
Abstract
Background
Diabetic nephropathy (DN) is the most common causes of end-stage renal disease. Long non-coding RNA cyclin-dependent kinase inhibitor 2B antisense RNA 1 (CDKN2B-AS1) is connected with the development of DN, but the role of CDKN2B-AS1 in DN has not been entirely elucidated.
Methods
Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out to measure CDKN2B-AS1 and miR-98-5p levels. Cell viability, proliferation, and apoptosis were analyzed with 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) or flow cytometry assays. Protein levels were measured by western blotting. The relationship between CDKN2B-AS1 or notch homolog 2 (NOTCH2) and miR-98-5p was verified via dual-luciferase reporter assay.
Results
CDKN2B-AS1 and NOTCH2 were upregulated in the serum of DN patients and high glucose-disposed human podocytes (HPCs) and human renal tubular cells (HK-2), whereas miR-98-5p was downregulated. High glucose repressed viability and accelerated apoptosis of HPCs and HK-2 cells. CDKN2B-AS1 knockdown impaired high glucose-induced apoptosis and fibrosis of HPCs and HK-2 cells. Mechanistically, CDKN2B-AS1 sponged miR-98-5p to regulate NOTCH2 expression. Also, CDKN2B-AS1 inhibition-mediated effects on apoptosis and fibrosis of high glucose-disposed HPCs and HK-2 cells were weakened by miR-98-5p inhibitor. Also, NOTCH2 knockdown partly reversed miR-98-5p inhibitor-mediated impacts on apoptosis and fibrosis of high glucose-disposed HPCs and HK-2 cells.
Conclusion
High glucose-induced CDKN2B-AS1 promoted apoptosis and fibrosis via the TGF-β1 signaling mediated by the miR-98-5p/NOTCH2 axis in HPCs and HK-2 cells.
Highlights
-
1.
CDKN2B-AS1 was upregulated in DN and high glucose-induced HPCs and HK-2 cells.
-
2.
Inhibition of CDKN2B-AS1 alleviated high glucose-induced apoptosis and fibrosis of HPCs and HK-2 cells.
-
3.
CDKN2B-AS1 regulated NOTCH2 expression via acting as a miR-98-5p sponge.
Similar content being viewed by others


Introduction
Diabetic nephropathy (DN), a progressive kidney disease caused by diabetes, is characterized by persistent albuminuria and a gradual decline in estimated glomerular filtration rate [1, 2]. It is reported that about 30–40% of patients with diabetes may develop DN, and approximately 50% of DN patients tend to develop end-stage renal disease [3]. Renal fibrosis is the main driving force for the occurrence of DN, and hyperglycemia in diabetic patients may trigger renal fibrosis [4]. Therefore, exploring the pathogenesis of DN is important to improve DN.
Long non-coding RNAs (lncRNAs) are a class of transcripts that do not have protein-encoding capabilities [5]. Studies have shown that lncRNAs are associated with a plethora of cellular functions [6, 7]. Also, lncRNAs play their roles in a series of diseases through interacting with microRNAs [8,9,10]. For instance, lncRNA GAS5 decreased pyroptosis and oxidative stress via sponging miR-452-5p in high glucose-induced renal tubular cells [11]. Cyclin-dependent kinase inhibitor 2B antisense RNA 1 (CDKN2B-AS1) is connected with the development of diabetes [12], coronary heart disease [13], atherosclerosis [14], and cancers [15]. Furthermore, CDKN2B-AS1 modulated extracellular matrix accumulation and proliferation of mesangial cells [16]. MiRNA-98-5p (miR-98-5p) plays vital roles in the advancement of numerous diseases. In diabetes, miR-98-5p accelerated cell apoptosis and impeded cell proliferation in keratinocytes through targeting PPP1R15B [17]. MiR-98-5p could improve OGD/R-induced neuronal injury via downregulating Bach1 [18]. Moreover, miR-98-5p mediated renal fibrosis and epithelial-to-mesenchymal in DN [19]. Nevertheless, it is unclear whether CDKN2B-AS1 mediates the development of DN via miR-98-5p.
It has been confirmed that the NOTCH pathway mediates renal fibrosis [20, 21]. Notch homolog 2 (NOTCH2) is one of the important receptors in the NOTCH pathway [22]. NOTCH2 was reported to be connected with high glucose-stimulated cardiac fibrosis and epithelial-to-mesenchymal in HAVECs [23]. Also, JNK is an upstream effector of NOTCH2 in TGF-β1-mediated renal fibrosis [24]. Furthermore, HG triggered EMT through the Notch2 pathway in NRK-52E cells [25]. However, the regulatory mechanisms associated with NOTCH2 in DN development have not been fully elucidated.
