
Abstract
Background
Hypocholesterolemia hallmarks critical illness though the underlying pathophysiology is incompletely understood. As low circulating cholesterol levels could partly be due to an increased conversion to cortisol/corticosterone, we hypothesized that glucocorticoid treatment, via reduced de novo adrenal cortisol/corticosterone synthesis, might improve cholesterol availability and as such affect adrenal gland and skeletal muscle function.
Methods
In a matched set of prolonged critically ill patients (n = 324) included in the EPaNIC RCT, a secondary analysis was performed to assess the association between glucocorticoid treatment and plasma cholesterol from ICU admission to day five. Next, in a mouse model of cecal ligation and puncture-induced sepsis, septic mice were randomized to receive either hydrocortisone (1.2 mg/day) (n = 17) or placebo (n = 15) for 5 days, as compared with healthy mice (n = 18). Plasma corticosterone, cholesterol, and adrenocortical and myofiber cholesterol were quantified. Adrenal structure and steroidogenic capacity were evaluated. Muscle force and markers of atrophy, fibrosis and regeneration were quantified. In a consecutive mouse study with identical design (n = 24), whole body composition was assessed by EchoMRI to investigate impact on lean mass, fat mass, total and free water.
Results
In human patients, glucocorticoid treatment was associated with higher plasma HDL- and LDL-cholesterol from respectively ICU day two and day three, up to day five (P < 0.05). Plasma corticosterone was no longer elevated in hydrocortisone-treated septic mice compared to placebo, whereas the sepsis-induced reduction in plasma HDL- and LDL-cholesterol and in adrenocortical cholesterol was attenuated (P < 0.05), but without improving the adrenocortical ACTH-induced CORT response and with increased adrenocortical inflammation and apoptosis (P < 0.05). Total body mass was further decreased in hydrocortisone-treated septic mice (P < 0.01) compared to placebo, with no additional effect on muscle mass, force or myofiber size. The sepsis-induced rise in markers of muscle atrophy and fibrosis was unaffected by hydrocortisone treatment, whereas markers of muscle regeneration were suppressed compared to placebo (P < 0.05). An increased loss of lean body mass and total and free water was observed in hydrocortisone-treated septic mice compared to placebo (P < 0.05).
Conclusions
Glucocorticoid treatment partially attenuated critical illness-induced hypocholesterolemia, but at a cost of impaired adrenal function, suppressed muscle regeneration and exacerbated loss of body mass.
Graphical abstract

Similar content being viewed by others


Background
Critical illness is hallmarked by an immediate and sustained decrease in plasma total-, high-density lipoprotein (HDL-) and low-density lipoprotein (LDL-) cholesterol concentrations, in proportion to illness severity and risk of death [1,2,3,4]. This hypocholesterolemia is considered a marker of poor prognosis and is most pronounced in intensive care unit (ICU) patients suffering from sepsis as compared with ICU patients admitted after surgery or trauma [4,5,6]. However, the underlying mechanisms of this critical illness-induced hypocholesterolemia remain poorly understood.
A potential contributor to the low cholesterol levels in critically ill patients might be an increased conversion of cholesterol to cortisol, the main glucocorticoid in humans [corticosterone (CORT) in rodents], in the adrenal cortex [7,8,9]. Plasma (free) cortisol levels are typically elevated in patients with sepsis and septic shock in face of low plasma cholesterol concentrations. While hypercortisolism in critical illness is largely explained by a reduced expression and activity of the cortisol metabolizing enzymes in liver and kidney with consequently reduced cortisol breakdown, cortisol production rates in ICU patients suffering from systemic hyperinflammation were found to be twice that of healthy subjects [10]. Importantly, in long-stay patients free of glucocorticoid treatment who require intensive care for more than 28 days, plasma cortisol concentrations were no longer elevated [9]. Moreover, in adrenal glands harvested post-mortem from long-stay ICU patients, a disturbed adrenocortical structure and depletion of cholesterol esters was observed as well as suppressed expression of the adrenocorticotropic hormone (ACTH)—regulated genes encoding cholesterol uptake transporters and the steroidogenic enzymes [11]. In addition, a study of critically ill patients reported a positive correlation between HDL-cholesterol concentrations and the incremental cortisol response to an intravenous bolus of ACTH [8]. A recent study of prolonged septic mice reported that treatment with a stress dose of hydrocortisone reversed the sepsis-induced reduction in adrenal cholesterol ester content [12].
Remarkably, critically ill patients suffering from ICU-acquired weakness (ICUAW), a debilitating complication characterized by limb and respiratory muscle weakness and/or muscle wasting, have lower serum cholesterol concentrations than patients without ICUAW [13]. Also, in our mouse model of prolonged sepsis-induced critical illness, the severity of muscle weakness inversely correlated with plasma cholesterol concentrations [13]. Patients who require a prolonged stay in the ICU are particularly prone to develop ICUAW [14]. The most important risk factors for ICUAW are sepsis, multiple organ failure and mechanical ventilation [14, 15]. Recently, in our mouse model of prolonged sepsis-induced critical illness, supplementation with 3-hydroxybutyrate, which can serve as a substrate for cholesterogenesis, increased myofiber cholesterol content and increased markers of muscle regeneration [13]. In contrast, in patients with sepsis, use of corticosteroids has been associated with higher risk of ICUAW, possibly via aggravating muscle atrophy and wasting [16,17,18,19,20,21].
Both ICU-acquired adrenal insufficiency (ICU-AI) and ICUAW are major complications in prolonged critically ill patients that might be related to reduced cholesterol availability [9, 22,23,24]. Whether administration of glucocorticoids can increase systemic and tissue cholesterol availability and hereby affect adrenal function during sepsis has not yet been thoroughly investigated. Also, whether increasing cholesterol availability could counteract the expected muscle wasting with glucocorticoid treatment remains to be investigated.
We here hypothesized that administration of stress doses of hydrocortisone can improve critical illness-induced hypocholesterolemia, via reduced de novo adrenal CORT synthesis, whereby adrenal function could be improved. In addition, although hydrocortisone treatment expectedly aggravates muscle wasting, muscle function could be beneficially affected via increased cholesterol availability. These hypotheses were tested in prolonged critically ill patients [25] and in two consecutive mouse studies using a validated and clinically relevant mouse model of prolonged sepsis-induced critical illness, which has previously shown to be characterized by both ICU-AI [12, 26] and ICUAW [13, 27, 28].
