
Abstract
Background
Equivalent efficacy and comparable pharmacokinetic, immunogenicity, and safety profiles of the biosimilar BAT1806/BIIB800 and reference tocilizumab (TCZ) in participants with moderate-to-severe rheumatoid arthritis (RA) have been reported up to week 24 (treatment period [TP] 1) of the phase 3 study. Here we present results for TP2 (study weeks 24–48).
Methods
In this phase 3, multicenter, multiregional, double-blind, active-controlled, equivalence study, participants with active RA despite methotrexate were randomized (1:1:2) to intravenous administration of 8 mg/kg TCZ every 4 weeks to week 48 (TCZ group), or TCZ to week 24 followed by BAT1806/BIIB800 to week 48 (TCZ to BAT1806/BIIB800 group), or BAT1806/BIIB800 to week 48 (BAT1806/BIIB800 group). Efficacy in TP2 was evaluated using American College of Rheumatology (ACR) response criteria (ACR20/50/70) and change from baseline in Disease Activity Score on 28 joints (DAS28). Pharmacokinetics (trough levels), safety, and immunogenicity were also evaluated.
Results
Of 621 randomized participants, 577 (92.9%) completed TP1 and entered TP2 (TCZ: N = 145 [93.5%]; TCZ to BAT1806/BIIB800: N = 142 [92.2%]; BAT1806/BIIB800: N = 290 [92.9%]). Proportions of ACR20 responders were similar between treatment groups throughout TP2 (87.8%, 90.3%, and 90.4%, respectively, at week 48), as were proportions of ACR50 and ACR70 responders, and reduction in DAS28. Drug trough levels and antidrug antibody incidences were comparable between the treatment groups. Adverse events were balanced across the treatment groups and no fatal events were reported.
Conclusion
In TP2, efficacy, safety, immunogenicity, and pharmacokinetic profiles were comparable between the TCZ, TCZ to BAT1806/BIIB800, and BAT1806/BIIB800 groups.
Trial registration
NCT03830203 and EudraCT 2018-002202-31.
Similar content being viewed by others
Background
Tocilizumab was the first humanized anti–interleukin-6 receptor (IL-6R) monoclonal antibody to be approved for the treatment of rheumatoid arthritis (RA). BAT1806/BIIB800 is an IL-6R–targeted, recombinant, humanized, monoclonal immunoglobulin G1 antibody developed in accordance with regulatory guidance as a biosimilar of the tocilizumab reference product (TCZ; Actemra™/RoActemra™) [1,2,3].
The development of biosimilars follows a stepwise approach to demonstrate similarity between the biosimilar and the reference product in terms of physicochemical characteristics, biological activity and pharmacokinetics (PK)/pharmacodynamics. For BAT1806/BIIB800, a phase 1 clinical study in healthy volunteers demonstrated equivalent PK and comparable safety and immunogenicity profiles following intravenous administration of a single dose of BAT1806/BIIB800 or TCZ [4]. A phase 3, multicenter, multiregional, randomized, double-blind, active-controlled equivalence study was conducted to demonstrate the clinical similarity of BAT1806/BIIB800 and TCZ administered intravenously in a population with moderate-to-severe RA with inadequate response to methotrexate (MTX). Findings from the initial 24 weeks (treatment period [TP] 1) of that trial demonstrated equivalent efficacy and similar safety, immunogenicity, and PK profiles of BAT1806/BIIB800 compared with TCZ at weeks 12 and 24 after initiation of study treatment [5].
Here the efficacy results for TP2 (i.e., week 24 to week 48), and safety, PK, and immunogenicity findings from week 24 to week 52 are reported.
Methods
Study design
This was a phase 3, multicenter, multiregional, randomized, double-blind, active-controlled clinical trial [5]. The study comprised a ≤ 28-day screening period, an initial 24-week period (TP1), and a subsequent 24-week period (TP2).
