
Abstract
Background
Neonatal Marfan syndrome (nMFS) is a rare condition characterized by severe phenotype and poor prognosis. nMFS is caused by mutations in a specific region of the fibrillin 1 gene (FBN1). Prompt recognition of typical signs of neonatal presentation, such as characteristic facial anomalies with senile appearance, arthrogryposis, and campto-arachnodactyly, is fundamental for performing an early cardiological examination. This usually reveals rapidly progressive cardiovascular disease due to severe atrioventricular valve dysfunction.
Case presentation
Herein, we report the case of an early-onset cardiac failure in a neonate with Marfan syndrome, with a brief review of the literature of cases with cardiac involvement in neonatal age. Clinical exome sequencing identified the novel heterozygous de novo missense variant c.3152T > G in FBN1 gene (NM_000138.4), causing the aminoacidic change p.Phe1051Cys. Phenotype-genotype correlation led to a multidisciplinary diagnostic and management workflow.
Conclusion
The prompt recognition of a typical phenotype such as that of Marfan syndrome should lead to a detailed evaluation and close follow-up of cardiac morphology and function. Indeed, multi-disciplinary evaluation based on genotype-phenotype correlations of nMFS cases is essential to finding out the best medical and surgical approach, predicting the relevant impact on patient prognosis, and adequately counseling their families.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
Neonatal Marfan syndrome (nMFS) is a rare condition characterized by severe phenotype and poor prognosis, caused by mutations in the specific “neonatal region” of the fibrillin 1 gene (FBN1) [1]. Prompt recognition of typical signs of neonatal presentation, such as characteristic facial anomalies with senile appearance, arthrogryposis, and campto-arachnodactyly, is fundamental for performing an early cardiological examination. This usually reveals rapidly progressive cardiovascular disease due to severe atrioventricular valve dysfunction.
Herein, we report the case of an early-onset cardiac failure in a neonate with Marfan syndrome, with a brief review of the literature of cases with cardiac involvement in neonatal age. Clinical exome sequencing revealed a de novo missense variant of the FBN1 gene. Phenotype-genotype correlation led to a multidisciplinary diagnostic and management workflow.
Case presentation
A female neonate spontaneously conceived was born in a 2nd-level hospital at 39 weeks of gestational age (GA), to a 32-year-old primigravida through spontaneous delivery. Oligohydramnios and multiple complex choroid cysts were noticed in the last two weeks of pregnancy. No invasive prenatal testing was performed. Family history revealed a first-degree cousin (from the paternal side) affected by de novo Baraitser-Winter syndrome, and a paternal aunt whose pregnancy was interrupted because of a not-specified chromosomal disorder.
Apgar score was 8 and 9 at the 1st and 5th minutes, respectively. Birth weight was 2750 gr (16th centile, z-score: -1,01 SDS according to INeS charts [2]), length 49 cm (47th centile, z-score: -0.07 SDS), and head circumference 35 cm (86th centile, z-score: 1.06 SDS). At birth, several dysmorphic features were noticed, including brachycephaly, triangular and asymmetric face with a typical “senile” appearance (Fig. 1.A) and hypertelorism, down-slanted palpebral fissures, blepharophimosis, blue sclerae, anteverted nares, narrow mouth, micrognathia, and low-set ear. A distal arthrogryposis of the upper (Fig. 1.B) and lower limbs and severe arachnodactyly of hands (Fig. 1.C) and feet were evident. In particular, the Steinberg sign (well-known as the “thumb” sign) was positive in both hands (Fig. 1.C).

Dysmorphic features of our patient: A) senile facial appearance; B) distal arthrogryposis of upper limbs; C) severe arachnodactyly of hands and positive Steinberg sign (or thumb sign) of left hand
Cerebral ultrasound (CUS) performed within the first days of life confirmed multiple microcysts of the choroid plexus and increased echogenicity in the periventricular white matter. Brain magnetic resonance imaging (MRI) revealed a dysplastic appearance of cerebellar vermis and hemispheres, with a markedly thickened cerebellar cortex and loss of normal arborization of white matter. Hypoxic-ischemic lesions were noticed in the right frontal area. Chest computed tomography (CT), performed because of respiratory distress, showed a right posterolateral diaphragmatic relaxation with ipsilateral atelectasis of lung tissue. Skeletal X-ray showed no significant malformations. Echocardiography within the first 5 days of life yielded patent foramen ovale and patent ductus arteriosus (both with left-to-right shunt), mild tricuspid insufficiency, and moderate mitral insufficiency. A mild dilatation of the aortic bulb was also observed.