Methods
Human study of adult critically ill patients
This is a secondary analysis of the EPaNIC randomized controlled trial (RCT) in which patients were randomized to early supplementary parenteral nutrition or withholding parenteral nutrition until after the first week of ICU stay [25]. We aimed to document plasma cholesterol concentrations in patients who received glucocorticoids during the first five ICU days and compare these with glucocorticoid-free patients, who were matched for type and severity of illness and for demographic characteristics. Of the original 4640 EPaNIC patients, 2936 patients were not eligible because they were admitted after organ transplantation, or because they had an ICU stay < 5 days (Additional file 1: Supplemental Fig. 1 (Figure S1)). Of the remaining 1704 patients, 178 patients received glucocorticoids during the first five ICU days. Of these 178 patients, 162 patients could be matched—by logistic regression estimated propensity scores with use of baseline risk factors including the randomized nutritional intervention, as covariates—with glucocorticoid-free patients, yielding two matched cohorts of 162 patients.

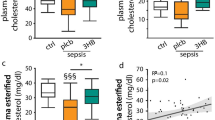
Plasma HDL- and LDL-cholesterol concentrations in critically ill adult patients. Plasma HDL- (a) and LDL- (b) cholesterol concentrations from admission day until day five in matched glucocorticoid-treated patients (n = 162) and glucocorticoid-free patients (n = 162). Data are presented as mean ± SEM. The solid and dashed line connect the mean of each day. Reference control values from Laboratory diagnostic measurements: HDL-cholesterol = 48.4 mg/dL; LDL-cholesterol = 132.5 mg/dL. SEM, standard error of the mean; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MANOVA, multivariate analysis of variance; n, sample size
Plasma cholesterol concentrations were measured in stored plasma samples from admission day up to day five with commercially available assays for HDL- and LDL-cholesterol (Diazyme Laboratories).
Mouse studies
In a first study (animal study 1), we investigated the impact of hydrocortisone treatment on adrenal function and muscle weakness in prolonged sepsis-induced critically ill mice (Additional file 1: Figure S2). Male 24-week-old C57BL/6J mice (Janvier Labs, Le Genest-Saint-Isle, France) were randomized to two interventional sepsis groups or a healthy control group. Mice in the sepsis groups were anaesthetized and implanted with a subcutaneous osmotic pump (ALZET Osmotic Pumps, Cupertino, CA, USA), delivering either hydrocortisone (1.2 mg/day) (Solu-Cortef®, Pfizer) or placebo (PlasmaLyte) [29]. Subsequently, a central venous catheter was implanted, followed by cecal ligation and puncture (CLP) to induce sepsis [30]. After surgery, septic mice received intravenous fluid resuscitation for the first 24 h, followed by continuous parenteral nutrition. Septic mice received twice daily broad-spectrum antibiotics and analgesics. Mice randomized to the healthy control group received ad libitum water and standard chow. All animals were sacrificed after a 5-day study period by cardiac puncture, whereafter blood and tissue samples were collected. From the 58 animals included in the survival analysis, 50 animals survived until the end of the study period (healthy control group, n = 18/18; placebo-treated sepsis, n = 15/17; hydrocortisone-treated sepsis, n = 17/23). Additional information on materials and methods is provided in Additional file 1.
To assess the impact of hydrocortisone treatment on whole body composition, a second animal study (animal study 2) was consecutively performed, of which the experimental setup was identical to animal study 1. In addition, immediately before CLP and immediately before sacrifice, lean mass, fat mass and total and free water content were measured in each animal and in healthy control animals using Magnetic Resonance Imaging (MRI) (EchoMRI-100H, Whole Body Magnetic Resonance Analyser, Zinsser Analytic GmbH, Germany). From the 30 animals included in the survival analysis, 24 animals survived until the end of the study period (healthy control group, n = 8/8; placebo-treated sepsis, n = 8/11; hydrocortisone-treated sepsis, n = 8/11).
All animals were treated according to the Principles of Laboratory Animal Care (US National Society of Medical Research) and to the European Union Directive 2010/63/EU concerning the welfare of laboratory animals. The animal study protocols were approved by the Institutional Ethical Committee for Animal Experimentation (P181-2018).
Adrenocortical response to ACTH and ex vivo muscle force measurements
The adrenocortical CORT response to ACTH was assessed in an in vitro adrenal explantation assay, as previously described, with use of adrenal glands harvested from mice in animal study 1 [12, 31, 32]. The CORT concentration in the incubation medium was measured with a commercially available ELISA (DRG) and adrenal glands were collected for further analysis. Immediately after sacrifice of mice from animal study 1, the hindlimb extensor digitorum longus (EDL) muscle was isolated, whereafter absolute and specific muscle force was measured (300C-LR Dual-Mode muscle lever, Aurora Scientific), as previously described [33]. Additional information on materials and methods is provided in Additional file 1.
Blood analyses
At sacrifice, whole blood glucose (animal study 1), Na+ and K+ (animal study 2) concentrations were measured using the Epoc® Blood Analysis System (Siemens Healthineers, The Hague, The Netherlands). After sacrifice of mice from animal study 1, plasma ACTH concentrations (Brahms Diagnostics), CORT (DRG), TNF-α (R&D Systems), aldosterone (LSBio), type I collagen (CTX-I) (RatLaps TM, Immunodiagnostic Systems), urea nitrogen (Invitrogen) and HDL- and LDL-cholesterol concentrations (Diazyme Laboratories) were measured with commercially available assays.
Gene and protein tissue expression
Total RNA was isolated from gastrocnemius, kidney (animal study 1) and adrenal gland (animal study 2) tissue, and reverse-transcribed into complementary DNA. Commercial TaqMan® assays (Applied Biosystems) were used for all gene expression analyses. Data were normalized to a stable housekeeping gene (Sdha or Rn18s, as appropriate) and expressed as fold change of the median of the healthy controls. A list of all used gene expression assays is provided in Additional file 1:Table S2. Protein content of mTOR (#2983, Cell Signaling; 1:1000) and Akt (#9272, Cell Signaling; 1:1000) was quantified by western blot analysis in the gastrocnemius muscle. Data were corrected for equal loading and presented as fold change of the median of the healthy controls. Cathepsin L enzymatic activity was measured on gastrocnemius muscle homogenates (Millipore, Merck, Calbiochem). Additional information on materials and methods is provided in Additional file 1.
Adrenal and intramuscular cholesterol content
Whole adrenal explants from animal study 1 were frozen in Tissue-Tek (Sakura Finetek) and cut at 8 µm thickness with a microtome-cryostat. Adrenal sections were stained overnight in 0.5% Oil-Red-O (ORO) (Sigma-Aldrich) in propylene glycol and counterstained with hematoxylin. Adrenocortical cholesterol ester content was quantified as the relative amount of redness in the adrenal cortex (ImageJ 53t).