Eligible participants were randomized (1:1:2) using an interactive web-response system to receive TCZ up to week 48 (TCZ group), or TCZ up to week 24 followed by BAT1806/BIIB800 up to week 48 (TCZ→BAT1806/BIIB800 group), or BAT1806/BIIB800 up to week 48 (BAT1806/BIIB800 group). All study drugs were administered intravenously at a dose of 8 mg/kg once every 4 weeks. Randomization was stratified by geographical region and by previous use of biological disease-modifying antirheumatic drugs (DMARDs), or targeted synthetic DMARDs [5].
Participants
All participants provided informed consent to participate, and the study was conducted in accordance with the Declaration of Helsinki and all relevant local regulations, in compliance with the International Council for Harmonisation Good Clinical Practice guidelines, and according to the appropriate regulatory requirements in the countries where the study was conducted.
Details of the study eligibility criteria are reported elsewhere [5]. In brief, eligible adults had an RA diagnosis of ≥ 6 months prior to screening, active disease (tender joint count ≥ 6 out of 68 joints and swollen joint count ≥ 6 out of 66 joints) and, at screening, a serum C-reactive protein (CRP) level greater than the upper limit of normal or erythrocyte sedimentation rate (ESR) ≥ 28 mm/h, despite receiving a stable dose of MTX (ranging between 10 and 25 mg/week). Prior treatment with more than two biological DMARDs or targeted synthetic DMARDs was not permitted, nor was treatment with any IL-6 inhibitor. Participants were expected to remain on their stable dose of MTX throughout the study.
Endpoints
Efficacy in TP2 was assessed as the proportion of participants achieving ≥ 20% improvement over time (week 28 to week 48) in American College of Rheumatology (ACR) response criteria (ACR20), ≥50% improvement in ACR response criteria (ACR50), and ≥ 70% improvement in ACR response criteria (ACR70), and change over time (week 28 to week 48) from baseline (week 0) in Disease Activity Score on 28 joints using ESR (DAS28-ESR) and Disease Activity Score on 28 joints using CRP (DAS28-CRP).
PK in TP2 was assessed as tocilizumab serum trough concentration (Ctrough), measured at weeks 28, 36, 44, and 48, and at the end-of-study follow-up visit (i.e., week 52) or at participant withdrawal, whichever occurred first.
Safety, assessed throughout TP2 and up to the end-of-study follow-up visit at week 52, was determined by monitoring vital signs, physical examination, laboratory assessments (hematology, chemistry with lipids panel, and urinalysis), 12-lead electrocardiogram, and reporting of adverse events (AEs) including treatment-emergent AEs (TEAEs), serious TEAEs, study-drug-related TEAEs, and study-drug-related serious TEAEs. Immunogenicity was assessed by the presence of antidrug antibodies (ADAs), including neutralizing antibodies (nAbs), and ADA titers at weeks 28, 36, and 48, and at the end-of-study follow-up visit at week 52 or 8 weeks after the last dose of study drug, whichever occurred first.
Statistical analysis
Efficacy analyses were performed by randomized treatment group sequence in participants receiving TCZ, TCZ→BAT1806/BIIB800, or BAT1806/BIIB800. The clinical efficacy endpoints (ACR20, ACR50, and ACR70, and change from baseline in DAS28-ESR and DAS28-CRP) for TP2 were analyzed on the full analysis set, which included all randomized participants who completed TP1 and entered TP2, using all observed outcome values. For DAS28 (ESR and CRP), least squares means for each group were estimated at each visit with a mixed model for repeated measures (MMRM) with the factors treatment arm, visit, randomized strata (region [Central Europe/Asia Pacific] and previous biologic or targeted synthetic DMARD use [Yes/No]), baseline, and treatment-by-visit interaction in the model, with and without missing data imputation, respectively. Missing data were imputed using baseline observation carried forward (BOCF) to reflect a nonresponder imputation. These MMRM analyses (for TP2) were not prespecified in the study statistical analysis plan, they were additionally requested by Regulatory Agencies during the scientific evaluation of the medicinal product.