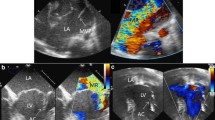
The neonate was admitted on the 15th day of life to our 3rd-level children’s hospital to perform a specialistic evaluation. Physical examination revealed fair general conditions with polypnea and mild dyspnea; a 3/6 systolic murmur was audible. Cardiomegaly and dilation of the left ventricle were detected by echocardiography, with a mild-to-moderate biventricular dysfunction. A severely dysplastic mitral valve showed severe multi-jet insufficiency (Fig. 2.A), and aortic valve showed mild insufficiency (Fig. 2.B) and moderate dilation of Valsalva sinuses. The right ventricle was mildly dilated, with mild-to-moderate insufficiency of the tricuspid valve, and a dysplastic pulmonary valve with moderate insufficiency was also observed. The ophthalmological examination did not reveal pathological signs. Multivalvular involvement required initial conservative medical treatment using intravenous furosemide (up to 3 mg/kg) associated with oral spironolactone and captopril. Medical treatment led to a gradual decreasing trend of brain natriuretic peptide (BNP) and troponin values. The baby was discharged in her 5th month of life.

Cardiac involvement in our patient: A) severe mitral insufficiency from a 4-chamber view; B) aortic valve insufficiency from long axis view
Blood karyotype and Chromosomal Microarray Analysis using platform Illumina® CytoSNP 850k showed no pathogenic results. After these negative results, at 2 months of life, clinical exome analysis of trios was performed on DNA extracted from circulating leukocytes using kit Twist Custom Panel (Twist Bioscience) on the Illumina sequencing platform (NovaSeq6000, San Diego, CA). Next Generation Sequencing (NGS) analysis identified the novel heterozygous de novo missense variant c.3152T > G in FBN1 gene (NM_000138.4), causing the aminoacidic change p.Phe1051Cys. The variant was never described in the literature; it was not on the Genome Aggregation Database (gnomAD) and was classified as probably pathogenetic (class 4) according to guidelines of the American College of Medical Genetics and Genomics (ACMG) [3].
Subsequently, severe feeding intolerance and gastroesophageal reflux led to progressive malnutrition and a significant impact on children’s physical growth; at the 6th month of life, the infant was admitted again to our hospital and required nutrition support through the placement of a percutaneous endoscopic gastrostomy (PEG) tube. Despite maximal medical treatment and PEG placement, the infant progressively developed symptoms of congestive heart failure resulting from severe multivalvular insufficiency. At about 7 months of life, she underwent cardiosurgical intervention with mitral valvuloplasty and the use of annuloplasty, tricuspid, and pulmonary valvuloplasty.
Unfortunately, at 9 months and 19 days, the infant died of complications related to cardiac failure and severe malnutrition status (weight at death time 5070 gr, z-score: -3.04 according to WHO charts [4]).
Methods
In order to review the literature about cardiac involvement in neonatal Marfan syndrome and compare other cases to ours, an extensive literature search in the MEDLINE database (via PubMed) has been performed up to December 31st, 2022. The following keywords, “neonatal,” “Marfan,” and “syndrome,” were searched as entree terms as well. All 239 retrieved articles of the last 20 years were screened, and then full texts of records deemed eligible for inclusion were assessed. References in the relevant papers were also reviewed. Papers written in languages other than English were excluded. Papers reporting a full description of the cases were included.
We systemically collected and summarized information on patients’ characteristics, cardiac involvement and procedures, and molecular findings.