Herein, we reported an accelerative influence of CDKN2B-AS1 on the pathogenesis of DN. Also, we found that CDKN2B-AS1 induced apoptosis and fibrosis through upregulating NOTCH2 via sponging miR-98-5p under high glucose treatment. Therefore, the research provided a novel mechanism to comprehend the pathogenesis of DN.
Materials and methods
Subjects
The research was authorized and supervised by the ethics committee of Qilu Hospital, Cheeloo College of Medicine, Shandong University. 30 patients with DN and 30 healthy controls were recruited from Qilu Hospital, Cheeloo College of Medicine, Shandong University. T2D patients with a urine albumin/creatinine ratio > 30 mg/g or estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 were defined to have DN. The participating patients were free of cardiovascular disease, chronic liver disease, or cerebrovascular disease. Participants in this study signed informed consent.
Cell culture, treatment, and transfection
Human podocytes (HPCs) and human renal tubular cells (HK-2) (Bena Culture Collection, Suzhou, China) were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma, St Louis, MO, USA) with 10% fetal bovine serum (FBS, Life Technologies, Carlsbad, CA, USA), streptomycin (100 μg/mL, Life Technologies), and penicillin (100 U/mL, Life Technologies) at 37 °C under an atmosphere containing 5% CO2. For high glucose treatment, HPCs and HK-2 cells were treated with 30 mM glucose for 24 h. 5 mM glucose acted as a normal glucose, whereas 5 mM glucose plus 25 mM mannitol acted as an osmotic control.
Small interference RNA targeting CDKN2B-AS1 (si-CDKN2B-AS1) and NOTCH2 (si-NOTCH2), as well as their matching controls (si-NC), were obtained from GenePharma (Shanghai, China). The sequences of CDKN2B-AS1 and NOTCH2 were cloned into the plenti-GIII-CMV-2A-Puro-GFP vector (vector) (ABM, Canada) or pcDNA3.1 vector (pc-NC) (Invitrogen, Carlsbad, CA, USA) to obtain plenti-GIII-CMV-2A-Puro-GFP-CDKN2B (CDKN2B-AS1) and pcDNA3.1-NOTCH2 (pc-NOTCH2) vectors, respectively. MiR-98-5p mimic and inhibitor, as well as their corresponding controls (miRNA NC and inhibitor NC), were also bought from GenePharma. When cell confluence reached 80%, HPCs and HK-2 cells were transfected with the designated plasmids or oligonucleotides using Lipofectamine 3000 reagent (Life Technologies).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted through TRIzol reagent (Life Technologies). Total RNA was reverse-transcribed by PrimeScript RT reagent Kit (Takara, Dalian, China) or miRNA First-Strand Synthesis Kit (Takara). QPCR was conducted through the SYBR Premix Ex Taq (Takara) with specific primers (Table1, β-actin and U6 were utilized as house-keeping genes). Expression levels of CDKN2B-AS1 and miR-98-5p were figured with the 2−ΔΔCt method.
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay
Cell viability and proliferation were determined with an MTT assay kit (Beyotime, Shanghai, China). After a period of incubation, the MTT solution (20 μL, 5 mg/mL) was added to each well and incubated for 4 h. After moving the medium, the dimethylsulfoxide (150 μL) was used to solubilize the crystals. The optical density at 490 nm was analyzed through a Microplate Reader (Bio-Rad, Richmond, CA, USA).
Cell apoptosis analysis
After collection, digestion, and centrifugation, the cells were re-suspended in binding buffer (1×). Cell apoptosis was analyzed using the Annexin V-fluorescein isothiocyanate (FITC)/PI apoptosis detection kit (KeyGen, Jiangsu, China). Cell fluorescence was analyzed through a FACScan flow cytometry (Beckman Coulter, Brea, CA, USA).
Dual-luciferase reporter assay
The sequences of wild type (WT) CDKN2B-AS1, mutant (MUT) CDKN2B-AS1, WT-NOTCH2-3ʹUTR, and MUT-NOTCH2-3ʹUTR were inserted into the pmirGLO luciferase vectors (GeneCreat, Wuhan, China), respectively. HPCs and HK-2 cells were transfected with a luciferase reporter together with miRNA NC or miR-98-5p mimic. The luciferase intensities were assessed via the luciferase reporter assay kit (Promega, Madison, WI, USA).
