescence
Cells were fixed with PHEMO buffer (0.025 M HEPES, 0.068 M PIPES, 0.003 M MgCl2·6H2O, 0.015 M EGTA·Na2, 10% DMSO, pH adjusted to 6.8. Additional reagents were added before use, with a final concentration as follows: 0.05% glutaraldehyde, 0.5% Triton X-100, 3.7% formaldehyde) for 10 min at room temperature before washing with PBS for 3 times. Then, cells were incubated with blocking buffer (3% BSA) for 30 min at room temperature. Afterward, cells were incubated with primary antibodies against CFLARL (Santa Cruz, dilution at 1:500) for 1 h at room temperature. After washing with PBS for 3 times, cells were incubated with another primary antibodies GMEB1 (Santa Cruz, dilution at 1:500) or USP40 (Santa Cruz, dilution at 1:500) for 1 h. Alex Fluor 488 (Green) and Alex Fluor 568 (Red)-conjugated secondary antibodies were then applied and incubated at room temperature for 1 h. Cell nuclei were stained with DAPI. Images were captured using a confocal microscope (ZEISS, LSM700).
Flow cytometry analysis
Annexin V-FITC Apoptosis Detection Kit (Biobox Biotech, Nanjing, China) was used for cell apoptosis analysis according to the manufacture’s protocol.
CASP8 activity detection
A549 cells were prepared for CASP8 activity according CASP8 Activity Apoptosis Assay Kit protocol (Sangon Biotech, Shanghai, China).
Tumor xenograft model
Fifteen female athymic nu/nu 4-week-old mice were purchased from Vital SPF Biotechnology (Beijing, China). For tumor xenograft establishments, A549 cells overexpressing green fluorescent protein or shGMEB1 RNA (1 × 106 cells/100 μL) were subcutaneously injected into the right side of the abdominal region of mice. Weight of mice and tumor size were detected every 2 days. The tumor volume was calculated as V = π × (length×width2)/6.
Statistical analysis
GraghPad Prism version 5.00 was used for statistical analysis. All data are presented as the mean ± SD. Differences between groups were identified using Student’s t-test. P < 0.05 was considered statistically significant.
Results
SAHA treatment reduced GMEB1 and CFLARL protein levels in NSCLC cells
To examine the interaction between GMEB1 and CFLARL, we measured the protein levels in six NSCLC lines: A549, H1792, Calu-1, H1299, H157, and H460. Western blot assay showed GMEB1 protein levels were high in A549, H1792, Calu-1 and H1299 cells. CFLARL protein levels were high in A549 and Calu-1 cells (Fig. 1a). CFLARL protein levels positively correlated with GMEB1 (Additional file 1: Figure S1A). SAHA is an inhibitor of histone deacetylase and enhances TRAIL, which induces apoptosis and decreases CFLAR protein [17,18,19]. To detect whether SAHA also decreases GMEB1, we treated NSCLC cells with SAHA at different concentrations and time points. The results show SAHA decreased GMEB1 in a dose-dependent (Fig. 1b and Additional file 1: Figure S1D) and time-dependent manner (Fig. 1c and Additional file 1: Figure S1E). This is consistent with the effect of SAHA on CFLARL. These data indicate SAHA treatment affects GMEB1 and CFLARL similarly in NSCLC cells.

SAHA treatment reduced GMEB1 and CFLARL protein levels in NSCLC cells. a The protein levels of GMEB1, CFLARL and USP40 were measured in 6 non-small cell lung cell lines and HEK293FT cell line by western blot. GMEB1 and CFLARL protein levels decreased after SAHA treatment in dose-dependent (b) and time-dependent manner (c) in different NSCLCs
GMEB1 enhanced the stability of CFLARL
We then characterized the biological function of the interaction between GMEB1 and CFLARL. GMEB1 was originally identified as a transcription factor, so we first determined if GMEB1 regulates CFLARL at the transcriptional level. GMEB1 and CFLARL relative mRNA levels were detected using q-PCR in A549 cells; GMEB1 siRNA was transfected for 24 h. Results show that GMEB1 knock-down did not affect the relative mRNA level of CFLARL (Fig. 2a). NSCLC cells with GMEB1 knock-down were treated with CHX [10 μg/ml] for various time points. WB data show GMEB1 knockdown decreased the stability of CFLARL (Fig. 2b), while overexpression of GMEB1 increased the stability of CFLARL (Fig. 2c). This confirms GMEB1 enhances the stability of CFLARL at post-translational level. Next, we knocked down GMEB1 by siRNA in A549, H1299 and Calu-1 cell lines and treated cells with SAHA [2.0 μM] for 6 h. Results show GMEB1 knockdown decreased CFLARL protein level (Fig. 2d). Overexpression of GMEB1 upregulated CFLARL protein (Fig. 2e). To confirm the effect of GMEB1 on CFLARL, we knocked down GMEB1 using GMEB1 shRNA in A549 cell lines and overexpressed GMEB1 using plasmid. We found that GMEB1 overexpression rescued the reduced CFLARL protein level caused by GMEB1 knockdown (Fig. 2f). These data indicate GMEB1 plays a role in maintaining the protein level of CFLARL.