Results
A brief review of the literature on nMFS cases with cardiac involvement in the last 20 years is displayed in Table 1 [1, 5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. Beyond our case, we included other 27 cases, of whom three were born preterm. All 28 cases except one (reported by Postma et al.) had a birthweight greater than 2500 g. The mitral valve was involved in all cases, whereas the tricuspid valve was in 20/28 patients (71.4%). Aortic structures were involved in 23/28 cases (82.1%). Fifteen patients (53.6%) underwent cardiac procedures. FBN1 was the involved gene in all patients where a genetic diagnosis was available (21/28: 75%). Thirteen patients (46.4%) died at the time of writing.
Discussion and conclusion
We compared the severe cardiac involvement of a neonate with Marfan syndrome to the cases available in the literature. Cardiac involvement is the main determinant in the prognosis of neonates with Marfan syndrome, and it is usually life-threatening. Severe valvular disease affecting mostly mitral and tricuspid valves quickly progresses to congestive heart failure and premature death within the first 2 years of life [1, 21, 22, 29, 30]. Indeed, cardiac involvement in neonates is usually characterized by severe multivalvular insufficiency instead of aortic structures involvement, which is a typical feature in adults and older children [31, 32].
Congenital pulmonary emphysema is also often detected in nMFS [33]. Rarely, patients with nMFS may develop progressive but not fatal heart failure; some young adults have been described [34].
Molecular analysis shows that most nMFS mutations are sporadic and occur in the so-called “neonatal region” of FBN1 gene mapping between exons 24 and 32 [35, 36].
In our case, we identified a novel missense variant, p.Phe1051Cys, located in exon 26 of the FBN1 gene inside the “neonatal critical region” that was not previously reported in the literature.
Interesting genotype-phenotype associations for both cardiovascular and extra-cardiovascular manifestations were identified in the pediatric population [37]. Previous studies and case reports demonstrated that mutations located in FBN1 “neonatal region” are usually associated with a rapidly worsening cardiac disease, poor response to medications [36], and severe congestive heart failure, which represent the main cause of early death. In particular, patients carrying variants in a specific region (amino acids 1028–1088, corresponding to exon 25 and a few residues from exon 26) show a worse prognosis with heart failure-related death within the first year of life, irrespective of gender [38].
Early genotype analysis and prompt phenotype recognition can potentially drive accurate genetic counseling. Indeed, the prompt recognition of a typical phenotype such as that of Marfan syndrome should lead to a detailed evaluation and close follow-up of cardiac morphology and function. Timely diagnosis is increasingly important in looking for genotype-phenotype characterization and improving early therapeutic strategies.
Despite their low quantity in skeletal matrices, MFS causes severe skeletal defects, highlighting the importance of fibrillin-1 and microfibrils in bone formation and function [39]. The extent of musculoskeletal disease is quite significant in older patients with Marfan syndrome: scoliosis, pectus deformity, and deformity of the foot. Many will need a specific follow-up, requiring corrective surgery during their life span [40].
Similarly, fibrillin-1-containing microfibrils are ubiquitous in the normal eye. Ectopia lentis in MFS patients is likely caused by an FBN1 mutation that prevents fibrillin-1 production. If they survive, MFS patients experience different ocular features depending on the mutation and severity of the illness. Patients with MFS typically acquire lenticular and/or axial myopia before the age of ten and should see an ophthalmologist to examine their near-sightedness [39].
Multi-disciplinary evaluation based on genotype-phenotype correlations of nMFS cases is essential to determine the best medical and surgical approach, predict the relevant impact on patient prognosis, and adequately counsel their families. MFS is an example of a syndrome where an early personalized approach to address a dynamic, genetically determined condition can make a difference in outcome [41].
In light of this, a careful evaluation of all clinical signs by neonatologists is mandatory: in particular, the Steinberg sign (also known as the thumb sign) presence should be considered a potential handle sign for diagnostic suspicion, and every neonatologist should rule out Marfan syndrome in cases like this. This can be useful to perform a correct differential diagnosis, like, for example, with distal arthrogryposis syndromes or other congenital defects with cardiac involvement, with the aim of timely reaching the correct diagnosis [42, 43].