Descriptive statistics of the observed clinical outcomes (ACR, ADAs, and intercurrent events) are provided.
Safety, immunogenicity, and PK endpoints were summarized descriptively. The safety set, as defined in the study statistical analysis plan, included all randomized participants who completed TP1, entered TP2, and who received at least one dose of study drug. All safety and immunogenicity endpoint analyses were performed on the safety set. Analysis of PK data was performed on the PK set, which included all randomized participants who had received at least one dose of study drug and had ≥ 1 evaluable PK assessment postbaseline.
All statistical analyses were conducted using SAS statistical software version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
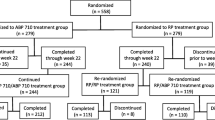
Between December 19, 2018 and January 5, 2021, 935 participants were screened and 621 eligible participants were randomized to study treatment (TCZ, N = 155; TCZ→BAT1806/BIIB800, N = 154; BAT1806/BIIB800, N = 312; Fig. 1). Of the 621 randomized participants, 577 (92.9%) completed TP1 and entered TP2 (N = 145 [93.5%], N = 142 [92.2%], and N = 290 [92.9%], respectively); overall, 89.4% (555/621) of participants completed the study. Demographic and clinical characteristics remained balanced at week 24 (Table 1).
The treatment groups were similar prior to the start of TP2 also in terms of intercurrent events (type and occurrence percentage) (Supplemental Fig. 1) [5, 6].
Proportions of ACR20, ACR50, and ACR70 responders were similar between treatment groups throughout TP2 (Fig. 2). At week 48, the proportions of participants who achieved ACR20 in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups were 87.8% (122/139), 90.3% (121/134), and 90.4% (253/280), respectively. The proportions of participants who achieved ACR50 at week 48 were 61.9% (86/139), 70.1% (94/134), and 70.4% (197/280), respectively, and for ACR70 were 38.8% (54/139), 49.3% (66/134), and 46.1% (129/280), respectively.
DAS28 (ESR and CRP) responses at each time point in TP2 were similar across the three treatment groups (Fig. 3, Supplemental Table 1).
The treatment groups displayed similar longitudinal trajectories in terms of DAS28-ESR and DAS28-CRP responses (Supplemental Fig. 2A–D). From the MMRM analyses, no differences between treatment arms with regards to DAS28-ESR and DAS28-CRP were observed, with overlapping 95% CIs at all time points. At week 48, mean (standard deviation [SD]) changes in DAS28-ESR were − 3.7 (1.5), − 4.1 (1.4), and − 4.2 (1.5) in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, respectively. At week 48, mean (SD) changes in DAS28-CRP were − 3.1 (1.1), − 3.4 (1.2), and − 3.4 (1.2), respectively.
In the PK analysis, Ctrough geometric mean values for the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups were 12.4 μg/mL (coefficient of variation [CV]% 180.7), 12.7 μg/mL (CV% 157.0), and 13.1 µg/mL (CV% 126.5), respectively, at week 28, and 13.5 µg/mL (CV% 121.0), 13.4 µg/mL (CV% 125.1), and 12.3 µg/mL (CV% 150.6), respectively, at week 44.