GMEB1 enhanced the stability of CFLARL. a Relative mRNA levels of GMEB1 and CFLARL were determined by quantitative reverse transcription-polymerase chain reaction (q-PCR) in A549 cell line when the cell transfected with GMEB1 siRNA for 24 h. Error bars represent s.d. ***P < 0.001. b, c Calu-1 and A549 cells were seeded in 6-well plates. GMEB1 siRNA or plasmid were transfected for 24 h and a non-sense siRNA or empty vector was transfected as control. Cells were treated with cycloheximide (CHX) [10 μg/ml] and harvested at different time points (0, 4, 8, 12 h) for western blot analysis. The band intensity of CFLARL was quantified by Photoshop CS6 and plotted. Experiments were repeated three times, a representative experiment is presented. Every experimental group was compared with the negative control group. Error bars represent s.d. *P < 0.05. d, e A549, H1299, Calu-1 and H1792 cells were seeded in 6-well plates. GMEB1 siRNA or plasmid were transfected for 24 h. Cells were treated with SAHA [2.0 μM] for 6 h and harvested for western blot analysis. f A549-LUC, A549-shGMEB1–1# and A549-shGMEB1–2# were seeded in 6-well plates. GMEB1 plasmid was transfected for 24 h. Cells were harvested for western blot analysis. g H1299 cells were seeded in 6-well plates. GMEB1 siRNA was transfected for 24 h. Cells were treated with DMSO, MG132 [20 μM] and E64D [15 μM] for 6 h. Cells were harvested for western blot analysis. h HEK293FT cells were prepared for GST pull down assay using GST-CFLARL, FLAG-GMEB1 and HA-Ub plasmids to detect the function of GMEB1 on the ubiquitination of CFLARL
Most protein degradation occurs in either the ubiquitination-proteasome or ubiquitination-lysosome pathway. As such, de-ubiquitinases inhibit the effective degradation of proteins. Therefore, we focused how GMEB1 affects the ubiquitination of CFLARL. First, we knocked down GMEB1 in H1299 cells and treated them with DMSO, MG132 [20 μM] and E64D [15 μM] for 6 h. MG132 inhibits the degradation of proteins by blocking proteasomes, and E64D inhibits the degradation of proteins via lysosomes. Western blot analysis shows MG132 treatment rescued the reduced CFLARL protein level caused by GMEB1 knockdown. This indicates CFLARL is degraded through the proteasome pathway when GMEB1 protein levels are decreased (Fig. 2g). In addition, we designed a co-IP assay to determine whether GMEB1 affects the ubiquitination of CFLARL. Data show overexpression of GMEB1 decreased the ubiquitination of CFLARL (Fig. 2h). Thus, we propose GMEB1 enhances the stability of CFALRL by modulating its ubiquitination level.
GMEB1 physically interacted with CFLARL in NSCLC cells
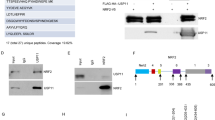
GMEB1 interacts with CASP8 and inhibits its activation. CFLARL gene has high homology with CASP8 gene, and the proteins display similar structures that may confer interaction with each other through DED domains. Thus, we determined if GMEB1 and CFLARL bind one another via a co-immunoprecipitation (co-IP) assay in HEK293FT cells. The data show that HA-tagged GMEB1 interacted with FLAG-tagged CFLARL (Fig. 3a and b). After GST-tagged CFLARL was pulled down with Glutathione Sepharose beads, GMEB1 was detected using WB assay, indicating GMEB1 physically interacted with CFLARL (Fig. 3c). An additional IP assay using A549 and H1299 cells (Fig. 3d) shows that endogenous CFLARL interacted with endogenous GMEB1. To further evaluate the interaction between GMEB1 and CFLARL, immunofluorescence staining experiments were conducted in Calu-1 cells. Results show GMEB1 localized in the cytosol. GMEB1 and CFLARL were co-localized in the cytosol (Fig. 3e). We determined which domains of CFLARL are required for this binding. Our data indicated that DED domains of CFLARL were not necessary for interaction with GMEB1. However, P20 and P12 fragments of CFLARL interacted with GMEB1 (Additional file 1: Figure S2A, B and C). Additional results show the N-terminal of GMEB1 was essential for interaction with CFLARL (Additional file 1: Figure S2D and E). And, the fragment 325–573 of GMEB1, which doesn’t interact with CFLARL, didn’t increase the protein level of CFLARL in A549 cell lines.