Western blot analysis
Total protein was extracted with the RIPA buffer containing a protease inhibitor cocktail (Sigma). 30 μg total protein was isolated via the sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Subsequently, the PVDF membranes were incubated with primary antibodies, including rabbit anti-NOTCH2 (ab137665, 1:500, Abcam, Cambridge, MA, USA), rabbit anti-TGF-β1 (ab92486, 1:500, Abcam), rabbit anti-Bax (ab32503, 1:1000, Abcam), rabbit anti-Bcl-2 (ab182858, 1:2000, Abcam), rabbit anti-fibronectin (FN) (ab32419, 1:1000, Abcam), rabbit anti-collagen I (Col.l) (ab34710, 1:2000, Abcam) and rabbit anti-β-actin (ab8227, 1:1000, Abcam). GAPDH was used as a loading control. Next, the PVDF membranes were incubated with goat anti-rabbit IgG (ab97051, 1:5000, Abcam). Protein bands were visualized with an ImmunoStar LD (Wako Pure Chemical, Osaka, Japan). Densitometric analysis was carried out using ImageJ software 1.6.0 (NIH, MD, USA).
Statistical analysis
The data were expressed as mean ± standard deviation, which was derived from 3 replicate experiments. GraphPad Prism 6.0 software was utilized for statistical analysis. Differences were deemed significant if P < 0.05. Student’s t test was used to analyze the differences between two groups. One-way variance analysis with Turkey’s test was utilized for the comparison of the differences among more groups.
Results
CDKN2B-AS1 was upregulated in DN and high glucose-stimulated HPCs and HK-2 cells
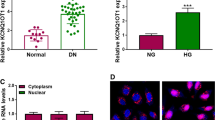
Considering the abnormal expression of CDKN2B-AS1 in DN, we detected the expression level of CDKN2B-AS1 in serums from 30 DN patients and 30 normal controls. QRT-PCR manifested that CDKN2B-AS1 expression levels were increased in the serum of DN patients compared to the control group (Fig. 1A). Subsequently, we assessed the viability of HPCs and HK-2 cells treated with different concentrations of glucose. MTT assay presented that high glucose (30 mM and 40 mM) led to a decrease in the viability of HPCs and HK-2 cells (Fig. 1B, C). And the HPCs and HK-2 cells treated with 30 mM glucose were chose for subsequent analysis. We observed that CDKN2B-AS1 expression levels were elevated in high glucose-treated HPCs and HK-2 cells (Fig. 1D). These results indicated that CDKN2B-AS1 might be involved in the development of DN.

Expression levels of CDKN2B-AS1 in DN and high glucose-disposed HPCs and HK-2 cells. A QRT-PCR was performed to analyze the expression levels of CDKN2B-AS1 in the serum of 30 DN patients and 30 normal controls. B, C MTT assay was conducted for the evaluation of the viability of HPCs and HK-2 cells disposed with different concentrations of glucose (0, 10, 20, 30, and 40 mM). D QRT-PCR was executed to assess the expression levels of CDKN2B-AS1 in HPCs and HK-2 cells treated with normal glucose, 5 mM glucose plus 25 mM, and 30 mM glucose. *P < 0.05
CDKN2B-AS1 regulated apoptosis and fibrosis of HPCs and HK-2 cells under high glucose treatment
Given that the upregulation of CDKN2B-AS1 in DN and high glucose-disposed HPCs and HK-2 cells, we further investigated the role of CDKN2B-AS1 in DN through loss-of-function experiments. Compared to the control groups, CDKN2B-AS1 was overexpressed in HPCs and HK-2 cells after transfection with CDKN2B-AS1 under high glucose treatment and decreased in HPCs and HK-2 cells after transfection with si-CDKN2B-AS1 under high glucose treatment (Fig. 2A, B). Moreover, CDKN2B-AS1 elevation aggravated proliferation inhibition and apoptosis of high glucose-stimulated HPCs and HK-2 cells, but CDKN2B-AS1 downregulation impaired proliferation inhibition and apoptosis of high glucose-stimulated HPCs and HK-2 cells (Fig. 2C–F). Western blotting displayed that CDKN2B-AS1 elevation resulted in a decrease in Bcl-2 protein levels and an increase in Bax protein levels in high glucose-disposed HPCs and HK-2 cells, while CDKN2B-AS1 silencing played an opposing impact (Fig. 2G, H). In addition, CDKN2B-AS1 overexpression elevation protein levels of TGF-β1, FN, Col.I in high glucose-disposed HPCs and HK-2 cells, but CDKN2B-AS1 silencing decreased protein levels of TGF-β1, FN, Col.I in high glucose-disposed HPCs and HK-2 cells. Collectively, these findings demonstrated that CDKN2B-AS1 regulated apoptosis and fibrosis of HPCs and HK-2 cells under high glucose treatment.