The current databases should be updated with the genomic and phenotypic findings of the present patient in order to provide a better characterization of such a rare disease. Additional patients and the identification of new mutations will increase the knowledge of the molecular bases and the pathogenic mechanisms underlying Marfan syndrome with neonatal onset.
Finally, clinicians must be aware of the possibility that neonates may have a severely poor outcome, even in the absence of symptoms in the first weeks of life.
Data availability
All data generated or analysed during this study are included in this published article.
Abbreviations
- ACMG:
-
American College of Medical Genetics
- CT:
-
Computed tomography
- CUS:
-
Cerebral ultrasound
- FBN1:
-
Fibrillin 1
- GA:
-
Gestational age
- gnomAD:
-
Genome Aggregation Database
- MRI:
-
Magnetic resonance imaging
- NGS:
-
Next Generation Sequencing
- nMFS:
-
Neonatal Marfan syndrome
- PEG:
-
Percutaneous endoscopic gastrostomy
- SDS:
-
Standard deviation
- WHO:
-
World Health Organization
References
Amado M, Calado MA, Ferreira R, Lourenço T. Neonatal Marfan syndrome: a successful early multidisciplinary approach. Case Rep. 2014;bcr2013202438. https://doi.org/10.1136/bcr-2013-202438
Bertino E, Spada E, Occhi L, Coscia A, Giuliani F, Gagliardi L, Gilli G, Bona G, Fabris C, De Curtis M, Milani S. Neonatal anthropometric charts: the Italian neonatal study compared with other European studies. J Pediatr Gastroenterol Nutr. 2010;51:353–61. https://doi.org/10.1097/MPG.0b013e3181da213e
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30
The WHO Child Growth Standards. https://www.who.int/tools/child-growth-standards/standards. (Accessed 11 Jan 2023).
Jacobs AM, Toudjarska I, Racine A, Tsipouras P, Kilpatrick MW, Shanske A. A recurring FBN1 gene mutation in neonatal Marfan syndrome. Arch Pediatr Adolesc Med. 2002;156(11):1081–5. https://doi.org/10.1001/archpedi.156.11.1081
Shinawi M, Boileau C, Brik R, Mandel H, Bentur L. Splicing mutation in the fibrillin-1 gene associated with neonatal Marfan syndrome and severe pulmonary emphysema with tracheobronchomalacia. Pediatr Pulmonol. 2005;39(4):374–8. https://doi.org/10.1002/ppul.20174
Ramaswamy P, Lytrivi ID, Nguyen K, Gelb BD. Neonatal Marfan syndrome: in utero presentation with aortic and pulmonary artery dilatation and successful repair of an acute flail mitral valve leaflet in infancy. Pediatr Cardiol. 2006;27(6):763–5. https://doi.org/10.1007/s00246-006-1378-0
Sutherell J, Zarate Y, Tinkle BT, Markham LW, Cripe LH, Hyland JC, Witte D, Hopkin RJ, Hinton RB. Novel fibrillin 1 mutation in a case of neonatal Marfan syndrome: the increasing importance of early recognition. Congenit Heart Dis. 2007;2(5):342–6. https://doi.org/10.1111/j.1747-0803.2007.00123.x
Tekin M, Cengiz FB, Ayberkin E, Kendirli T, Fitoz S, Tutar E, Ciftçi E, Conba A. Familial neonatal Marfan syndrome due to parental mosaicism of a missense mutation in the FBN1 gene. Am J Med Genet A. 2007;143A(8):875–80. https://doi.org/10.1002/ajmg.a.31660
Kochilas L, Gundogan F, Atalay M, Bliss JM, Vatta M, Pena LS, Abuelo D. A novel mutation of the fibrillin-1 gene in a newborn with severe Marfan syndrome. J Perinatol. 2008;28(4):303–5. https://doi.org/10.1038/sj.jp.7211915
Brito-Filho SL, Oporto V, Campos O, Alvares AB, Carvalho AC. A case of neonatal Marfan syndrome with good late follow-up: is it possible to avoid an early unfavourable outcome? Cardiol Young. 2013;23(2):301–3. https://doi.org/10.1017/S104795111200090X
Sípek A Jr, Grodecká L, Baxová A, Cibulková P, Dvořáková M, Mazurová S, Magner M, Zeman J, Honzík T, Freiberger T. Novel FBN1 gene mutation and maternal germinal mosaicism as the cause of neonatal form of Marfan syndrome. Am J Med Genet A. 2014;164A(6):1559–64. https://doi.org/10.1002/ajmg.a.36480
Elshershari H, Harris C. Paternal fibrillin-1 mutation transmitted to an affected son with neonatal marfan syndrome: the importance of early recognition. Cardiol Young. 2014;24(4):735–8. https://doi.org/10.1017/S1047951113001029
Ozyurt A, Baykan A, Argun M, Pamukcu O, Halis H, Korkut S, Yuksel Z, Gunes T, Narin N. Early onset marfan syndrome: atypical clinical presentation of two cases. Balkan J Med Genet. 2015;18(1):71–6. https://doi.org/10.1515/bjmg-2015-0008
Buthia E, Kumar P, Kishore S, Yadav DK. Unremitting congestive heart failure: neonatal Marfan syndrome. J Clin Neonatol. 2016;5(2):128–30.