GMEB1 physically interacted with CFLARL in NSCLC cells. a, b Co-IP assays were conducted in HEK293FT cells using FLAG-CFLARL and HA-GMEB1 plasmids. c GST-pull down assay was conducted in A549 cells using GST-CFLARL plasmids. d IP assays was conducted in A549 and H1299 cells using anti-FLIPL (Santa Cruz, US) antibody. e Calu-1 cells were fixed and subjected to indirect immunofluorescence staining with anti-FLIPL (Santa Cruz, US) and GMEB1 (Santa Cruz, US). The red signal (CFLARL) was obtained with anti-rabbit IgG Alexa 568-conjugated secondary Ab, and the green signals (GMEB1) were obtained with anti-mouse IgG Alexa 488-conjugated secondary Ab. Nuclei were stained with DAPI
USP40 interacted with GMEB1 and CFLARL
We next aimed to identify the de-ubiquitination enzyme that targets CFLARL. We examined protein level of several de-ubiquitinases (USP4, USP40, USP7, USP8 and USP22) in six NSCLC cell lines and found USP40 was positively correlated with CFLARL (Fig. 1a and Additional file 1: Figure S1B) and GMEB1 (Fig. 1a and Additional file 1: Figure S1C). We proposed that USP40 modulated the ubiquitination of CFLARL via its de-ubiquitinase activity. We then examined the interaction between GMEB1 and USP40 using Co-IP assay and found that tagged GMEB1 directly interacted with tagged USP40, which was confirmed by a reverse experiment (Additional file 1: Figure S3A and B). Given that de-ubiquitination enzymes interact with substrates, we then detected if USP40 directly interacted with CFLARL using Co-IP assay in 293FT cells and found that tagged USP40 did in fact interact with tagged CFLARL. This was also confirmed by a reverse experiment (Additional file 1: Figure S3C and D). IP assay using anti-FLIPL showed that endogenous CFLARL interacted with endogenous USP40 in A549 and H1299 cells (Fig. 2d). Immunofluorescence staining results indicate that USP40 also co-localized with CFLARL in cytosol (Additional file 1: Figure S3E). We next aimed to identify the domains of CFLARL that were required for binding with USP40. Our data show that DED domains of CFLARL did not interact with USP40. P20 and P12 fragments of CFLARL interacted with USP40 (Additional file 1: Figures S2A, S3F and S3G).
USP40 targets CFLARL for de-ubiquitination
Previous work showed ITCH targets CFLARL as an E3 ligase, and USP8 is one of the de-ubiquitination enzymes of CFLARL. Here, we found USP40 was positively correlated with and interacted with CFLARL in NSCLC cells. Therefore, USP40 may target CFLARL for de-ubiquitination. To confirm, we first evaluated the function of USP40 on CFLARL in NSCLC cell lines treated with CHX. Results show that knocking down USP40 decreased the stability of CFLARL (Fig. 4a). Furthermore, we knocked down USP40 in H1299, A549 and H157 cell lines using siRNA and treated cells with SAHA [2.0 μM] for 6 h. Data show protein level of CFLARL decreased after knockdown of USP40 (Fig. 4b). Conversely, overexpression of USP40 in H1299 and A549 cell lines increased the protein level of CFLARL (Fig. 4c). These findings demonstrate USP40 promotes the stability of CFLARL at the protein level. We used GST pull-down assays to determine the role of USP40 during CFALRL ubiquitination. Results show that overexpression of USP40 decreased the ubiquitination of CFLARL (Fig. 4d), and knockdown USP40 increased the ubiquitination of CFLARL (Fig. 4e). Additional results show that USP40 targeted CFLARL for K48-linked de-ubiquitination (Fig. 4f). To confirm our proposal, we cloned mutant USP40 (C62A) (Additional file 1: Figure S4A) without enzyme activity [20]. Data show mutant USP40 (C62A) did not increase the protein level of CFLARL in A549 cell lines (Additional file 1: Figure S4B), but it still interacted with CFLARL (Additional file 1: Figure S4C). GST pull-down assay results show mutant USP40 (C62A) did not de-ubiquitinate CFLARL (Additional file 1: Figure S4D). Taken together, these data suggest that USP40 is a de-ubiquitinase of CFLARL.

USP40 targeted CFLARL for de-ubiquitination. a A549 cells were seeded in 6-well plates. USP40 siRNA was transfected for 24 h and a non-sense siRNA was transfected as control. Cells were treated with CHX [10 μg/ml] and harvested at different time points (0, 4, 8, 12 h) for western blot analysis. The band intensity of CFLARL was quantified by Photoshop CS6 and plotted. Experiments were repeated three times, a representative experiment is presented. Every experimental group was compared with the negative control group. Error bars represent s.d. *P < 0.05. b, c H1299, A549 and H157 cells were seeded in 6-well plates. USP40 siRNA or plasmid were transfected for 24 h. Cells were treated with SAHA [2.0 μM] for 6 h and harvested for western blot analysis. d, e HEK293FT cells were prepared for GST pull down assays using GST-CFLARL, HIS-USP40 and HA-Ub plasmids or USP40 siRNA to detect the de-ubiquitination of USP40 targeted on CFLARL. f HA-Ub-K48WT and HA-Ub-K63WT were cloned from HA-Ub wild type plasmid. Only the 48th or 63rd lysine was natural while all other lysine amino acids were mutated to alanine. GST pull down assay was conducted in HEK293FT cells and the de-ubiquitination model of USP40 targeted on CFLARL was detected
GMEB1 promoted the interaction between USP40 and CFLARL
Our findings show that GMEB1, USP40 and CFLARL interact to form a complex. We looked into the function of GMEB1 in this complex. Data from co-IP assay in 293FT cells show that knock down of GMEB1 weakened the interaction between CFLARL and USP40; overexpression of GMEB1 enhanced the interaction (Fig. 5a and b, respectively). In addition, we found that overexpression of HA-GMEB1 (325–573), which did not interact with CFLARL, had no significant impact on the interaction between CFLARL and USP40 (Fig. 5c). Our data indicate that GMEB1 acts as an adaptor protein in the complex, and GMEB1 is essential for the interaction between CFLARL and GMEB1.