Effects of CDKN2B-AS1 on apoptosis and fibrosis of HPCs and HK-2 cells under high glucose treatment. A, B The expression of CDKN2B-AS1 in HPCs and HK-2 cells transfected with vector, CDKN2B-AS1, si-NC, or si-CDKN2B-AS1 under high glucose stimulation was analyzed with qRT-PCR. C–F Effects of CDKN2B-AS1 overexpression and inhibition on the proliferation and apoptosis of high glucose-disposed HPCs and HK-2 cells were determined through MTT or flow cytometry assays. G–J Effects of CDKN2B-AS1 overexpression and inhibition on protein levels of Bax, Bcl-2, TGF-β1, FN, and Col.I in high glucose-disposed HPCs and HK-2 cells were analyzed by western blot analysis.*P < 0.05
CDKN2B-AS1 was identified as a sponge for miR-98-5p
To explore the underlying molecular mechanism of CDKN2B-AS1 in DN, we predicted the miRNAs that might interact with CDKN2B-AS1 through using the starBase database. MiR-98-5p was discovered to possess a complementary sequence with CDKN2B-AS1 (Fig. 3A). Subsequently, we performed the dual-luciferase reporter assay to verify this prediction. The results exhibited that miR-98-5p mimic repressed the luciferase intensity in HPCs and HK-2 cells with a luciferase reporter containing WT-CDKN2B-AS1, while there was no overt difference in the luciferase reporter containing MUT-CDKN2B-AS1 (Fig. 3B, C). And miR-98-5p expression was decreased in HPCs and HK-2 cells after transfection with miR-98-5p inhibitor (Fig. 3D). Also, CDKN2B-AS1 silencing elevated miR-98-5p expression in HPCs and HK-2 cells, but this suppression was reversed by miR-98-5p downregulation (Fig. 3E, F). Moreover, miR-98-5p was downregulated in the serum of DN patients relative to the normal controls (Fig. 3G). There was a marked reduction in miR-98-5p expression in high glucose-treated HPCs and HK-2 cells (Fig. 3H). These results suggested that CDKN2B-AS1 acted as a miR-98-5p sponge.

CDKN2B-AS1 acted as a sponge for miR-98-5p. A The binding sites between CDKN2B-AS1 and miR-98-5p were predicted by the starBase database. B, C Dual-luciferase reporter assay was executed for the assessment of the luciferase reporter with WT-CDKN2B-AS1 or MUT-CDKN2B-AS1 in HPCs and HK-2 cells transfected with miR-98-5p mimic or miRNA NC. D The expression of miR-98-5p in HPCs and HK-2 cells transfected with miR-98-5p inhibitor or inhibitor NC was detected via qRT-PCR. E, F Impact of miR-98-5p inhibitor on the expression of miR-98-5p in CDKN2B-AS1-inhibiting HPCs and HK-2 cells was analyzed through qRT-PCR. G, H The expression of miR-98-5p in serum of DN patients and high glucose-treated HPCs and HK-2 cells was determined via qRT-PCR. *P < 0.05
CDKN2B-AS1 regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells by sponging miR-98-5p
Subsequently, we further explored whether CDKN2B-AS1 regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells by sponging miR-98-5p. MiR-98-5p downregulation impaired CDKN2B-AS1 downregulated-mediated effects on proliferation and apoptosis of high glucose-treated HPCs and HK-2 cells (Fig. 4A–D). Also, miR-98-5p inhibitor partially overturned the upregulation of Bcl-2 and the downregulation of Bax, TGF-β1, FN, and Col.I in high glucose-treated HPCs and HK-2 cells caused by CDKN2B-AS1 inhibition (Fig. 4E–H). Together, these results indicated that CDKN2B-AS1 regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells by acting as a miR-98-5p sponge.

CDKN2B-AS1 regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells by sponging miR-98-5p. A–H HPCs and HK-2 cells were transfected with si-NC, si-CDKN2B-AS1, si-CDKN2B-AS1 + inhibitor NC, or si-CDKN2B-AS1 + miR-98-5p inhibitor and then treated with high glucose. A–D The proliferation and apoptosis of high glucose-treated HPCs and HK-2 cells was detected by MTT and flow cytometry assays. E–H Protein levels of Bax, Bcl-2, TGF-β1, FN, and Col.I in high glucose-disposed HPCs and HK-2 cells were measured by western blot analysis. *P < 0.05
NOTCH2 was a downstream target of miR-98-5p
We further predicted the underlying targets of miR-98-5p with the starBase database. And the results presented that NOTCH2 had the complementary base fragment with miR-98-5p (Fig. 5A). The luciferase activity of luciferase reporter with WT-NOTCH2-3ʹUntranslated Regions (UTR) was decreased by miR-98-5p mimic in HPCs and HK-2 cells, while the luciferase intensity of luciferase reporter with MUT-NOTCH2-3ʹUTR did not change (Fig. 5B, C). And the protein levels of NOTCH2 in HPCs and HK-2 cells were markedly restrained after transfection with si-NOTCH2 compared to the control si-NC (Fig. 5D). Moreover, miR-98-5p inhibitor elevated NOTCH2 protein levels in HPCs and HK-2 cells, while this elevation was weakened by NOTCH2 silencing (Fig. 5E, F). Furthermore, NOTCH2 protein levels were also increased in the serum of DN patients and high glucose-treated HPCs and HK-2 cells (Fig. 5G, H). Also, the level of activated NOTCH2 protein was significantly increased under HG conditions (Additional file 1: Figure S1). Collectively, these results indicated that NOTCH2 served as a target of miR-98-5p.