In TP2, 345 (59.8%) participants experienced a total of 1179 AEs, including 323 events in 91 (62.8%), 287 events in 92 (64.8%), and 569 events in 162 (55.9%) participants in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, respectively (Table 2). Overall, 344 (59.6%) participants reported 1163 TEAEs; 318 TEAEs in 90 (62.1%) participants in the TCZ group, 286 TEAEs in 92 (64.8%) participants in the TCZ→BAT1806/BIIB800 group, and 559 TEAEs in 162 (55.9%) participants in the BAT1806/BIIB800 group. The most common TEAEs in TP2 across treatment groups were upper respiratory tract infection, reported by 41 of 577 (7.1%) participants, and leukopenia, reported in 40 of 577 (6.9%) participants. Related TEAEs occurred in 59 (40.7%), 64 (45.1%), and 112 (38.6%) participants in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, respectively. Most were mild in severity (mild: 56 [38.6%] participants, 56 [39.4%] participants, and 101 [34.8%] participants, respectively; moderate: 14 [9.7%] participants, 18 [12.7%] participants, and 26 [9.0%] participants, respectively; severe: 2 [1.4%] participants, 0 [0%] participants, and 1 [0.3%] participants, respectively). Five serious TEAEs were reported in four (2.8%) participants in the TCZ group, five serious TEAEs in five (3.5%) participants in the TCZ→BAT1806/BIIB800 group, and nine serious TEAEs in eight (2.8%) participants in the BAT1806/BIIB800 group.
The only serious TEAE to occur in more than one participant in any one treatment group was tooth abscess, which occurred in two participants in each of the treatment groups. Of the serious TEAEs, one (pneumonia) in the TCZ group, one (laryngitis) in the TCZ→BAT1806/BIIB800 group, and two (pneumonia and salpingo-oophoritis) in the BAT1806/BIIB800 group were considered by the investigator to be possibly related to study treatment (Table 3). There were no fatal events in TP2.
In TP2, 18.6% (27/145), 16.2% (23/142), and 21.0% (61/290) of participants in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, respectively, had ≥ 1 positive ADA result at any time point. Across the treatment groups, the majority of ADA-positive participants reported titers of < 20 or 20. All ADA-positive tests were also positive for nAbs, with the exception of a single result in the TCZ group. The point prevalence for participants testing positive for ADAs remained stable in all treatment groups, and the majority of positive cases transitioned from positive to negative across the study period (Fig. 4). At week 52 (8 weeks after the last dose of study drug), 11 (7.9%), 2 (1.5%), and 16 (5.7%) participants were ADA-positive in the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, respectively; the majority of these participants were also positive for nAbs at this time point (10 [7.2%], 2 [1.5%], and 15 [5.4%], respectively).
Discussion
In TP2 BAT1806/BIIB800 showed comparable efficacy, safety, immunogenicity, and PK to TCZ, in a population of patients with RA and an inadequate response to MTX. These longer-term findings reinforce the results reported for TP1 and further support biosimilarity. The results do not suggest any detriment in terms of efficacy, safety, or immunogenicity following a switch from TCZ to BAT1806/BIIB800.
Efficacy as assessed by ACR response or change in DAS28 at 1 year was comparable across the treatment groups. No loss of efficacy was observed during TP2 in any treatment group [5], and was in line with reported efficacy at 1 year in published studies of the reference product [7, 8]. The safety profile of BAT1806/BIIB800 was comparable to that of TCZ up to week 52 and no new safety signals were identified.
Demonstrating PK similarity of a reference product and its biosimilar is a key aspect of a successful biosimilarity exercise [9]. No differences in PK parameters were observed between the treatment groups, with a similarly large magnitude of variability observed. These findings are consistent with results from the phase 1 PK study [4] and from TP1 of the present study [5]. Additionally, previous studies have reported that > 95% of the sIL-6R is bound at Ctrough levels of 1 µg/mL [10] and that trough concentrations above 1 µg/mL could normalize CRP, whereas lower trough levels were associated with reduced DAS28 responses [11].
While the prevalence of ADAs in TP2 was comparable between the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 groups, the overall prevalence was higher than historical values reported for the reference product in a pooled analysis of clinical studies (1.2%) [12]. This is likely to be due to the use of more sensitive and drug-tolerant ADA assays than those used previously [13]. Most ADA-positive participants had a titer of < 20 or 20, and transitioned between positive and negative status throughout the study, suggesting that the majority of the antibody responses were transient and not clinically meaningful. This is in line with the aforementioned pooled analysis of the reference product, which reported that for the majority of positive ADA results titers were low and ADAs were transient; that analysis also reported no impact of ADA positivity on efficacy [12]. Furthermore, the pooled analysis found no correlation between the presence of ADAs and either safety or PK events (including anaphylaxis, hypersensitivity, and injection-site reactions) [12].