GMEB1 promoted the interaction between USP40 and CFLARL. a, b GMEB1 siRNA or plasmid were co-transfected with FLAG-CFLARL and HIS-USP40 in HEK293FT cells. Cells were harvested after 24 h for co-IP assay analysis. c HA-GMEB1–325-573 plasmid were co-transfected with FLAG-CFLARL and HIS-USP40 in HEK293FT cells. Cells were harvested after 24 h for co-IP assay analysis. d H460 and A549 cells were seeded in 6-well plates. GMEB1 plasmid and USP40 siRNA were co-transfected for 24 h; non-sense siRNA and pcDNA3.1 were transfected as control. Cells were harvested for western blot analysis. e A549 cells were seeded in 6-well plates. USP40 plasmid and GMEB1 siRNA were co-transfected for 24 h; non-sense siRNA and pcDNA3.1 were transfected as control. Cells were harvested for western blot analysis
Further, we questioned whether GMEB1 affects CFLARL via USP40. We knocked down USP40 and overexpressed GMEB1 in H460 and A549 cell lines and found no impact on CFLARL (Fig. 5d). Using a reverse experiment in A549 cell lines, we showed that USP40 overexpression slightly increased CFLARL protein when GMEB1 was knocked down (Fig. 5e). These data indicate GMEB1 confers stability to CFLARL via USP40.
GMEB1 inhibited apoptosis via CFLARL and DISC formation upon TRAIL exposure
GMEB1 inhibits the activation of pro-caspases, but the molecular mechanism is still unclear. Our study demonstrates that GMEB1 interacts with CFLARL and promotes CFLARL stability via the de-ubiquitination enzyme USP40. We asked if GMEB1 inhibits apoptosis via CFLARL. GMEB1 siRNA was transfected in A549 and H1299 cell lines for 48 h, and then treated with TRAIL for 6 or 24 h. Western blot results show that GMEB1 knockdown increased the level of cleaved CASP8, CASP3 and PARP (Fig. 6a). Flow Cytometry analysis shows that GMEB1 knockdown enhanced apoptosis of A549 induced by TRAIL (Additional file 1: Figure S5B and C). Further, we transfected plasmid FLAG-CFLARL while knocking down GMEB1 in A549 cells. Western blot results show that overexpression of CFLARL decreased protein levels of cleaved CASP8, CASP3 and PARP that were induced by GMEB1 knockdown and TRAIL treatment (Additional file 1: Figure S5A). Flow Cytometry analysis show that CFLARL overexpression partially attenuated apoptosis induced by GMEB1 knockdown, indicating GMEB1 inhibited apoptosis through regulating CFLARL (Additional file 1: Figure S5D and E). GMEB1 knockdown increased the activation of CASP8 upon TRAIL treatment in A549 cells (Fig. 6b). And another similar experiment showed that CFLARL overexpression partially attenuated the activation of CASP8 upon knocking down GMEB1 and TRAIL treatment (Fig. 6c). We then evaluated the formation of DISC induced by TRAIL treatment. Data show GMEB1 knockdown promoted the interaction of FADD and CASP8 (Fig. 6d and e).

GMEB1 inhibited the formation of DISC upon TRAIL activation. a A549 cells and H1299 cells were seeded in 12-well plates. GMEB1 siRNA was transfected for 24 h and a non-sense siRNA was transfected as negative control. Cells were then treated with TRAIL with different concentrations for 6 h and harvested for western blot analysis. b A549 cells were seeded in 96-well plates. GMEB1 siRNA was transfected for 48 h and a non-sense siRNA was transfected as negative control. Cells were then treated with TRAIL [40 ng/ml] for 6 h and prepared for CASP8 activity detection. c A549 cells were seeded in 6-well plates. GMEB1 siRNA and FLAG-CFLARL plasmid were co-transfected for 48 h. Cells were then treated with TRAIL [40 ng/ml] for 6 h and prepared for CASP8 activity detection. d, e A549 cells were seeded in 96-well plates. GMEB1 siRNA was transfected for 48 h and a non-sense siRNA was transfected as negative control. Cells were then treated with TRAIL at different concentrations for 4 h and harvested for IP assay using FADD or CASP8 antibody
CFLARL is critical for the interaction of GMEB1 and CASP8
GMEB1 interacts with pro-caspase 8 and inhibits its activation, but the mechanism is not clear. Our experiments indicated that GMEB1 also interacted with CFLARL, which interacted with pro-caspase 8 and inhibited its activity. We questioned which protein plays the dominant role in the interaction with GMEB1, CASP8 or CFLARL. Co-IP assays were conducted to evaluate the interaction between GMEB1 and CFLARL while CASP8 was knocked down using siRNA. Results show no significant change (Fig. 7a). We conducted another co-IP assay to evaluate the interaction between GMEB1 and CASP8 (using a plasmid HIS-CASP8M that 374 and 384 Aspartic acids mutated to Alanine acids) while CFLARL was knocked down using siRNA. Results show CFLARL knockdown decreased the interaction between GMEB1 and CASP8 (Fig. 7b). These results confirm CFLARL directly interacts with GMEB1, and CFLARL plays a critical role in the interaction of CASP8 and GMEB1.