After sacrifice of mice from animal study 1, intramuscular cholesterol content was quantified in the soleus muscle. Therefore, complete soleus muscle was homogenized in H2O, whereafter 50% methanol, 1% acetic acid and hexane was added for lipid extraction. Afterwards, 2% Thesit (Sigma-Aldrich) was added and the hexane phase was evaporated using GeneVac EZ-2 (Genevac Limited). The evaporated lipid fractions were dissolved in assay buffer, whereafter total cholesterol was quantified using the Amplex™ Red Cholesterol Assay Kit (Thermo Fisher Scientific), as per manufacturer’s instructions. Additional information on materials and methods is provided in Additional file 1.
Histology and immunostaining
After sacrifice of mice from animal study 1, laminin immunofluorescence staining was performed on 5 µm-thick paraffin-embedded tibialis anterior muscle tissue sections to quantify myofiber cross-sectional area (CSA). Tissue sections were incubated overnight (4 °C) with an anti-laminin primary antibody (1:50) (ab11575, Abcam). Myofiber segmentation was performed using Cellpose and myofiber CSA was quantified using an available LabelsToRoi plugin in ImageJ 53t. In addition, hematoxylin & eosin and Trichrome-Masson (HT15, Sigma-Aldrich) staining was performed on tibialis anterior muscle sections and scored semi-quantitatively for the presence of inflammation and fibrosis, respectively, by two independent investigators blinded for randomization. Any discrepancy was resolved by consensus. Muscle regeneration was investigated using a Pax7 immunohistochemistry staining (1:50) (Mab1675, R&D Systems). The total number of Pax7 + cells was quantified and corrected for the total muscle surface area.
After sacrifice of mice from animal study 2, 5 µm-thick paraffin-embedded adrenal gland tissue sections were stained with hematoxylin & eosin to investigate structural integrity. To assess the presence of macrophages in the adrenal cortex, tissue sections were stained overnight with an anti-CD68 primary antibody (1:100) (#125212, Abcam). Furthermore, the presence of apoptosis (Tunel + cells) in the adrenal cortex was evaluated using the In Situ Cell Death Detection Kit, TMR Red (Roche), according to the manufacturer’s instructions. Tissue sections were counterstained with Hoechst for nuclei visualization. Structural integrity and the presence of CD68 + cells and Tunel + cells were semi-quantitatively scored by two independent investigators, blinded for randomization. Any discrepancy was resolved by consensus. All images were taken with a TissueFAXS i PLUS microscope (TissueGnostics, Vienna, Austria). Additional information on materials and methods is provided in Additional file 1.
Statistical analysis
For the human study, logistic regression propensity score matching with use of baseline risk factors as covariates (admission diagnosis, APACHE-II score, age, BMI, sepsis upon admission, infection upon admission, elective surgery, randomized nutritional intervention) was performed with IBM SPSS Statistics software. Differences in plasma cholesterol over time between patients treated with glucocorticoids and glucocorticoid-free patients (presented as mean ± standard error of the mean (SEM)) were analyzed with use of repeated-measures analysis of variance (MANOVA), after transformation to obtain a near-normal distribution. For the mouse studies, data are presented as box and whisker plots with median, interquartile range (25th–75th percentiles) and 10th and 90th percentiles. Differences between groups were analyzed with use of Mann–Whitney U or Fisher exact test, as appropriate. No corrections for multiple comparisons were done. A P-value ≤ 0.05 was considered statistically significant. All statistical analyses were performed with JMP Pro 17.0.0 (SAS Institute Inc., Cary, NC, USA).
Results
The association between glucocorticoid treatment and plasma cholesterol concentrations in adult critically ill patients
In the matched set of 324 human critically ill patients, the association between glucocorticoid treatment and plasma cholesterol concentrations was documented over time (Fig. 1a, b). Baseline characteristics are described in Table 1. Additional information on the type and dose of glucocorticoids administered can be found in Additional file 1: Table S1.
All included patients had an ICU stay of at least five days. Overall, mean plasma HDL- and LDL-cholesterol was low in both patient groups over the studied time period (Fig. 1a, b). Glucocorticoid-treated patients had higher plasma HDL- and LDL-cholesterol concentrations than glucocorticoid-free patients from, respectively, day two and day three, up to day five (P < 0.05) (Fig. 1a, b). Repeated-measures MANOVA showed a significant difference in plasma HDL- and LDL-cholesterol concentrations between the two groups (HDL-cholesterol: P = 0.04; LDL-cholesterol: P = 0.004) with a statistically significant interaction between glucocorticoid treatment and time (HDL-cholesterol: P = 0.0008; LDL-cholesterol: P = 0.004). An additional assessment in the patients with a diagnosis of sepsis only was also performed. In the 205 septic patients, glucocorticoid-treated patients had higher plasma HDL- and LDL-cholesterol concentrations than glucocorticoid-free patients from day four up to day five (P < 0.05) (Additional file 1: Figure S3).
The impact of hydrocortisone treatment on survival, illness severity and total body weight in prolonged sepsis-induced critically ill mice
On day five after sepsis-induced critical illness, survival of hydrocortisone-treated mice (74%, 17/23) was not different from placebo (88%, 15/17) (P = 0.3), but was lower as compared to healthy controls (100%, n = 18/18) (P < 0.05) (Fig. 2a). Cumulative illness severity scores tended to be somewhat decreased by hydrocortisone treatment (P = 0.07 versus placebo) (Fig. 2b). Blood glucose levels were similar in hydrocortisone- and placebo-treated septic mice (P = 0.4) (Fig. 2c). The sepsis-induced rise in plasma TNF-α concentrations (P ≤ 0.0001 versus healthy controls) tended to be somewhat attenuated in hydrocortisone- versus placebo-treated septic mice (P = 0.06) (Fig. 2d). Over five days of sepsis, hydrocortisone treatment aggravated the loss of total body weight as compared with placebo (P = 0.0004) (Fig. 2e).

Impact of hydrocortisone on survival, illness severity and body weight loss during sepsis. a Kaplan–Meier survival curves of healthy control mice (n = 18/18), placebo-treated septic mice (n = 15/17) and hydrocortisone-treated septic mice (n = 17/23) for the five-day study period. b Cumulative illness severity scores of hydrocortisone- and placebo-treated prolonged septic mice. c Whole blood glucose concentrations. d Plasma TNF-α concentrations. e Change in total body weight over five days. Data presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls. */**/*** P ≤ 0.05/P ≤ 0.01/P ≤ 0.0001 compared with healthy control mice. IQR, interquartile range; n, sample size
The impact of hydrocortisone treatment on plasma CORT, ACTH, cholesterol and on the adrenal CORT response to ACTH in prolonged sepsis-induced critically ill mice
Hydrocortisone treatment lowered plasma endogenous CORT concentrations as compared with placebo (P = 0.02), to levels no longer higher than those in healthy mice (Fig. 3a). In addition, as compared with placebo septic mice, hydrocortisone treatment further lowered plasma ACTH concentrations (P = 0.0004) (Fig. 3b), and attenuated the sepsis-induced fall in plasma HDL- and LDL-cholesterol concentrations (HDL-cholesterol, P = 0.02 versus placebo-treated sepsis; LDL-cholesterol, P = 0.06 versus placebo-treated sepsis) (Fig. 3c, d). Furthermore, hydrocortisone treatment increased adrenocortical cholesterol ester content as compared with placebo septic mice (P = 0.02) (Fig. 3e). In adrenal explants, CORT concentrations after ACTH stimulation (100 nM) were comparable for hydrocortisone- and placebo-treated septic mice (P = 0.4) (Fig. 3f).