Kitahara H, Aeba R, Takaki H, Shimizu H. Palliative mitral valve repair during infancy for neonatal Marfan syndrome. Ann Thorac Surg. 2016;101(5):1987–8. https://doi.org/10.1016/j.athoracsur.2015.06.115
Le Gloan L, Hauet Q, David A, Hanna N, Arfeuille C, Arnaud P, Boileau C, Romefort B, Benbrik N, Gournay V, Joram N, Baron O, Isidor B. Neonatal Marfan Syndrome: report of a case with an inherited splicing mutation outside the neonatal domain. Mol Syndromol. 2016;6(6):281–6. https://doi.org/10.1159/000443867
Maeda J, Kosaki K, Shiono J, Kouno K, Aeba R, Yamagishi H. Variable severity of cardiovascular phenotypes in patients with an early-onset form of Marfan syndrome harboring FBN1 mutations in exons 24–32. Heart Vessels. 2016;31(10):1717–23. https://doi.org/10.1007/s00380-016-0793-2
Peng Q, Deng Y, Yang Y, Liu H. A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum. BMC Pediatr. 2016;16:60. https://doi.org/10.1186/s12887-016-0598-6
Heo JS, Song JY, Choi EY, Kim EH, Kim JH, Park SE, Jeon JH. Atypical neonatal Marfan syndrome with p.Glu1073Lys mutation of FBN1: the first case in Korea. J Korean Med Sci. 2017;32(1):1–3. https://doi.org/10.3346/jkms.2017.32.1.1
Solé-Ribalta A, Rodríguez-Fanjul X, Carretero-Bellon JM, Pascual-Sala C, Martorell-Sampol L, Bobillo-Pérez S, Morillo-Palomo AM. Neonatal Marfan syndrome: a rare, severe, and life-threatening genetic disease. J Pediatr. 2019;211:221–e2212. https://doi.org/10.1016/j.jpeds.2019.03.033
Tognato E, Perona A, Aronica A, Bertola A, Cimminelli L, De Vecchi S, Eshraghy MR, Loperfido B, Vivenza C, Manzoni P. Neonatal Marfan syndrome. Am J Perinatol. 2019;36(02):S74–6. https://doi.org/10.1055/s-0039-1691770
Wojcik MH, Thiele K, Grant CF, Chao K, Goodrich J, O’Donnell-Luria A, Lacro RV, Tan WH, Agrawal PB. Genome sequencing identifies the pathogenic variant missed by prior testing in an infant with Marfan Syndrome. J Pediatr. 2019;213:235–40. https://doi.org/10.1016/j.jpeds.2019.05.029
Veiga-Fernández A, Joigneau Prieto L, Álvarez T, Ruiz Y, Pérez R, Gámez F, Ortega Abad V, Yllana F, De León-Luis J. Perinatal diagnosis and management of early-onset Marfan syndrome: case report and systematic review. J Matern Fetal Neonatal Med. 2020;33(14):2493–504. https://doi.org/10.1080/14767058.2018.1552935
Postma JK, Altamirano-Diaz L, Rupar CA, Siu VM. Symptomatic mosaicism for a novel FBN1 splice site variant in a parent causing inherited neonatal Marfan syndrome. Am J Med Genet A. 2021;185(8):2507–13. https://doi.org/10.1002/ajmg.a.62339
Yoon SH, Kong Y. Severe neonatal Marfan syndrome with a novel mutation in the intron of the FBN1 gene: a case report. Med (Baltim). 2021;100(6):e24301. https://doi.org/10.1097/MD.0000000000024301
Motonaga T, Ohnishi Y, Okada S, Suzuki Y, Furuta T, Kawamura M, Okayama N, Suehiro Y, Hasegawa S. Successful mitral valve replacement in an infant with neonatal Marfan syndrome due to a novel missense mutation of the FBN1 gene. Int Heart J. 2022;63(4):777–81. https://doi.org/10.1536/ihj.21-821
Kawamura J, Ueno K, Kawano Y. Neonatal Marfan syndrome with missense variant of c.3706T > C undergoing bilateral atrioventricular valve replacement. Cardiol Young. 2022;32(5):833–6. https://doi.org/10.1017/S1047951121003905