CFLARL is critical for the interaction of GMEB1 and CASP8. a CASP8 siRNA were transfected in HEK293FT cells with FLAG-CFLARL and HA-GMEB1 plasmids for a co-IP assay. b CFLARL siRNA were transfected in HEK293FT cells with HIS-CASP8M and FLAG-GMEB1 plasmids for a co-IP assay
Downregulation of GMEB1 inhibited A549 xenograft tumor growth in vivo
To evaluate whether the tumor growth of A549 is regulated by GMEB1 in vivo, A549-LUC, A549-shGMEB1–1# and A549-shGMEB1–2# were subcutaneously injected into the right side of the abdominal region of athymic nu/nu mice. GMEB1 and CFLARL protein levels were detected in A549 cell lines (Fig. 8a). Mice weights were measured using an electronic balance, results show no significant difference among the three groups (Fig. 8b). Tumor sizes were measured with calipers (Fig. 8c). Results show tumor growth was inhibited after GMEB1 knockdown compared with the control group (Fig. 8d). And, tumor weights also support this finding (Fig. 8e). These results suggest GMEB1 plays a key role in cellular mechanisms related to apoptosis and cancer progress.

Downregulation of GMEB1 inhibited A549 xenograft tumor growth in vivo. a A549 cells stably overexpressing shGMEB1 RNA were seeded in 6-well plates and harvested after 24 h for western blot analysis. b A549 cells stably overexpressing shGMEB1 RNA were subcutaneously injected into the right side of the abdominal region of athymic nu/nu mice. Mice weights were measured using electronic balance every 2 days. c Tumor sizes were measured with calipers. The error bars represent the SD, ** P < 0.01, and *** P < 0.001. d Tumors were dissected and pictures taken. e Tumor weights were measured using electronic balance. The error bars represent the SD, ** P < 0.01, and *** P < 0.001
Discussion
GMEB1 is found throughout many cell types with multiple functions that are now being uncovered. GMEB1 and GMEB2 bind to the promoter sequence of the TAT gene and modulate GR transactivation. In the cytosol, GMEB1 interacts with HSP27, a protein chaperone with many critical functions in cancer invasion, metastasis, proliferation and apoptosis [21,22,23,24,25,26,27,28,29]. HSP27 is related to CFLARL and the activation of caspases [30, 31]. However, the relationship between GMEB1 and HSP27 has not been well studied. Recently, GMEB1 was reported to inhibit cell apoptosis by binding to pro-caspases and inhibiting their activation in cytosol [6]. GMEB1 interacts with pro-caspases via DED domains, but the molecular mechanism is unknown.
CFLARL is an apoptotic inhibitor protein that interacts with pro-caspase 8 and inhibits its activation; DED domains play important roles in this interaction. The interaction between CFLARL and pro-caspase 8 inhibits the formation of DISC [15]. As an inhibitor of histone deacetylase, SAHA is effective in treating skin T cell lymphoma clinically. It enhances the acetylation of Ku70 and disrupts the CFLAR/Ku70 complex and then triggers CFLAR poly-ubiquitination and degradation by the proteasome [32, 33]. SAHA treatment in NSCLC cells shows that SAHA decreased CFLARL and GMEB1 protein levels in a dose-dependent and a time-dependent manner. The results indicate GMEB1 and CFLARL are positively correlated, which was confirmed by analyzing protein levels in six NSCLC cell lines and a HEK293FT cell line. In addition, GMEB1 protein reduction by SAHA suggests a relationship similar to SAHA and CFLARL.
Given that GMEB1 acts in the nucleus as a transcription factor, we first conducted q-PCR experiments to check the function of GMEB1. We found GMEB1 affected the transcription of CFLARL. We then turned our attention to the cytosol. Knock-down and overexpression experiments showed that GMEB1 promoted the stability and directly regulated the protein level of CFLARL. Our in vitro studies indicated that GMEB1 interacts with CFLARL outside the nucleus. DED domains of CFLARL were not necessary for this process; P20 and P12 domains of CFLARL accounted for the interaction with GMEB1. Our results also show that N-terminal of GMEB1 interacted with CFLARL, and GMEB1 (325–573) fragments, which did not interact with CFLARL, couldn’t increase the protein level of CFLARL. These findings suggest that the function of GMEB1 is dependent on the interaction with CFLARL. Our experiments also indicate GMEB1 affects the degradation of CFLARL through proteasome pathway. GMEB1 doesn’t have ubiquitin-related enzymatic activity that directly modulates the ubiquitination of CFLARL. This suggests that another enzyme is needed in this process. Therefore, USP40 was emphasized because its expression was positively correlated with GMEB1 and CFLARL in NSCLC cells.