Impact of hydrocortisone on plasma CORT, ACTH, cholesterol and the adrenal ACTH response during sepsis. a Plasma CORT concentrations. b Plasma ACTH concentrations. c Plasma HDL-cholesterol concentrations. d Plasma LDL-cholesterol concentrations. e Adrenal cholesterol ester content, represented as the relative amount of redness in the adrenal cortex. The right panel shows representative images for each group. f CORT increase measured in the incubation medium of explanted adrenal glands after overnight stimulation with either basal medium (no additive) or ACTH (100 nM). Data presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls. */**/*** P ≤ 0.05/P ≤ 0.01/P ≤ 0.0001 compared with healthy control mice. CORT, corticosterone; ACTH, adrenocorticotropic hormone; HDL, high-density lipoprotein; LDL, low-density lipoprotein; IQR, interquartile range; n, sample size
The impact of hydrocortisone treatment on adrenocortical structure, inflammation, apoptosis and on markers of adrenal regeneration in prolonged sepsis-induced critically ill mice
Hydrocortisone treatment partially prevented the sepsis-induced loss of normal architecture of the zona fasciculata, zona glomerulosa and reticularis that was present with placebo treatment (P = 0.01) (Fig. 4a). Furthermore, as compared with placebo, hydrocortisone treatment increased gene expression of Gli1, a marker of adrenal regeneration (P = 0.007), whereas expression levels of Yap and Taz, markers of stem cell maintenance, cell growth and differentiation, were unaffected (P > 0.05) (Fig. 4b). Immunohistochemical staining with CD68, a macrophage marker of inflammation, revealed a trend for more CD68 + cells in the adrenal cortex of hydrocortisone-treated septic mice as compared with placebo (P = 0.08) (Fig. 4c). In addition, hydrocortisone treatment increased the adrenocortical presence of Tunel + cells, revealing more apoptosis compared with placebo (P = 0.04) (Fig. 4d).

Impact of hydrocortisone on adrenal structure, regeneration, macrophage infiltration and apoptosis during sepsis. a Semi-quantitative scoring of adrenocortical structure, indicated as either “normal”, “moderately distorted” or “severely distorted”. Panels below show representative images for each condition. b Relative mRNA expression of adrenal regeneration markers (Gli1, Yap, Taz), normalized to Sdha housekeeping gene and expressed as fold change of the median of the healthy controls. c Semi-quantitative scoring of CD68 + cells in the adrenal cortex, indicated as either “not present”, “moderately present” or “highly present”. Panels below show representative images for each condition. d Semi-quantitative scoring of Tunel + cells in the adrenal cortex, indicated as either “not present”, “moderately present” or “highly present”. Panels below show representative images for each condition. All data were obtained from animal study 2 (n = 24). Gene expression data is presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls. */** P ≤ 0.05/P ≤ 0.01 compared with healthy control mice. A.u. arbitrary unit; n, sample size
The impact of hydrocortisone treatment on markers of muscle force and wasting in prolonged sepsis-induced critically ill mice
Hydrocortisone- and placebo-treated septic mice suffered from a similar reduction (P ≥ 0.1) in EDL absolute and specific muscle force as compared with healthy controls (P ≤ 0.05) (Fig. 5a, b). The sepsis-induced loss of tibialis anterior muscle weight (P ≤ 0.0001 versus healthy controls) tended to be more pronounced with hydrocortisone than with placebo treatment (P = 0.08) (Fig. 5c), whereas intramuscular cholesterol content in the soleus muscle was unaffected (P > 0.05) (Fig. 5d).

Impact of hydrocortisone on markers of muscle force and wasting during sepsis. a EDL absolute tetanic muscle force. b EDL specific muscle force. c Tibialis anterior muscle weight. d Myofiber cholesterol content per mg protein in soleus muscle. e Tibialis anterior muscle myofiber CSA. f Plasma urea concentrations. g Plasma CTX-I concentrations. h Muscle cathepsin L enzymatic activity per gram tissue. i Relative mRNA expression of atrogenes (Trim63, Fbxo32, Foxo3, Ubb) in the skeletal muscle. j Relative mRNA expression of GR-target genes (Fkbp5, Klf15, Ddit4, Pik3r1) in the skeletal muscle. Gene expression data are normalized to Sdha housekeeping gene and expressed as fold change of the median of the healthy controls. Data are presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls. */**/*** P ≤ 0.05/P ≤ 0.01/P ≤ 0.0001 compared with healthy control animals. EDL, extensor digitorum longus; GR, glucocorticoid receptor; CSA, cross-sectional area; IQR, interquartile range; a.u., arbitrary unit; n, sample size
To further assess muscle wasting, muscle fiber size and markers of protein breakdown were investigated. As compared with placebo, hydrocortisone treatment did not affect the sepsis-induced reduction of tibialis anterior myofiber CSA (P ≤ 0.01 versus healthy controls) (Fig. 5e). Also, plasma urea, a marker of protein degradation, was not affected by hydrocortisone treatment (P = 0.3 versus placebo) (Fig. 5f). Hydrocortisone did also not affect degradation products of type I collagen (CTX-I), a marker of bone turnover in plasma (P > 0.05 versus placebo) (Fig. 5g). The sepsis-induced rise in muscle cathepsin L enzymatic activity, a lysosomal endoproteinase, was attenuated by hydrocortisone treatment (P = 0.009 versus placebo) (Fig. 5h), while the sepsis-induced rise in markers of muscle atrophy (Trim63, Fbxo32, Foxo3, Ubb) was unaffected by hydrocortisone treatment (P > 0.05 versus placebo) (Fig. 5i).
We next investigated gene expression of glucocorticoid receptor (GR) target genes in the gastrocnemius muscle, that can counterregulate GR activity (Fkbp5) or can contribute to muscle wasting (Klf15, Ddit4, Pik3r1). Hydrocortisone treatment further increased the sepsis-induced rise in Fkbp5 expression (P = 0.005 versus placebo) (Fig. 5j), whereas hydrocortisone did not alter the sepsis-induced rise in Klf15 and Ddit4 expression and only moderately further increased Pik3r1 expression (P = 0.04 versus placebo) (Fig. 5j).