Heide Hter, Schrander- Stumpel CTRM, Pals G, Delhaas T. Neonatal Marfan syndrome: clinical report and review of the literature. Clin Dysmorphol. 2005;14:81–4.
Hennekam RCM. Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am J Med Genet A. 2005;139A:1–1. https://doi.org/10.1002/ajmg.a.30979
Ekhomu O, Naheed ZJ. Aortic involvement in pediatric Marfan syndrome: a review. Pediatr Cardiol. 2015;36(5):887–95. https://doi.org/10.1007/s00246-015-1101-0
Nucera M, Heinisch PP, Langhammer B, Jungi S, Mihalj M, Schober P, Luedi MM, Yildiz M, Schoenhoff FS. The impact of sex and gender on aortic events in patients with Marfan syndrome. Eur J Cardiothorac Surg. 2022;62(5):ezac305. https://doi.org/10.1093/ejcts/ezac305
Stheneur C, Faivre L, Collod-Béroud G, Gautier E, Binquet C, Bonithon-Kopp C, Claustres M, Child AH, Arbustini E, Adès LC, Francke U, Mayer K, Arslan-Kirchner M, De Paepe A, Chevallier B, Bonnet D, Jondeau G, Boileau C. Prognosis factors in probands with an FBN1 mutation diagnosed before the age of 1 year. Pediatr Res. 2011;69:265–70. https://doi.org/10.1203/PDR.0b013e3182097219
Hussain S, Geddes G, Darragh R, Parent JJ. Successful heart transplantation in a patient with neonatal Marfan syndrome. J Heart Lung Transpl. 2022;41:S515. https://doi.org/10.1016/j.healun.2022.01.1306
Booms P, Cisler J, Mathews KR, Godfrey M, Tiecke F, Kaufmann UC, Vetter U, Hagemeier C, Robinson PN. Novel exon skipping mutation in the fibrillin-1 gene: two ‘hot spots’ for the neonatal Marfan syndrome. Clin Genet. 1999;55:110–7. https://doi.org/10.1034/j.1399-0004.1999.550207.x
Tiecke F, Katzke S, Booms P, Robinson PN, Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner RE, Hövels-Gürich HH, Hagemeier C, Fuchs J, Skovby F, Rosenberg T. Classic, atypically severe and neonatal Marfan syndrome: twelve mutations and genotype-phenotype correlations in FBN1 exons 24–40. Eur J Hum Genet. 2001;9:13–21. https://doi.org/10.1038/sj.ejhg.5200582
Meester JAN, Peeters S, Van Den Heuvel L, Vandeweyer G, Fransen E, Cappella E, Dietz HC, Forbus G, Gelb BD, Goldmuntz E, Hoskoppal A, Landstrom AP, Lee T, Mital S, Morris S, Olson AK, Renard M, Roden DM, Singh MN, Selamet Tierney ES, Tretter JT, Van Driest SL, Willing M, Verstraeten A, Van Laer L, Lacro RV, Loeys BL. (2022) Molecular characterization and investigation of the role of genetic variation in phenotypic variability and response to treatment in a large pediatric Marfan syndrome cohort. Genet Med. 2022;24:1045–1053. https://doi.org/10.1016/j.gim.2021.12.015
Brogger MN, Fernandez Ferro G, Cardenas Reyes I, Ochoa JP, Garcia Hernandez S, Valverde M, Fernandez X, Garcia Giustiniani D, Lamounier A, De La Higuera Romero L, Ortiz Genga M, Monserrat L, McKenna WJ. Narrowing of the neonatal region in the FBN1 gene. Eur Heart J. 2021;42:ehab7241988. https://doi.org/10.1093/eurheartj/ehab724.1988
Milewicz DM, Braverman AC, De Backer J, Morris SA, Boileau C, Maumenee IH, Jondeau G, Evangelista A, Pyeritz RE. Marfan syndrome. Nat Rev Dis Primers. 2022;8(1):3. https://doi.org/10.1038/s41572-022-00338-w