CFLARL is an effective target for cancer therapy [9,10,11,12]. ITCH, which has important roles in cell immune regulation [34], was identified as an E3 ligase targeting CFLARL. It induces apoptosis by degrading CFLARL and activating pro-caspase 8. De-ubiquitination is an important protein modification that reverses the ubiquitination of proteins via E1/E2/E3 ligase. De-ubiquitination inhibits the degradation of proteins and drives the fate of substrate proteins [35]. USP8 regulates the morphology of the endosome by ubiquitinating proteins and is also involved in cargo sorting and membrane trafficking at the early endosome stage [36, 37]. USP8 is a de-ubiquitination enzyme of CFLARL that promotes the stability of CFLARL and inhibits the activation of pro-caspase 8. Changes in CFLARL protein levels also affect the formation of DISC and apoptosis induced by extrinsic ligands.
To find the de-ubiquitinase that regulates CFLARL by GMEB1, we focused on USP8 that was identified as a de-ubiquitinase of CFLARL. But our experiments showed that GMEB1 did not interact with USP8 (Additional file 1: Figure S4E). Western blot data from six NSCLC lines suggested USP40 protein levels are positively correlated with GMEB1 and CFLARL. This underscores the importance of USP40.
Several reports show that USP40 is correlated with late-onset Parkinson’s disease and USP24 [38, 39]. In addition, USP40 affects glomerular permeability in zebrafish [40]. Our results suggest USP40 interacts with both GMEB1 and CFLARL. GMEB1 promoted the binding of USP40 with CFLARL, conferring a role as an adaptor protein. Consequently, USP40 augmented the stability of CFLARL via its de-ubiquitinase activity. USP40 targeted CFLARL for K48-linked de-ubiquintion. USP40 knockdown did not increase the protein level of CFLARL even though GMEB1 protein level was overexpressed. And, overexpression of USP40 increased the protein level of CFLARL though GMEB1 was knocked down. Our data indicate that USP40 is the key protein affecting GMEB1 on CFLARL.
In addition, we also found that GMEB1 knockdown promoted the activation of pro-caspase 8 and apoptosis induced by TRAIL. CFLARL attenuated apoptosis that was induced by GMEB1 knockdown, which highlights the function of GMEB1 in inhibiting apoptosis via CFLARL. GMEB1 inhibited the formation of DISC upon TRAIL activation.
Previous studies show GMEB1 interacts with the DED domain of CASP8 and inhibits its activation. Our results show GMEB1 did not interact with DED domain of CFLARL which has a similar structure with CASP8. However, GMEB1 interacted with CFLARL via the P20 and P12 domains. Co-IP results showed that CASP8 knockdown didn’t affect the interaction between GMEB1 and CFLARL, while CFLARL knockdown decreased the interaction between GMEB1 and CASP8. These findings suggest that CFLARL is crucial for the interaction between GMEB1 and CASP8. Thus, GMEB1 interacts with CFLARL, and CFLARL interacts with CASP8 via DED domains. GMEB1 inhibits the activation of CASP8 via the function of CFLARL.
In vivo data showed GMEB1 knockdown inhibited the A549 xenograft tumor growth, which also confirmed our results.
Conclusions
We described the interaction among GMEB1, USP40 and CFLARL (Fig. 9). We found that GMEB1 promoted the stability of CFLARL by de-ubiquitinase USP40. USP40 targeted CFLARL for K48-linked de-ubiquitination. GMEB1 inhibited the activation of CASP8 and apoptosis in NSCLC via CFLARL. CFLARL promoted the interaction between GMEB1 and CASP8. GMEB1 knockdown inhibited tumor growth in vivo. These findings provide more in-depth knowledge that serves as potential therapies for cancer.

Regulatory mechanism of CFLARL by GMEB1 and USP40. GMEB1 acts as an adaptor protein to improve the interaction of USP40 and CFLARL. USP40 targets CFLARL for de-ubiquitination to stabilize CFLARL protein levels. GMEB1 inhibits the activation of pro-caspase 8 via CFLARL and inhibits apoptosis
Abbreviations
- CFLAR:
-
Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein
- DISC:
-
Death-inducing signaling complex
- HSP27:
-
Heat Shock Protein Family B (Small) Member 1
- ITCH:
-
Itchy E3 Ubiquitin Protein Ligase
- NSCLC:
-
Non-small cell lung cancer
- PARP:
-
Poly ADP-ribose polymerase
References
Zeng H, Jackson DA, Oshima H, Simons SJ. Cloning and characterization of a novel binding factor (GMEB-2) of the glucocorticoid modulatory element. J Biol Chem. 1998;273:17756–62.
Theriault JR, Charette SJ, Lambert H, Landry J. Cloning and characterization of hGMEB1, a novel glucocorticoid modulatory element binding protein. FEBS Lett. 1999;452:170–6.
Oshima H, Szapary D, Simons SJ. The factor binding to the glucocorticoid modulatory element of the tyrosine aminotransferase gene is a novel and ubiquitous heteromeric complex. J Biol Chem. 1995;270:21893–901.