The impact of hydrocortisone treatment on muscle inflammation, fibrosis and on markers of protein synthesis and regeneration in prolonged sepsis-induced critically ill mice
Hydrocortisone treatment did not alter muscle gene expression of the pro-inflammatory marker Tnf-α (P = 0.3 versus placebo) (Fig. 6a) and did not affect muscle fibrosis, indicated by a lack of impact on the sepsis-induced increased expression of fibrogenes (Ctgf, Tgf-β1, Mmp9) (P ≤ 0.01 versus healthy controls) (Fig. 6a). The presence of inflammation and fibrosis in muscle was further investigated in tibialis anterior muscle tissue sections using hematoxylin & eosin staining and Trichrome-Masson staining, respectively. A comparable increase in inflammation and fibrosis (P ≤ 0.05 versus healthy controls) was observed with hydrocortisone and placebo treatment (Fig. 6b).

Impact of hydrocortisone on muscle inflammation, fibrosis, protein synthesis and regeneration during sepsis. a Relative mRNA expression of markers of inflammation and fibrosis (Tnf-α, Ctgf, Tgf-β1, Mmp9) in the skeletal muscle. b Semi-quantitative scoring of inflammation and fibrosis in the tibialis anterior muscle, indicated as either “not present”, “moderately present” or “highly present”. Panels show representative images for each condition. Arrows indicate presence of inflammation. Blue staining indicates presence of fibrosis. c Relative mRNA expression of markers of protein synthesis (Acta1, Myh1, Myh2) in the skeletal muscle. d Relative protein expression of mTOR in the skeletal muscle. e Relative protein expression of Akt in the skeletal muscle. f Representative images of western blot analysis of muscle mTOR (~ 289 kDa) and Akt (~ 57 kDa) protein content. g Relative mRNA expression of regeneration markers (Myf5, Mylip, Myog, Myod1, Pax7, Mstn) in the skeletal muscle. h Number of Pax7 + cells per total muscle area. Panels show representative images for each group. Arrows indicate Pax7 + cells. Gene expression data are normalized to Sdha housekeeping gene. Gene and protein expression data are expressed as fold change of the healthy controls. Data are presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls */**/*** P ≤ 0.05/P ≤ 0.01/P ≤ 0.0001 compared with healthy control animals. IQR, interquartile range; kDa, kilodalton; a.u. arbitrary unit; n, sample size
Muscle mass is determined by protein breakdown and protein synthesis. To evaluate protein synthesis, gene expression of the contractile proteins actin and myosin was quantified. Gene expression of Acta1 was decreased in hydrocortisone-treated septic mice (P ≤ 0.01 versus healthy controls), whereas mRNA expression of Myh1 and Myh2 was unaffected by sepsis (P > 0.05) (Fig. 6c). Sepsis did not alter protein expression of mTOR whereas it increased protein expression of Akt (P ≤ 0.01 versus healthy controls), both unaffected by hydrocortisone treatment (Fig. 6d, e, f). In contrast, as compared with placebo, hydrocortisone treatment reduced gene expression of Myf5, Mylip and Myog, markers of muscle regeneration (P < 0.05) (Fig. 6g). Gene expression of Myod1 was unaffected, whereas expression of Pax7 was decreased during sepsis (P ≤ 0.01 versus healthy controls), both unaffected by hydrocortisone treatment. In contrast, as compared with placebo, hydrocortisone treatment increased gene expression of Mstn, a negative regulator of muscle growth (P = 0.0003) (Fig. 6g). Muscle regeneration was further investigated in tibialis anterior muscle tissue sections using a Pax7 immunohistochemical staining, but no differences were found between the treatment groups (P > 0.05 versus healthy controls) (Fig. 6h).
The impact of hydrocortisone treatment on whole body composition, blood electrolytes, aldosterone and renal urea and water transporters in prolonged sepsis-induced critically ill mice
As indicated by the whole body MRI, hydrocortisone and placebo treatment resulted in a comparable loss of fat mass (P = 0.6) (Fig. 7a). In contrast, as compared with placebo, hydrocortisone treatment further increased the loss of lean body mass and reduced total and free water content (P < 0.05) (Fig. 7a). Hydrocortisone treatment also further increased blood Na+ concentrations (P = 0.01) and decreased blood K+ concentrations (P = 0.01) compared to placebo (Fig. 7b, c). In addition, a trend for a further increase in plasma aldosterone concentrations was observed with hydrocortisone treatment (P = 0.07 versus placebo) (Fig. 7d). Furthermore, hydrocortisone treatment also lowered the gene expression of the urea transporter Slc14a2 in kidney (P = 0.002 versus placebo), and increased mRNA expression of Aqp1 (P = 0.05) (Fig. 7e), without an effect on Aqp2, Aqp3 and Aqp4 expression (P > 0.05) (data not shown).

Impact of hydrocortisone on body composition, blood electrolytes, aldosterone and renal transporters during sepsis. a Change in fat mass, lean body mass and total and free water, based on EchoMRI measurements. b Whole blood Na+ concentrations. c Whole blood K+ concentrations. d Plasma aldosterone concentrations. e Relative mRNA expression of respectively the Slc14a2 and Aqp1 urea and water transporter in the kidney, normalized to Rn18s housekeeping gene and expressed as fold change of the median of the healthy controls. Data are presented as box and whisker plots with median, IQR and 10th and 90th percentiles. Gray area represents IQR of the healthy controls. Data from whole body composition and whole blood electrolyte concentrations were obtained from animal study 2 (n = 24). */**/*** P ≤ 0.05/P ≤ 0.01/P ≤ 0.0001 compared with healthy control animals. MRI, Magnetic Resonance Imaging; IQR, interquartile range; a.u. arbitrary unit; n, sample size; ∆, delta (day 5–day 0)
Discussion
We demonstrated that, in the face of the known critical illness-induced hypocholesterolemia, prolonged treatment with glucocorticoids was associated with increased plasma cholesterol levels in adult critically ill patients, but remained below normal reference ranges. Causality of this association is suggested by its confirmation in the mouse model of prolonged sepsis-induced critical illness. Hydrocortisone treatment attenuated critical illness-induced hypocholesterolemia in prolonged sepsis-induced critically ill mice, without improving adrenal and muscle function and at a cost of exacerbated loss of total body mass. Five days treatment of septic mice with hydrocortisone resulted in increased adrenocortical cholesterol ester content, but did not improve the blunted ACTH-induced CORT response and exerted a pro-inflammatory and pro-apoptotic effect in the adrenal cortex of prolonged septic mice. The sepsis-induced loss of muscle mass, muscle force and myofiber size was not further affected by hydrocortisone treatment and no additional effect on expression of atrogenes or markers of muscle inflammation and fibrosis was observed. Whether any potential beneficial effect of an increased cholesterol availability on muscle function was blunted because of glucocorticoid-induced wasting and suppressed muscle regeneration mechanisms remains unclear.