The findings of this study must be considered in the context of several potential limitations. A single indication (RA) was chosen for the study population; this was selected as being the most sensitive population eligible for TCZ treatment. Based on the principle of extrapolation of indications, a demonstration of clinical similarity in one sensitive indication can be extrapolated to all labeled indications of the reference product, without the need for additional clinical studies, if adequately justified [12]. The study population was predominantly female; however, global prevalence data show that 70% of the population diagnosed with RA are women, [14] indicating that the study sample is broadly representative. The geographic locations of the study sites included China and four European countries, with limited enrollment of different ethnicities; as there is no evidence that ethnicity or race affects the PK of the reference product this is not considered likely to lessen the validity of the study results [15, 16]. Disease progression was not measured radiographically, however the clinical endpoints used in this study (ACR, DAS28) were agreed with regulatory agencies during the study design stage as being appropriately sensitive to detect differences between the biosimilar and reference product, should these exist. This study was designed to assess the possible impact of a single treatment switch from TCZ to BAT1806/BIIB800 after week 24. In clinical practice, multiple treatment switches may occur; post-approval studies may provide additional data about the use of biosimilars in real-world settings.
Conclusion
In conclusion, the efficacy, safety, immunogenicity, and PK profiles in TP2 were comparable between the TCZ, TCZ→BAT1806/BIIB800, and BAT1806/BIIB800 treatment groups.

Trial disposition for TP2. a Eligible participants were randomized (1:1:2) to receive TCZ up to week 48 (TCZ), TCZ up to week 24 followed by BAT1806/BIIB800 up to week 48 (TCZ→BAT1806/BIIB800), or BAT1806/BIIB800 up to week 48 (BAT1806/BIIB800). b Participants were noted as having completed the study if they completed the follow-up visit, regardless of whether they completed TP2 (defined as completing week 48/EOS visit). EOS End of study, TB Tuberculosis, TCZ Tocilizumab reference product, TP2 Treatment period 2

Proportions of participants achieving ACR20, ACR50, and ACR70 during TP2 (FAS). Participants were considered as entered into TP2 if the last study drug administration visit was on or after week 24, or participants received the first dose of BAT1806/BIIB800 for the switched group (TCZ→BAT1806/BIIB800). ACR response rates were calculated based on the number of participants with evaluable data at each time point, with data as observed; participants with missing data were considered as nonresponders. FAS included all randomized participants. ACR20/50/70 ≥ 20%/50%/70% response in the American College of Rheumatology criteria, FAS Full analysis set, TCZ Tocilizumab reference product, TP2 Treatment period 2

DAS28 (CRP [A] and ESR [B]) for the treatment groups by TP2 visits (FAS). CRP C-reactive protein, DAS28 Disease Activity Score on 28 joints, ESR Erythrocyte sedimentation rate, FAS Full analysis set, SD Standard deviation, TCZ Tocilizumab reference product, TP2 Treatment period 2

Sankey plot of ADA status at each visit during TP2: for (A) the TCZ group, (B) the TCZ→BAT1806/BIIB800 group, and (C) the BAT1806/BIIB800 group. ADA Antidrug antibody, TCZ Tocilizumab reference product, TP2 Treatment period 2
Data availability
The data that support the findings of this study are available from the authors on request. Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further information on data sharing is available via Biogen’s clinical transparency and data sharing policy.