Zeng H, Kaul S, Simons SJ. Genomic organization of human GMEB-1 and rat GMEB-2: structural conservation of two multifunctional proteins. Nucleic Acids Res. 2000;28:1819–29.
Tsuruma K, Nakagawa T, Shirakura H, Hayashi N, Uehara T, Nomura Y. Regulation of procaspase-2 by glucocorticoid modulatory element-binding protein 1 through the interaction with caspase recruitment domain. Biochem Biophys Res Commun. 2004;325:1246–51.
Tsuruma K, Nakagawa T, Morimoto N, Minami M, Hara H, Uehara T, Nomura Y. Glucocorticoid modulatory element-binding protein 1 binds to initiator procaspases and inhibits ischemia-induced apoptosis and neuronal injury. J Biol Chem. 2006;281:11397–404.
Nakagawa T, Tsuruma K, Uehara T, Nomura Y. GMEB1, a novel endogenous caspase inhibitor, prevents hypoxia- and oxidative stress-induced neuronal apoptosis. Neurosci Lett. 2008;438:34–7.
Kawabe K, Lindsay D, Braitch M, Fahey AJ, Showe L, Constantinescu CS. IL-12 inhibits glucocorticoid-induced T cell apoptosis by inducing GMEB1 and activating PI3K/Akt pathway. Immunobiology. 2012;217:118–23.
Safa AR, Day TW, Wu CH. Cellular FLICE-like inhibitory protein (C-FLIP): a novel target for cancer therapy. Curr Cancer Drug Targets. 2008;8:37–46.
Safa AR. c-FLIP, a master anti-apoptotic regulator. Exp Oncol. 2012;34:176–84.
Safa AR, Pollok KE. Targeting the anti-apoptotic protein c-FLIP for Cancer therapy. Cancers (Basel). 2011;3:1639–71.
Yu JW, Jeffrey PD, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci U S A. 2009;106:8169–74.
Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, Liu YC, Karin M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–13.
Murata E, Hashimoto M, Aoki T. Interaction between cFLIP and itch, a ubiquitin ligase, is obstructed in Trypanosoma cruzi-infected human cells. Microbiol Immunol. 2008;52:539–43.
Jeong M, Lee EW, Seong D, Seo J, Kim JH, Grootjans S, Kim SY, Vandenabeele P, Song J. USP8 suppresses death receptor-mediated apoptosis by enhancing FLIPL stability. Oncogene. 2017;36:458–70.
Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst. 2004;96:1769–80.
Yerbes R, Lopez-Rivas A. Itch/AIP4-independent proteasomal degradation of cFLIP induced by the histone deacetylase inhibitor SAHA sensitizes breast tumour cells to TRAIL. Investig New Drugs. 2012;30:541–7.
Al-Yacoub N, Fecker LF, Mobs M, Plotz M, Braun FK, Sterry W, Eberle J. Apoptosis induction by SAHA in cutaneous T-cell lymphoma cells is related to downregulation of c-FLIP and enhanced TRAIL signaling. J Invest Dermatol. 2012;132:2263–74.
Lauricella M, Ciraolo A, Carlisi D, Vento R, Tesoriere G. SAHA/TRAIL combination induces detachment and anoikis of MDA-MB231 and MCF-7 breast cancer cells. Biochimie. 2012;94:287–99.
Quesada V, Diaz-Perales A, Gutierrez-Fernandez A, Garabaya C, Cal S, Lopez-Otin C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem Biophys Res Commun. 2004;314:54–62.
Lianos GD, Alexiou GA, Mangano A, Mangano A, Rausei S, Boni L, Dionigi G, Roukos DH. The role of heat shock proteins in cancer. Cancer Lett. 2015;360:114–8.
Bakthisaran R, Tangirala R, Rao C. Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta. 2015;1854:291–319.
Pavan S, Musiani D, Torchiaro E, Migliardi G, Gai M, Di Cunto F, Erriquez J, Olivero M, Di Renzo MF. HSP27 is required for invasion and metastasis triggered by hepatocyte growth factor. Int J Cancer. 2014;134:1289–99.
Zhang X, Shi J, Tian J, Robinson AC, Davidson YS, Mann DM. Expression of one important chaperone protein, heat shock protein 27, in neurodegenerative diseases. Alzheimers Res Ther. 2014;6:78.
Zhang S, Hu Y, Huang Y, Xu H, Wu G, Dai H. Heat shock protein 27 promotes cell proliferation through activator protein-1 in lung cancer. Oncol Lett. 2015;9:2572–6.
Tan JGL, Lee YY, Wang T, Yap MGS, Tan TW, Ng SK. Heat shock protein 27 overexpression in CHO cells modulates apoptosis pathways and delays activation of caspases to improve recombinant monoclonal antibody titre in fed-batch bioreactors. Biotechnol J. 2015;10:790–800.
Kim J, Kim SY, Kang S, Yoon HR, Sun BK, Kang D, Kim JH, Song JJ. HSP27 modulates survival signaling networks in cells treated with curcumin and TRAIL. Cell Signal. 2012;24:1444–52.