The immediate and sustained decrease in circulating cholesterol levels during the course of critical illness is suggested to be linked to endotoxin scavenging mechanisms, or to an increased production of cortisol in the adrenal cortex, however, the exact pathophysiology remains unclear [7, 8, 35,36,37]. We observed that hydrocortisone treatment reduced endogenous plasma CORT levels in prolonged septic mice and reversed the sepsis-induced reduction in adrenocortical cholesterol ester content, thereby partially increasing circulating cholesterol levels. Although glucocorticoids are frequently used in patients with acute respiratory distress syndrome (ARDS), to reduce pulmonary inflammation or as an adjunctive vasopressor therapy for patients with septic shock [38, 39], the mortality benefit of glucocorticoids across randomized controlled trials is inconsistent [20, 40,41,42,43,44,45]. In the current mice study of sepsis-induced critical illness, no survival benefit from glucocorticoid treatment was observed. In addition, a pro-inflammatory and pro-apoptotic effect of glucocorticoid treatment within the adrenal cortex was observed [46, 47].
In septic mice, five-day continuous infusion with hydrocortisone lowered plasma ACTH levels, likely through increased hypothalamic and pituitary negative feedback inhibition, as previously documented in a similar experimental setup of seven-day continuous hydrocortisone infusion [12]. It is suggested that ligands other than ACTH might contribute to adrenocortical steroidogenesis, at least partially offering an explanation for the normal range of plasma endogenous CORT concentrations after hydrocortisone treatment [12, 26]. Although hydrocortisone treatment reversed the sepsis-induced reduction in adrenocortical cholesterol ester content in prolonged septic mice, ex vivo overnight stimulation with ACTH did not result in increased CORT concentrations in the incubation medium of explanted adrenal glands. Such failure could be due to impaired ACTH signaling and subsequent impaired adrenocortical steroidogenesis or to suppressed adrenal regeneration mechanisms [36].
Recent findings in both critically ill patients and septic mice suggest that an altered cholesterol homeostasis could be involved in the development of ICUAW [13]. We here demonstrated that continuous treatment with hydrocortisone in prolonged septic mice partially attenuated the sepsis-induced reduction in circulating cholesterol levels without improving muscle force. We cannot exclude that the effect on circulating cholesterol levels was too small to induce a protective effect inside the myofibers. Furthermore, hydrocortisone treatment did not affect myofiber cholesterol content. Moreover, muscle mass, myofiber size and gene expression markers of muscle atrophy and fibrosis were similarly negatively affected in hydrocortisone- and placebo-treated prolonged septic mice. This suggests an already maximally activated GR in sepsis which could explain the lack of difference in muscle wasting between hydrocortisone- and placebo-treated prolonged septic mice. Indeed, gene expression of Fkbp5, a GR-regulated co-chaperone couterregulating GR activity, was substantially increased in prolonged septic mice receiving hydrocortisone treatment. Importantly, main gene expression markers of muscle regeneration were suppressed in prolonged septic mice receiving hydrocortisone treatment, whereas Mstn, a negative regulator of muscle growth, was increased in hydrocortisone-treated prolonged septic mice. This suppression of regeneration might suggest potential complications in recovery from ICUAW, but this could not be studied with the current experimental design. However, use of corticosteroids during ICU stay was found to be associated with a worse long-term post-ICU outcome [48].
In addition to the exacerbated loss of total body weight in prolonged septic mice receiving hydrocortisone, the whole body MRI measurements demonstrated increased loss of lean body mass and body water content with hydrocortisone treatment, and this in the presence of hyperaldosteronemia and hypokalemia. Also, decreased renal expression of Slc14a2, encoding the UT-A urea transporter, was observed with hydrocortisone treatment. High doses of glucocorticoids can cause increased electrolyte-free water loss, however, the exact mechanisms are unclear and need to be further clarified, but are beyond the scope of this paper [49,50,51]. Of note, on top of the further increase in plasma aldosterone, high doses of glucocorticoids also have direct mineralocorticoid activity which explains the severe hypokalemia. Persistent hypokalemia can cause arginine vasopressin (AVP) resistance, resulting in nephrogenic diabetes insipidus, at least partially explaining the observed loss in body water content [12, 52, 53].
This study has some limitations. First, we performed a secondary analysis in a matched set of glucocorticoid-treated and glucocorticoid-free critically ill patients to assess the association between glucocorticoid treatment and plasma cholesterol concentrations, however we did not correct for the indication of glucocorticoid treatment as a potential confounding factor. Second, although our validated mouse model of prolonged sepsis-induced critical illness mimics several aspects of the human clinical setting, translation to the human setting has to be done with caution. Our septic mice were not mechanically ventilated and were not fully immobilized. However, septic mice received intravenous fluid resuscitation, parenteral nutrition, broad-spectrum antibiotics and analgesics, mimicking the clinical setting as much as possible. Also, septic mice typically have higher plasma HDL-cholesterol concentrations and lower plasma LDL-cholesterol concentrations in comparison to humans [54]. Therefore, extrapolations for these lipoproteins between species must be done carefully. Furthermore, healthy control mice did not undergo any intervention in order to serve as a true reference group for the ‘healthy state’. The study drugs (hydrocortisone or placebo) were given only in septic mice and not in the healthy controls. As such, the comparison of hydrocortisone or placebo with untreated healthy controls has to be interpret within the context of sepsis. Therefore, we cannot exclude that the observed effect of glucocorticoids on plasma cholesterol concentrations is partly confounded by other indirect factors on the course of sepsis.
Conclusion
In critically ill patients, glucocorticoid treatment was associated with higher circulating cholesterol concentrations. In septic mice, treatment with stress doses of hydrocortisone for five days partially attenuated critical illness-induced hypocholesterolemia, without improving adrenal or muscle function and at a cost of exacerbated loss of lean body mass, suppressed muscle regeneration mechanisms and increased adrenocortical inflammation and apoptosis. Whether these harmful effects counteracted any potential beneficial effect of an increased cholesterol availability on adrenal and muscle function remains unclear. Future studies should consider the use of direct cholesterol substitution therapy to improve altered cholesterol availability during critical illness.