NOTCH2 was a target of miR-98-5p. A The binding sites of NOTCH2 in miR-98-5p were predicted by the starBase database. B, C The luciferase intensity of a luciferase reporter with WT-NOTCH2-3ʹUTR or MUT-NOTCH21-3ʹUTR in HPCs and HK-2 cells transfected with miR-98-5p mimic or miRNA NC was determined through dual-luciferase reporter assay. D The protein level of NOTCH2 in HPCs and HK-2 cells transfected with si-NC or si-NOTCH2 was detected with western blot analysis. E, F Effect of NOTCH2 knockdown on the level of NOTCH2 protein in miR-98-5p-inhibiting HPCs and HK-2 cells was analyzed through western blot analysis. G, H The protein level of NOTCH2 in the serum of DN patients and high glucose-treated HPCs and HK-2 cells was assessed via western blot analysis
MiR-98-5p targeted NOTCH2 to regulate apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells
To determine whether miR-98-5p regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells through NOTCH2, HPCs and HK-2 cells were transfected with inhibitor NC, miR-98-5p inhibitor, miR-98-5p inhibitor + si-NC, and miR-98-5p inhibitor + si-NOTCH2. The results exhibited that miR-98-5p inhibitor repressed the proliferation of high glucose-treated HPCs and HK-2 cells, while this influence caused by miR-98-5p inhibitor were offset by NOTCH2 knockdown (Fig. 6A, B). In addition, miR-98-5p inhibitor promoted apoptosis, decreased protein levels of Bcl-2, and elevated protein levels of Bax in high glucose-disposed HPCs and HK-2 cells, but these impacts were overturned after NOTCH2 knockdown (Fig. 6C–F). Also, miR-98-5p inhibitor elevated TGF-β1, FN, and Col.I protein levels in high glucose-disposed HPCs and HK-2 cells, but these increases were restored by NOTCH2 silencing (Fig. 6G, H). Taken together, these findings revealed that miR-98-5p regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells through targeting NOTCH2.

MiR-98-5p regulated apoptosis and fibrosis of high glucose-treated HPCs and HK-2 cells by targeting NOTCH2. A–H HPCs and HK-2 cells were transfected with inhibitor NC, miR-98-5p inhibitor, miR-98-5p inhibitor + si-NC, or miR-98-5p inhibitor + si-NOTCH2 and then treated with high glucose. A–F The proliferation, apoptosis, and apoptosis-related proteins of HPCs and HK-2 cells under high glucose treatment were evaluated via MTT assay, flow cytometry assay, and western blotting. G, H Protein levels of TGF-β1, FN, and Col.I in HPCs and HK-2 cells under high glucose treatment were measured by western blot analysis. *P < 0.05
CDKN2B-AS1 regulated NOTCH2 expression through sponging miR-98-5p
Based on the above findings, we investigated whether CDKN2B-AS1 sponged miR-98-5p to regulate NOTCH2 expression. The results manifested that CDKN2B-AS1 silencing reduced NOTCH2 protein levels in HPCs and HK-2 cells under high glucose treatment, while this decrease was overturned by pc-NOTCH2 introduction (Fig. 7A). Furthermore, miR-98-5p inhibitor weakened the suppressive influence of CDKN2B-AS1 knockdown on NOTCH2 protein levels in HPCs and HK-2 cells under high glucose treatment (Fig. 7B). These results indicated that CDKN2B-AS1 modulated NOTCH2 expression via miR-98-5p.