Ghosh A, Lai C, McDonald S, Suraweera N, Sengupta N, Propper D, Dorudi S, Silver A. HSP27 expression in primary colorectal cancers is dependent on mutation of KRAS and PI3K/AKT activation status and is independent of TP53. Exp Mol Pathol 2013;94: 103–108.
Qi S, Xin Y, Qi Z, Xu Y, Diao Y, Lan L, Luo L, Yin Z. HSP27 phosphorylation modulates TRAIL-induced activation of Src-Akt/ERK signaling through interaction with beta-arrestin2. Cell Signal. 2014;26:594–602.
Lee SW, Cho JM, Cho HJ, Kang JY, Kim EK, Yoo TK. Expression levels of heat shock protein 27 and cellular FLICE-like inhibitory protein in prostate cancer correlate with Gleason score sum and pathologic stage. Korean J Urol. 2015;56:505–14.
Kim SS, Cho HJ, Cho JM, Kang JY, Yang HW, Yoo TK. Dual silencing of Hsp27 and c-FLIP enhances doxazosin-induced apoptosis in PC-3 prostate cancer cells. ScientificWorldJournal. 2013;2013:174392.
Kerr E, Holohan C, McLaughlin KM, Majkut J, Dolan S, Redmond K, Riley J, McLaughlin K, Stasik I, Crudden M, Van Schaeybroeck S, Fenning C, O'Connor R, Kiely P, Sgobba M, Haigh D, Johnston PG, Longley DB. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012;19:1317–27.
Kim MJ, Hong KS, Kim HB, Lee SH, Bae JH, Kim DW, Dao TT, Oh WK, Kang CD, Kim SH. Ku70 acetylation and modulation of c-Myc/ATF4/CHOP signaling axis by SIRT1 inhibition lead to sensitization of HepG2 cells to TRAIL through induction of DR5 and down-regulation of c-FLIP. Int J Biochem Cell Biol. 2013;45:711–23.
Aki D, Zhang W, Liu YC. The E3 ligase itch in immune regulation and beyond. Immunol Rev. 2015;266:6–26.
Kalderon D. Protein degradation: de-ubiquitinate to decide your fate. Curr Biol. 1996;6:662–5.
Niendorf S, Oksche A, Kisser A, Lohler J, Prinz M, Schorle H, Feller S, Lewitzky M, Horak I, Knobeloch KP. Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol Cell Biol. 2007;27:5029–39.
MacDonald E, Urbe S, Clague MJ. USP8 controls the trafficking and sorting of lysosomal enzymes. Traffic. 2014;15:879–88.
Li Y, Schrodi S, Rowland C, Tacey K, Catanese J, Grupe A. Genetic evidence for ubiquitin-specific proteases USP24 and USP40 as candidate genes for late-onset Parkinson disease. Hum Mutat. 2006;27:1017–23.
Wu YR, Chen CM, Chen YC, Chao CY, Ro LS, Fung HC, Hsiao YC, Hu FJ, Lee-Chen GJ. Ubiquitin specific proteases USP24 and USP40 and ubiquitin thiolesterase UCHL1 polymorphisms have synergic effect on the risk of Parkinson's disease among Taiwanese. Clin Chim Acta. 2010;411:955–8.
Takagi H, Nishibori Y, Katayama K, Katada T, Takahashi S, Kiuchi Z, Takahashi SI, Kamei H, Kawakami H, Akimoto Y, Kudo A, Asanuma K, Takematsu H, Yan K. USP40 gene knockdown disrupts glomerular permeability in zebrafish. Am J Physiol Renal Physiol. 2017;312:F702–15.
Acknowledgements
We thank Dr. Lingqiang Zhang and Dr. Ping Wang for their kindly provided USP40 plasmids. We also thank Dr. Austin Cape at ASJ Editors for careful review and feedback.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (31571422, 81672291, 81672855, 31771526 and 31371402,) and the Science and technology development plan of Shandong Province (2016GSF201153).
Availability of data and materials
The datasets used/analyzed to support the conclusions of this article are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
Xiangguo Liu, Ling Su and Yidan Lin conceived the study. Xiangguo Liu, Wentao An and Ling Su designed the experiments. Wentao An performed most of the experiments. Wentao An, Shun Yao and Xiaoyang Sun performed the animal experiments. Zhaoyuan Hou provided GMEB1 shRNA plasmids. Wentao An and Xiangguo Liu interpreted the data and wrote the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All the animal experiments were carried out in accordance with the approval of the Animal Research Committee of Shandong University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing of interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. CFLARL protein level positively correlated with GMEB1 and USP40 protein levels in NSCLC cell lines. Figure S2. CFLARL directly interact with GMEB1. Figure S3. USP40 interacted with GMEB1 and CFLARL. Figure S4. USP40 targeted CFLARL for de-ubiquitination. Figure S5. GMEB1 inhibited apoptosis via CFLARL. (DOCX 1283 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
An, W., Yao, S., Sun, X. et al. Glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the de-ubiquitinase USP40 to stabilize CFLARL and inhibit apoptosis in human non-small cell lung cancer cells. J Exp Clin Cancer Res 38, 181 (2019). https://doi.org/10.1186/s13046-019-1182-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1182-3