Abbreviations
- ACR:
-
American College of Rheumatology
- ADA:
-
Antidrug antibody
- AE:
-
Adverse event
- ALT:
-
Alanine aminotransferase
- bDMARD:
-
Biological disease-modifying antirheumatic drug
- BOCF:
-
Baseline observation carried forward
- CCP:
-
Cyclic citrullinated peptide
- CRP:
-
C-reactive protein
- Ctrough :
-
Serum trough concentration
- CV:
-
Coefficient of variation
- DAS28:
-
Disease Activity Score on 28 joints
- DMARD:
-
Disease-modifying antirheumatic drug
- EOS:
-
End of study
- ESR:
-
Erythrocyte sedimentation rate
- FAS:
-
Full analysis set
- GA:
-
Global Assessment
- HAQ-DI:
-
Health Assessment Questionnaire – Disability Index
- IL-6R:
-
Interleukin-6 receptor
- MMRM:
-
Mixed model for repeated measures
- MTX:
-
Methotrexate
- nAb:
-
Neutralizing antibody
- PK:
-
Pharmacokinetic
- RA:
-
Rheumatoid arthritis
- RF:
-
Rheumatoid factor
- SD:
-
Standard deviation
- SJC68:
-
Swollen joint count of 68 joints
- TB:
-
Tuberculosis
- TCZ:
-
Tocilizumab reference product
- TEAE:
-
Treatment-emergent adverse event
- TP:
-
Treatment period
- tsDMARD:
-
Targeted synthetic disease-modifying antirheumatic drug
- VAS:
-
Visual analogue scale
References
Center for Drug Evaluation of the National Medical Products Administration. Technical guidelines for the development and evaluation of biosimilar drugs (in Chinese). [Internet]. 2015. https://www.cde.org.cn/zdyz/domesticinfopage?zdyzIdCODE=f044cdf4b7d7286aa12ffb85fc81a74c. Accessed January 12, 2023.
European Medicines Agency. Guideline on similar biological medicinal products [Internet]. 2014. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Accessed August 16, 2022.
US Food and Drug Administration. Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations [Internet]. 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-therapeutic-protein-biosimilars-comparative-analytical-assessment-and-other-quality. Accessed August 16, 2022.
Zhang H, Wang H, Wei H, Chen H, Liu J, Li C, et al. A phase I clinical study comparing the tolerance, immunogenicity, and pharmacokinetics of proposed biosimilar BAT1806 and reference tocilizumab in healthy Chinese men. Front Pharmacol. 2020;11:609522.
Leng X, Leszczynski P, Jeka S, Liu SY, Liu H, Miakisz M, et al. Comparing tocilizumab biosimilar BAT1806/BIIB800 with reference tocilizumab in patients with moderate-to-severe rheumatoid arthritis with an inadequate response to methotrexate: a phase 3, randomised, multicentre, double-blind, active-controlled clinical trial. Lancet Rheumatol. 2024;6(1):e40–50.
US Department of Health and Human Services, US Food and Drug Administration. E9(R1): Statistical principles for clinical trials: addendum: estimands and sensitivity analysis in clinical trials [Internet]. 2021. https://www.fda.gov/media/148473/download. Accessed December 4, 2023.
Kaneko Y, Atsumi T, Tanaka Y, Inoo M, Kobayashi-Haraoka H, Amano K, et al. Comparison of adding tocilizumab to methotrexate with switching to tocilizumab in patients with rheumatoid arthritis with inadequate response to methotrexate: 52-week results from a prospective, randomised, controlled study (SURPRISE study). Ann Rheum Dis. 2016;75(11):1917–23.
Dougados M, Kissel K, Conaghan PG, Mola EM, Schett G, Gerli R, et al. Clinical, radiographic and immunogenic effects after 1 year of tocilizumab-based treatment strategies in rheumatoid arthritis: the ACT-RAY study. Ann Rheum Dis. 2014;73(5):803–9.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–34.
Nishimoto N, Terao K, Mima T, Nakahara H, Takagi N, Kakehi T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti–IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood. 2008;112(10):3959–64.