CDKN2B-AS1 modulated NOTCH2 expression through miR-98-5p. A Effect of NOTCH2 overexpression on the level of NOTCH2 protein in CDKN2B-AS1-inhibiting HPCs and HK-2 cells under high glucose stimulation was assessed through western blot analysis. B Influence of miR-98-5p inhibitor on the level of NOTCH2 protein in CDKN2B-AS1-inhibiting HPCs and HK-2 cells under high glucose treatment was detected through western blot analysis. *P < 0.05
Discussion
Persistent proteinuria with or without decreased glomerular filtration rate has been used to define DN [26]. In this study, T2D patients with a urine albumin/creatinine ratio > 30 mg/g or estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 were defined to have DN. In addition, the inclusion of DN patients without renal biopsy was a major limitation of the study.
Podocytes, which constitute the glomerular filtration barrier, have limited regeneration and repair capabilities [27]. Renal tubular injury is an important manifestation of DN [28]. Studies have confirmed that the deregulation of lncRNAs is closely related to the progress of DN [8]. Report of Zhang et al. revealed that lncRNA MALAT1 was overexpressed in high glucose-treated HK-2 cells, resulting in accelerating cell epithelial-to-mesenchymal transition and injury [29]. Another report pointed out that lncRNA MALAT1 increased SIRT1 expression via targeting miR-9, thus alleviating podocyte damage via boosting cell viability and repressing cell apoptosis under high glucose treatment [30]. Lv et al. manifested that lncRNA GAS5 silencing mitigated high glucose-induced viability suppression and apoptosis acceleration of HK-2 cells via sponging miR-27a [28]. In the current study, CDKN2B-AS1 was upregulated in the serum of DN patients and high glucose-treated HPCs and HK-2 cells. Moreover, CDKN2B-AS1 silencing elevated cell viability and decreased cell apoptosis in HPCs and HK-2 cells under high glucose treatment. Thomas et al. demonstrated that CDKN2B-AS1 downregulation protected decreased urine albumin levels and urine volume in diabetic mice [31]. A recent research indicated that CDKN2B-AS1 knockdown inhibited extracellular matrix accumulation and proliferation of high glucose-treated HGMC cells through repressing HMGA2 expression by adsorbing miR-424-5p [16].
TGF-β1 is considered to be the main regulator of pro-fibrosis [32]. Increasing evidence has demonstrated that the TGF-β1 signaling exerts a vital role in DN pathogenesis [33,34,35,36]. Moreover, TGF-β1 can contribute to glomerular filtration disorder, fibrosis, and sclerosis [37]. Also, Sitagliptin can block the TGF-beta1/Smad pathway, thus ameliorating diabetic nephropathy [38]. Herein, CDKN2B-AS1 silencing decreased protein levels of TGF-β1, FN and Col.I in high glucose-disposed HPCs and HK-2 cells, indicating that CDKN2B-AS1 silencing decreased the fibrosis of HPCs and HK-2 under high glucose treatment. Thus, we concluded that high glucose-induced apoptosis and fibrosis of HPCs and HK-2 were partly dependent on CDKN2B-AS1.
LncRNAs usually exert their roles through acting as a sponge for miRNAs in DN [16, 28]. A previous study revealed that miR-98-5p repressed human endothelial cell growth through targeting cyclinD2 [39]. Another research reported that miR-98-5p mitigated renal fibrosis and epithelial-to-mesenchymal via modulating HMGA2 expression in DN [19]. Herein, miR-98-5p was downregulated in the serum of DN patients and high glucose-treated HPCs and HK-2 cells. CDKN2B-AS1 was validated as a sponge for miR-98-5p, and the impacts of CDKN2B-AS1 inhibition on proliferation, apoptosis, and fibrosis of high glucose-treated HPCs and HK-2 cells were overturned by miR-98-5p inhibitor. Thus, we concluded that CDKN2B-AS1 played its influence on high glucose-treated HPCs and HK-2 cells via sponging miR-98-5p.
Additionally, NOTCH2 was identified as a miR-98-5p target in the research. Also, NOTCH2 silencing abolished miR-98-5p inhibitor-mediated impacts on proliferation, apoptosis, and fibrosis of high glucose-treated HPCs and HK-2 cells. It was reported that miR-18a-5p targeted NOTCH2 in high glucose-induced HAVECs, thus impeding cardiac fibrosis and epithelial-to-mesenchymal [23]. Furthermore, De-Glycyrrhizinated Licorice Extract blocked the NOTCH2 pathway in high glucose-treated NRK-52E cells, thereby attenuating the epithelial-to-mesenchymal of NRK-52E cells [25]. Importantly, CDKN2B-AS1 regulated NOTCH2 expression via sponging miR-98-5p in the study. Therefore, we inferred that CDKN2B-AS1 silencing could relieve high glucose-induced apoptosis and fibrosis by regulating the miR-98-5p/NOTCH2 axis.