Andersen NH, Hauge EM, Baad-Hansen T, Groth KA, Berglund A, Gravholt CH, Stochholm K. Musculoskeletal diseases in Marfan syndrome: a nationwide registry study. Orphanet J Rare Dis. 2022;17(1):118. https://doi.org/10.1186/s13023-022-02272-2
Baban A, Parlapiano G, Cicenia M, Armando M, Franceschini A, Pacifico C, Panfili A, Zinzanella G, Romanzo A, Fusco A, Caiazza M, Perri G, Galletti L, Digilio MC, Buonuomo PS, Bartuli A, Novelli A, Raponi M, Limongelli G. Unique features of cardiovascular involvement and progression in children with Marfan syndrome justify dedicated multidisciplinary care. J Cardiovasc Dev Dis. 2024;11(4):114. https://doi.org/10.3390/jcdd11040114
Serra G, Antona V, Cannata C, Giuffrè M, Piro E, Schierz IAM, Corsello G. Distal arthrogryposis type 5 in an Italian family due to an autosomal dominant gain-of-function mutation of the PIEZO2 gene. Ital J Pediatr. 2022;48(1):133. https://doi.org/10.1186/s13052-022-01329-z
Serra G, Felice S, Antona V, Di Pace MR, Giuffrè M, Piro E, Corsello G. Cardio-facio-cutaneous syndrome and gastrointestinal defects: report on a newborn with 19p13.3 deletion including the MAP 2 K2 gene. Ital J Pediatr. 2022;48(1):65. https://doi.org/10.1186/s13052-022-01241-6
Acknowledgements
None.
Funding
This work was supported by the Italian Ministry of Health with “Current Research funds”.
Author information
Authors and Affiliations
Contributions
FP took care of the patient, reviewed the literature, and drafted the first version of the manuscript. DUDR took care of the patient, reviewed the literature, collected the clinical data, and revised the manuscript. MCD and MM contributed to the acquisition of genetic data and revised the manuscript. AB and AD took care of the patient and revised the manuscript. LV performed a surgical assessment and follow-up. AT performed cardiological assessment and follow-up. ADP conceptualized the report, revised the manuscript, and gave final approval of the version to be submitted. All authors approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Clinical data were obtained in accordance with the ethical standards of our hospital and Helsinki Declaration. Personal data were restricted to essential information, and were treated in order to guarantee the respect of privacy of the involved patients, as specifically stated by Italian Law D.Lgs n.196 of 2003 about personal data protection. Therefore, the study did not require preliminary evaluation by the local Ethical Committee. Written informed consent was obtained from the parents for publication of this case report and accompanying images.
Consent for publication
Written informed consent was obtained from the parents for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pugnaloni, F., De Rose, D., Digilio, M. et al. Neonatal Marfan syndrome: a case report of a novel fibrillin 1 mutation, with genotype-phenotype correlation and brief review of the literature. Ital J Pediatr 50, 183 (2024). https://doi.org/10.1186/s13052-024-01756-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-024-01756-0