Availability of data and materials
Some or all datasets generated during and/or analyzed during the current study are not publicly available but are available from the corresponding author on reasonable request. Data are located in controlled access data storage at KU Leuven.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- APACHE:
-
Acute Physiology And Chronic Health Evaluation
- ARDS:
-
Acute respiratory distress syndrome
- AVP:
-
Arginine vasopressin
- BSA:
-
Bovine serum albumin
- CLP:
-
Cecal ligation and puncture
- CORT:
-
Corticosterone
- CSA:
-
Cross-sectional area
- DAB:
-
3,3’-Diaminobenzidine
- GR:
-
Glucocorticoid receptor
- HDL:
-
High-density lipoprotein
- ICU:
-
Intensive care unit
- ICU-AI:
-
ICU-acquired adrenal insufficiency
- ICUAW:
-
ICU-acquired weakness
- LDL:
-
Low-density lipoprotein
- MANOVA:
-
Multivariate Analysis of Variance
- MRI:
-
Magnetic resonance imaging
- ORO:
-
Oil-red O
- RCT:
-
Randomized controlled trial
- SEM:
-
Standard error of the mean
References
Mesotten D, Swinnen JV, Vanderhoydonc F, Wouters PJ, Van den Berghe G. Contribution of circulating lipids to the improved outcome of critical illness by glycemic control with intensive insulin therapy. J Clin Endocrinol Metab. 2004;89(1):219–26.
Dunham CM, Fealk MH, Sever WE 3rd. Following severe injury, hypocholesterolemia improves with convalescence but persists with organ failure or onset of infection. Crit Care. 2003;7(6):R145–53.
Gordon BR, Parker TS, Levine DM, Saal SD, Wang JC, Sloan BJ, et al. Relationship of hypolipidemia to cytokine concentrations and outcomes in critically ill surgical patients. Crit Care Med. 2001;29(8):1563–8.
Tanaka S, Labreuche J, Drumez E, Harrois A, Hamada S, Vigue B, et al. Low HDL levels in sepsis versus trauma patients in intensive care unit. Ann Intensive Care. 2017;7(1):60.
Chiarla C, Giovannini I, Giuliante F, Zadak Z, Vellone M, Ardito F, et al. Severe hypocholesterolemia in surgical patients, sepsis, and critical illness. J Crit Care. 2010;25(2):361e7-e12.
Bakalar B, Hyspler R, Pachl J, Zadak Z. Changes in cholesterol and its precursors during the first days after major trauma. Wien Klin Wochenschr. 2003;115(21–22):775–9.
Marik PE. Adrenal insufficiency: the link between low apolipoprotein A-I levels and poor outcome in the critically ill? Crit Care Med. 2004;32(9):1977–8.
van der Voort PHJ, Gerritsen RT, Bakker AJ, Boerma EC, Kuiper MA, de Heide L. HDL-cholesterol level and cortisol response to synacthen in critically ill patients. Intensive Care Med. 2003;29(12):2199–203.
Peeters B, Meersseman P, Vander Perre S, Wouters PJ, Vanmarcke D, Debaveye Y, et al. Adrenocortical function during prolonged critical illness and beyond: a prospective observational study. Intensive Care Med. 2018;44(10):1720–9.
Boonen E, Vervenne H, Meersseman P, Andrew R, Mortier L, Declercq PE, et al. Reduced cortisol metabolism during critical illness. N Engl J Med. 2013;368(16):1477–88.
Boonen E, Langouche L, Janssens T, Meersseman P, Vervenne H, De Samblanx E, et al. Impact of duration of critical illness on the adrenal glands of human intensive care patients. J Clin Endocrinol Metab. 2014;99(11):4214–22.
Teblick A, De Bruyn L, Van Oudenhove T, Vander Perre S, Pauwels L, Derde S, et al. Impact of hydrocortisone and of CRH infusion on the hypothalamus-pituitary-adrenocortical axis of septic male mice. Endocrinology. 2022;163(1):bqab222.
Goossens C, Weckx R, Derde S, Vander Perre S, Derese I, Van Veldhoven PP, et al. Altered cholesterol homeostasis in critical illness-induced muscle weakness: effect of exogenous 3-hydroxybutyrate. Crit Care. 2021;25(1):252.
Vanhorebeek I, Latronico N, Van den Berghe G. ICU-acquired weakness. Intensive Care Med. 2020;46(4):637–53.
Schefold JC, Wollersheim T, Grunow JJ, Luedi MM, Z’Graggen WJ, Weber-Carstens S. Muscular weakness and muscle wasting in the critically ill. J Cachexia Sarcopenia Muscle. 2020;11(6):1399–412.
Tiao G, Fagan J, Roegner V, Lieberman M, Wang JJ, Fischer JE, et al. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest. 1996;97(2):339–48.
Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129(1S Suppl):227S-S237.
Schmidt F, Kny M, Zhu X, Wollersheim T, Persicke K, Langhans C, et al. The E3 ubiquitin ligase TRIM62 and inflammation-induced skeletal muscle atrophy. Crit Care. 2014;18(5):545.
Rochwerg B, Oczkowski SJ, Siemieniuk RAC, Agoritsas T, Belley-Cote E, D’Aragon F, et al. Corticosteroids in sepsis: an updated systematic review and meta-analysis. Crit Care Med. 2018;46(9):1411–20.
Pirracchio R, Annane D, Waschka AK, Lamontagne F, Arabi YM, Bollaert PE, et al. Patient-level meta-analysis of low-dose hydrocortisone in adults with septic shock. NEJM Evid. 2023;2(6):EVIDoa2300034.
Yang T, Li Z, Jiang L, Xi X. Corticosteroid use and intensive care unit-acquired weakness: a systematic review and meta-analysis. Crit Care. 2018;22(1):187.
Teblick A, Peeters B, Langouche L, Van den Berghe G. Adrenal function and dysfunction in critically ill patients. Nat Rev Endocrinol. 2019;15(7):417–27.
Teblick A, Gunst J, Van den Berghe G. Critical illness-induced corticosteroid insufficiency: what it is not and what it could be. J Clin Endocrinol Metab. 2022;107(7):2057–64.
Lauwers C, De Bruyn L, Langouche L. Impact of critical illness on cholesterol and fatty acids: insights into pathophysiology and therapeutic targets. Intensive Care Med Exp. 2023;11(1):84.
Casaer MP, Mesotten D, Hermans G, Wouters PJ, Schetz M, Meyfroidt G, et al. Early versus late parenteral nutrition in critically ill adults. N Engl J Med. 2011;365(6):506–17.
Teblick A, Vander Perre S, Pauwels L, Derde S, Van Oudenhove T, Langouche L, et al. The role of pro-opiomelanocortin in the ACTH-cortisol dissociation of sepsis. Crit Care. 2021;25(1):65.
Goossens C, Marques MB, Derde S, Vander Perre S, Dufour T, Thiessen SE, et al. Premorbid obesity, but not nutrition, prevents critical illness-induced muscle wasting and weakness. J Cachexia Sarcopenia Muscle. 2017;8(1):89–101.
Goossens C, Weckx R, Derde S, Dufour T, Vander Perre S, Pauwels L, et al. Adipose tissue protects against sepsis-induced muscle weakness in mice: from lipolysis to ketones. Crit Care. 2019;23(1):236.
Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31.
Derde S, Thiessen S, Goossens C, Dufour T, Van den Berghe G, Langouche L. Use of a central venous line for fluids, drugs and nutrient administration in a mouse model of critical illness. J Vis Exp. 2017;123:e55553.
Richter HG, Torres-Farfan C, Garcia-Sesnich J, Abarzua-Catalan L, Henriquez MG, Alvarez-Felmer M, et al. Rhythmic expression of functional MT1 melatonin receptors in the rat adrenal gland. Endocrinology. 2008;149(3):995–1003.
Uschold-Schmidt N, Nyuyki KD, Fuchsl AM, Neumann ID, Reber SO. Chronic psychosocial stress results in sensitization of the HPA axis to acute heterotypic stressors despite a reduction of adrenal in vitro ACTH responsiveness. Psychoneuroendocrinology. 2012;37(10):1676–87.
Weckx R, Goossens C, Derde S, Pauwels L, Vander Perre S, Van den Berghe G, et al. Identification of the toxic threshold of 3-hydroxybutyrate-sodium supplementation in septic mice. BMC Pharmacol Toxicol. 2021;22(1):50.
Meersseman P, Boonen E, Peeters B, Vander Perre S, Wouters PJ, Langouche L, et al. Effect of early parenteral nutrition on the HPA axis and on treatment with corticosteroids in intensive care patients. J Clin Endocrinol Metab. 2015;100(7):2613–20.
Levels JH, Abraham PR, van den Ende A, van Deventer SJ. Distribution and kinetics of lipoprotein-bound endotoxin. Infect Immun. 2001;69(5):2821–8.
Bhakdi S, Tranum-Jensen J, Utermann G, Fussle R. Binding and partial inactivation of Staphylococcus aureus alpha-toxin by human plasma low density lipoprotein. J Biol Chem. 1983;258(9):5899–904.
Parker TS, Levine DM, Chang JC, Laxer J, Coffin CC, Rubin AL. Reconstituted high-density lipoprotein neutralizes gram-negative bacterial lipopolysaccharides in human whole blood. Infect Immun. 1995;63(1):253–8.
Chaudhuri D, Sasaki K, Karkar A, Sharif S, Lewis K, Mammen MJ, et al. Corticosteroids in COVID-19 and non-COVID-19 ARDS: a systematic review and meta-analysis. Intensive Care Med. 2021;47(5):521–37.
Annane D, Pastores SM, Arlt W, Balk RA, Beishuizen A, Briegel J, et al. Critical Illness-Related Corticosteroid Insufficiency (CIRCI): a narrative review from a multispecialty task force of the society of critical care medicine (SCCM) and the European Society of Intensive Care Medicine (ESICM). Crit Care Med. 2017;45(12):2089–98.
Annane D, Renault A, Brun-Buisson C, Megarbane B, Quenot JP, Siami S, et al. Hydrocortisone plus fludrocortisone for adults with septic shock. N Engl J Med. 2018;378(9):809–18.
Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288(7):862–71.
Sprung CL, Annane D, Keh D, Moreno R, Singer M, Freivogel K, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;358(2):111–24.
Venkatesh B, Finfer S, Cohen J, Rajbhandari D, Arabi Y, Bellomo R, et al. Adjunctive glucocorticoid therapy in patients with septic shock. N Engl J Med. 2018;378(9):797–808.
Tongyoo S, Permpikul C, Mongkolpun W, Vattanavanit V, Udompanturak S, Kocak M, et al. Hydrocortisone treatment in early sepsis-associated acute respiratory distress syndrome: results of a randomized controlled trial. Crit Care. 2016;20(1):329.
Villar J, Ferrando C, Martinez D, Ambros A, Munoz T, Soler JA, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med. 2020;8(3):267–76.
Spiga F, Zhao Z, Lightman SL. Prolonged treatment with the synthetic glucocorticoid methylprednisolone affects adrenal steroidogenic function and response to inflammatory stress in the rat. Brain Behav Immun. 2020;87:703–14.
Finco I, Lerario AM, Hammer GD. Sonic hedgehog and WNT signaling promote adrenal gland regeneration in male mice. Endocrinology. 2018;159(2):579–96.
Hermans G, Van Aerde N, Meersseman P, Van Mechelen H, Debaveye Y, Wilmer A, et al. Five-year mortality and morbidity impact of prolonged versus brief ICU stay: a propensity score matched cohort study. Thorax. 2019;74(11):1037–45.
Imaizumi T, Nakatochi M, Fujita Y, Yamamoto R, Watanabe K, Maekawa M, et al. Glucocorticoid treatment is associated with ICU-acquired hypernatremia: a nested case-control study. Clin Exp Nephrol. 2021;25(2):131–9.
Thunhorst RL, Beltz TG, Johnson AK. Glucocorticoids increase salt appetite by promoting water and sodium excretion. Am J Physiol Regul Integr Comp Physiol. 2007;293(3):R1444–51.
Li C, Wang W, Summer SN, Falk S, Schrier RW. Downregulation of UT-A1/UT-A3 is associated with urinary concentrating defect in glucocorticoid-excess state. J Am Soc Nephrol. 2008;19(10):1975–81.
Khositseth S, Uawithya P, Somparn P, Charngkaew K, Thippamom N, Hoffert JD, et al. Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci Rep. 2015;5:18311.
Correa TD, Takala J, Jakob SM. Angiotensin II in septic shock. Crit Care. 2015;19(1):98.
Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2012;53(1):51–65.
Acknowledgements
We thank Caroline Lauwers for the technical assistance with the animal experiments.
Funding
This work was supported by the European Research Council Advanced Grant (AdvG-2017–785806 to GVdB) from European Union’s Horizon 2020 research and innovation program and the Methusalem program of the Flemish Government (METH/14/06 to GVdB and LL via the KU Leuven) and the Research Foundation—Flanders, Belgium (Grant G069421N to LL and GVdB).
Author information
Authors and Affiliations
Contributions
LDB, AT, TVO, SVP, ID, LP, SD, GDV, GVdB and LL generated, analyzed and interpreted the data. LDB, GVdB and LL drafted the first manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All animals were treated according to the Principles of Laboratory Animal Care (US National Society of Medical Research) and to the European Union Directive 2010/63/EU concerning the welfare of laboratory animals. The study was approved by the Institutional Ethical Committee for Animal Experimentation (P181-2018). The study protocol of the human study was approved by the Institutional Ethical Review Board (ML4190). Written informed consent was obtained from all patients or their next of kin.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
De Bruyn, L., Téblick, A., Van Oudenhove, T. et al. Glucocorticoid treatment increases cholesterol availability during critical illness: effect on adrenal and muscle function. Crit Care 28, 295 (2024). https://doi.org/10.1186/s13054-024-05079-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-024-05079-8