In conclusion, high glucose-mediated CDKN2B-AS1 elevated NOTCH2 expression via adsorbing miR-98-5p, leading to facilitating cell apoptosis and fibrosis in HPCs and HK-2 cells. The study offered a novel mechanism by which CDKN2B-AS1 participated in the pathogenesis of DN.
Availability of data and materials
The analyzed data sets generated during the present study are available from the corresponding author on reasonable request.
References
Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–40.
Sharma D, Bhattacharya P, Kalia K, Tiwari V. Diabetic nephropathy: new insights into established therapeutic paradigms and novel molecular targets. Diabetes Res Clin Pract. 2017;128:91–108.
Gregg EW, Sattar N, Ali MK. The changing face of diabetes complications. Lancet Diabetes Endocrinol. 2016;4(6):537–47.
Matoba K, Takeda Y, Nagai Y, Kawanami D, Utsunomiya K, Nishimura R. Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int J Mol Sci. 2019;20(14):3393.
Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10(6):925–33.
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21.
Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73(13):2491–509.
Li Y, Xu K, Xu K, Chen S, Cao Y, Zhan H. Roles of identified long noncoding rna in diabetic nephropathy. J Diabetes Res. 2019;2019:5383010.
Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339(2):159–66.
Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, Zhang Q, Yan G, Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41(Database issue):D983–6.
Xie C, Wu W, Tang A, Luo N, Tan Y. lncRNA GAS5/miR-452-5p reduces oxidative stress and pyroptosis of high-glucose-stimulated renal tubular cells. Diabetes Metab Syndr Obes. 2019;12:2609–17.
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JRB, Rayner NW, Freathy RM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336–41.
Huang K, Zhong J, Li Q, Zhang W, Chen Z, Zhou Y, Wu M, Zhong Z, Lu S, Zhang S. Effects of CDKN2B-AS1 polymorphisms on the susceptibility to coronary heart disease. Mol Genet Genomic Med. 2019;7(11):e955.
Li H, Han S, Sun Q, Yao Y, Li S, Yuan C, Zhang B, Jing B, Wu J, Song Y, et al. Long non-coding RNA CDKN2B-AS1 reduces inflammatory response and promotes cholesterol efflux in atherosclerosis by inhibiting ADAM10 expression. Aging (Albany NY). 2019;11(6):1695–715.
Huang Y, Xiang B, Liu Y, Wang Y, Kan H. LncRNA CDKN2B-AS1 promotes tumor growth and metastasis of human hepatocellular carcinoma by targeting let-7c-5p/NAP1L1 axis. Cancer Lett. 2018;437:56–66.
Li Y, Zheng L-L, Huang D-G, Cao H, Gao Y-H, Fan Z-C. LNCRNA CDKN2B-AS1 regulates mesangial cell proliferation and extracellular matrix accumulation via miR-424–5p/HMGA2 axis. Biomed Pharmacother. 2020;121:109622.
Khan R, Kadamkode V, Kesharwani D, Purkayastha S, Banerjee G, Datta M. Circulatory miR-98-5p levels are deregulated during diabetes and it inhibits proliferation and promotes apoptosis by targeting PPP1R15B in keratinocytes. RNA Biol. 2020;17(2):188–201.
Sun X, Li X, Ma S, Guo Y, Li Y. MicroRNA-98-5p ameliorates oxygen-glucose deprivation/reoxygenation (OGD/R)-induced neuronal injury by inhibiting Bach1 and promoting Nrf2/ARE signaling. Biochem Biophys Res Commun. 2018;507(1–4):114–21.
Zhu Y, Xu J, Liang W, Li J, Feng L, Zheng P, Ji T, Bai S. miR-98-5p Alleviated epithelial-to-mesenchymal transition and renal fibrosis via targeting Hmga2 in diabetic nephropathy. Int J Endocrinol. 2019;2019:4946181.
Li B, Zhu C, Dong L, Qin J, Xiang W, Davidson AJ, Feng S, Wang Y, Shen X, Weng C, et al. ADAM10 mediates ectopic proximal tubule development and renal fibrosis through Notch signalling. J Pathol. 2020;252(3):274–89.
Huang S, Park J, Qiu C, Chung KW, Li SY, Sirin Y, Han SH, Taylor V, Zimber-Strobl U, Susztak K. Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol. 2018;16(9):e2005233.
Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97(4):1235–94.
Geng H, Guan J. MiR-18a-5p inhibits endothelial-mesenchymal transition and cardiac fibrosis through the Notch2 pathway. Biochem Biophys Res Commun. 2017;491(2):329–36.
Tung CW, Hsu YC, Cai CJ, Shih YH, Wang CJ, Chang PJ, Lin CL. Trichostatin A ameliorates renal tubulointerstitial fibrosis through modulation of the JNK-dependent Notch-2 signaling pathway. Sci Rep. 2017;7(1):14495.