Kneepkens EL, van den Oever I, Plasencia CH, Pascual-Salcedo D, de Vries A, Hart M, et al. Serum tocilizumab trough concentration can be used to monitor systemic IL-6 receptor blockade in patients with rheumatoid arthritis: a prospective observational cohort study. Scand J Rheumatol. 2017;46(2):87–94.
Burmester GR, Choy E, Kivitz A, Ogata A, Bao M, Nomura A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–85.
Gehin JE, Goll GL, Brun MK, Jani M, Bolstad N, Syversen SW. Assessing immunogenicity of biologic drugs in inflammatory joint diseases: progress towards personalized medicine. BioDrugs. 2022;36(6):731–48.
World Health Organization. Rheumatoid arthritis [Internet]. 2023. https://www.who.int/news-room/fact-sheets/detail/rheumatoid-arthritis#:~:text=In%202019%2C%2018%20million%20people,benefit%20from%20rehabilitation%20(2). Accessed August 18, 2023.
European Medicines Agency. Actemra (tocilizumab). Prescribing information [Internet]. https://www.ema.europa.eu/en/documents/product-information/roactemra-epar-product-information_en.pdf. Accessed August 30, 2022.
US Food and Drug Administration. Actemra (tocilizumab). Prescribing information [Internet]. 2023. https://www.gene.com/download/pdf/actemra_prescribing.pdf. Accessed July 7, 2023.
Acknowledgements
The authors would like to thank all participants who participated in this study, as well as the investigators, study coordinators, study teams and nurses at each study site. Research was sponsored by Bio-Thera Solutions Ltd, Guangzhou, China; ClinicalTrials.gov Identifier (NCT03830203) and EudraCT (2018-002202-31). Hans Ebbers of Biogen contributed to data interpretation and contributed to manuscript content. Medical writing assistance was provided by Simon Rhead, PhD and Jacqueline Kolston, PhD of Parexel International and was funded by Biogen International GmbH, Baar, Switzerland.
Funding
Bio-Thera Solutions Ltd funded this study. Biogen International GmbH funded the development of the manuscript.
Author information
Authors and Affiliations
Contributions
All external authors contributed to data collection (as study investigators). XY, YZ, QD, (employees of Bio-Thera Solutions Ltd) contributed to the trial design as well as data analysis, and reporting. MMit, JA, and MFR were involved in data analysis, reporting, and interpretation, and reviewed and revised the manuscript (as employees of Biogen). All authors had full access to all study results and contributed to interpretation. All authors participated in writing the manuscript, with the support of medical writing services provided by the funder. All authors read and approved the submitted version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki and/or all relevant local regulations, in compliance with the International Council for Harmonisation Good Clinical Practice guidelines, and according to the appropriate regulatory requirements in the countries where the study was conducted.
Consent for publication
Not applicable.
Competing interests
XL, SJ, SL, HL, MMia, JG, and XZ have no conflicts of interest. PL has received speaker fees from Novartis, AbbVie, UCB, MSD, and GSK; has received support for attending meetings from AbbVie, AstraZeneca, and Medac, and has received a grant for this study from Bio-Thera Solutions Ltd. LK has received speaker fees from Sandoz, Amgen, and Takeda. MS has received speaker fees from Pfizer, Orion, and Boehringer Ingelheim. XY, YZ, and QD are Bio-Thera Solutions Ltd employees and might hold stock, stock options, or both in Bio-Thera Solutions Ltd. MMit, JA, and MFR are Biogen employees and may hold stock, stock options, or both in Biogen.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Leng, X., Leszczyński, P., Jeka, S. et al. A phase 3, randomized, double-blind, active-controlled clinical trial to compare BAT1806/BIIB800, a tocilizumab biosimilar, with tocilizumab reference product in participants with moderate-to-severe rheumatoid arthritis with inadequate response to methotrexate: treatment period 2 analysis (week 24 to week 48). Arthritis Res Ther 26, 157 (2024). https://doi.org/10.1186/s13075-024-03375-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13075-024-03375-w