Hsu Y-C, Chang P-J, Tung C-W, Shih Y-H, Ni W-C, Li Y-C, Uto T, Shoyama Y, Ho C, Lin C-L. De-glycyrrhizinated licorice extract attenuates high glucose-stimulated renal tubular epithelial-mesenchymal transition via suppressing the Notch2 signaling pathway. Cells. 2020;9(1):125.
Bhalla V, Zhao B, Azar KM, Wang EJ, Choi S, Wong EC, Fortmann SP, Palaniappan LP. Racial/ethnic differences in the prevalence of proteinuric and nonproteinuric diabetic kidney disease. Diabetes Care. 2013;36(5):1215–21.
Zhou L, Liu Y. Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol. 2015;11(9):535–45.
Lv L, Li D, Tian F, Li X, Jing Z, Yu X. Silence of lncRNA GAS5 alleviates high glucose toxicity to human renal tubular epithelial HK-2 cells through regulation of miR-27a. Artif Cells Nanomed Biotechnol. 2019;47(1):2205–12.
Zhang J, Jiang T, Liang X, Shu S, Xiang X, Zhang W, Guo T, Xie W, Deng W, Tang X. lncRNA MALAT1 mediated high glucose-induced HK-2 cell epithelial-to-mesenchymal transition and injury. J Physiol Biochem. 2019;75(4):443–52.
Zhang Y, Chang B, Zhang J, Wu X. LncRNA SOX2OT alleviates the high glucose-induced podocytes injury through autophagy induction by the miR-9/SIRT1 axis. Exp Mol Pathol. 2019;110:104283.
Thomas AA, Feng B, Chakrabarti S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in diabetic complications. Am J Physiol Endocrinol Metab. 2018;314(3):E191–200.
Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–35.
Liu L, Wang Y, Yan R, Li S, Shi M, Xiao Y, Guo B. Oxymatrine inhibits renal tubular EMT induced by high glucose via upregulation of SnoN and inhibition of TGF-β1/Smad signaling pathway. PLoS ONE. 2016;11(3):e0151986.
Tang F, Hao Y, Zhang X, Qin J. Effect of echinacoside on kidney fibrosis by inhibition of TGF-β1/Smads signaling pathway in the db/db mice model of diabetic nephropathy. Drug Des Devel Ther. 2017;11:2813–26.
Hu W, Han Q, Zhao L, Wang L. Circular RNA circRNA_15698 aggravates the extracellular matrix of diabetic nephropathy mesangial cells via miR-185/TGF-β1. J Cell Physiol. 2019;234(2):1469–76.
Zhu QJ, Zhu M, Xu XX, Meng XM, Wu YG. Exosomes from high glucose-treated macrophages activate glomerular mesangial cells via TGF-β1/Smad3 pathway in vivo and in vitro. FASEB J. 2019;33(8):9279–90.
Chang AS, Hathaway CK, Smithies O, Kakoki M. Transforming growth factor-β1 and diabetic nephropathy. Am J Physiol Renal Physiol. 2016;310(8):F689–96.
Wang D, Zhang G, Chen X, Wei T, Liu C, Chen C, Gong Y, Wei Q. Sitagliptin ameliorates diabetic nephropathy by blocking TGF-β1/Smad signaling pathway. Int J Mol Med. 2018;41(5):2784–92.
Li X-X, Liu Y-M, Li Y-J, Xie N, Yan Y-F, Chi Y-L, Zhou L, Xie S-Y, Wang P-Y. High glucose concentration induces endothelial cell proliferation by regulating cyclin-D2-related miR-98. J Cell Mol Med. 2016;20(6):1159–69.
Acknowledgements
Not applicable.
Funding
No funding was received.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology: SB and JC; Formal analysis and data curation: YL, SZ and ZH; Validation and investigation: MX and SB; Writing—original draft preparation and writing—review and editing: MX, SB, JC and YL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present study was approved by the ethical review committee of Qilu Hospital, Cheeloo College of Medicine, Shandong University. Written informed consent was obtained from all enrolled patients.
Patient consent for publication
The results presented in this paper have not been published preciously in whole or in part.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Western blotting was executed to detection the protein levels of activated NOTCH2 in HPCs and HK-2 cells with normal glucose, osmotic treatment, and HG treatment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiao, M., Bai, S., Chen, J. et al. CDKN2B-AS1 participates in high glucose-induced apoptosis and fibrosis via NOTCH2 through functioning as a miR-98-5p decoy in human podocytes and renal tubular cells. Diabetol Metab Syndr 13, 107 (2021). https://doi.org/10.1186/s13098-021-00725-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13098-021-00725-5