
Abstract
Colorectal cancer (CRC) is a common malignant tumor with the third and second highest incidence and mortality rates among various malignant tumors. Despite significant advancements in the present therapy for CRC, the majority of CRC cases feature proficient mismatch repair/microsatellite stability and have no response to immunotherapy. Therefore, the search for new treatment options holds immense importance in the diagnosis and treatment of CRC. In recent years, clinical research on immunotherapy combined with epigenetic therapy has gradually increased, which may bring hope for these patients. This review explores the role of epigenetic regulation in exerting antitumor effects through its action on immune cell function and highlights the potential of certain epigenetic genes that can be used as markers of immunotherapy to predict therapeutic efficacy. We also discuss the application of epigenetic drug sensitization immunotherapy to develop new treatment options combining epigenetic therapy and immunotherapy.
Similar content being viewed by others

Introduction
By 2040, it is estimated that there will be 3.2 million cases of CRC, making it the third most prevalent cancer and the second leading cause of death among cancers [1]. In recent years, immunotherapy has achieved remarkable accomplishments. Over the past few years, immunotherapy has achieved remarkable accomplishments in the treatment of CRC with dMMR/MSI-H, such as Checkmate142 and Keynote177 [2, 3]. The FDA has also approved nivolumab (a PD-1 antibody) for treating dMMR/MSI-H CRC. Nevertheless, most CRC (approximately 85%) features proficient mismatch repair (pMMR)/microsatellite stability (MSS) tumors, which do not exhibit any response to immune checkpoint inhibitors (ICIs) [4]. Several clinical trials suggest that the combined regimen with ICIs may be promising for these patients. In addition, it is vital to differentiate between those who can benefit from immunotherapy and those who do not respond, and biomarkers are a key factor in this process. Biomarkers have been used to guide CRC immunotherapy, such as MSS status, MMR expression, PD-L1 expression, POLD1/POLE mutations and tumor mutation burden (TMB) [5, 6]. However, these markers can only screen out a small number of applicable populations. Hence, there is an urgent need to carry out new combination therapies and establish new biomarkers to enhance the effectiveness of immunotherapy in CRC.
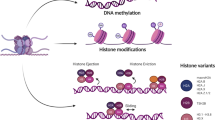
The development of various cancers, including CRC, is significantly influenced by epigenetics. Epigenetics involves modifications in gene function that do not change the DNA sequence but ultimately result in phenotypic changes [7, 8]. Epigenetic modifications includes DNA methylation, histone modifications (such as histone methylation, acetylation and lactylation), chromatin remodeling and epitranscriptomic modifications (such as N6-methyladenosine modification, m6A). These modifications primarily rely on the coordinated action of "writer," "reader," and "eraser" enzymes (Additional file 1: Table S1), which add, recognize and remove chemical modifications on DNA, histones or RNA, enabling cells to dynamically regulate gene expression [9, 10]. For instance, TET1 (DNA demethylase) participates in DNA demethylation to activate silenced genes. Conversely, EZH2 is part of the Polycomb repressive complex 2 (PRC2), which mediates trimethylation of lysine 27 on histone H3 (H3K27me3) to suppress transcription of certain genes. KMT2D (histone methyltransferase) modifies histone H3 lysine 4 (H3K4) to promote gene activation. ARID1A is a component of the SWI/SNF chromatin remodeling complex, regulating gene expression by altering chromatin structure. Additionally, histone lactylation, as a novel histone modification, primarily involves adding lactyl groups to specific histone lysine residues, thereby modulating gene expression and cellular metabolism [11, 12]. These epigenetic regulatory mechanisms and their key enzymes are crucial in the development of tumors, immune evasion and drug resistance (Fig. 1).

Epigenetic modifications
In this review, we focus on the connection between epigenetic regulation (DNA and histone modifications) and the tumor microenvironment in CRC patients and emphasize the application of epigenetic drug-sensitizing immunotherapy. We also discuss some epigenetic genes as immunotherapeutic markers to guide better immunotherapy in CRC patients. Finally, we summarize the clinical trials of epi-immunotherapy that have been reported or are ongoing in CRC. Through this review, we hope clinicians or researchers will pay attention to the great potential of epigenetic mechanisms in enhancing the effectiveness of immunotherapy in CRC and provide new ideas for immunotherapy for CRC.
The full names of the proteins involved in the epigenetic modifications are provided in Additional file 1.
Epigenetic regulation of the tumor immune microenvironment
Immune checkpoint
Immune checkpoint expression can be hijacked by tumor cells and myeloid cells, leading to the inhibition of the antitumor immune response of T cells. Immune checkpoints include PD-1, CTLA-4, LAG3, TIM3, TIGIT and BTLA [13]. The regulation of immune checkpoints and their receptors is significantly influenced by epigenetic mechanisms. High expression of JMJD2D in CRC may indirectly suppress the functional capabilities of CD8 + T cells that infiltrate the tumor by enhancing the expression of PD-L1, thereby promoting the occurrence of CRC. The results show a positive correlation between the expression of PD-L1 and JMJD2D expression [14]. Studies have shown that ARID3B also enhances phosphorylation at STAT3 Y705, leading to the upregulation of PD-L1 expression, which promotes immune escape [15]. Moreover, a study showed that the upregulation of CTLA-4 was associated with DNA hypomethylation, as well as the involvement of H3K9me3 and H3K27me3 [16]. Additionally, multiple histone modifications contribute to the upregulation of PD-1 and TIM-3 genes in CRC tumor tissue. Epigenetic alterations are capable of having the potential to function as diagnostic biomarkers for CRC. Blocking immune checkpoints reactivates T-cell functions, enhancing sustained antitumor effects and strengthening the host's immune response to cancer [17]. Therefore, in-depth study of epigenetic regulatory mechanisms contributes to enhancing immune checkpoint inhibitor effectiveness.
Interferon-γ pathway (IFN-γ)
Epigenetic regulation is closely linked to the IFN-γ pathway, which has an important impact on tumor immunity and immunotherapy. In CRC, the IFN-γ pathway is regulated by Polycomb repressive complex 2 (PRC2) and switch/sucrose non-fermentable (SWI/SNF) complexes [18]. This regulation happens in part through the control of chemokines, which help effector T-cell recruitment to the tumor microenvironment. In tumors, EZH2, a component of PRC2, enhances the trimethylation of histone H3 lysine 27 and inhibits the production of chemokines [19, 20]. In contrast, ARID1A (an SWI/SNF complex) contributes to the expression of chemokines in CRC cells, thereby enhancing immune cell recruitment [21]. In CD8 + T cells, DNA methylation was also found to negatively regulate IFN-γ expression. Nevertheless, continuous IFN-γ signaling boosts the programmed PD-L1 checkpoint pathway to suppress the antitumor immune response. Therefore, epigenetic regulation of the interferon response may play a diametrically opposite role at different stages of tumor progression.
T-Cell exhaustion
Throughout the process of T-cell exhaustion, CD8 + T cells gradually decrease the generation of effector cytokines while simultaneously enhancing the expression of their inhibitory receptors like PD-1, Tim-3 and Large-3 [21]. ARID1A loss leads to a decrease in the expression of exhaustion-related genes in tumor-infiltrating T cells [22]. De novo DNA methylation by DNMT3A has been demonstrated to play a role in promoting T-cell exhaustion [23]. Thus, the renewal of exhausted CD8 + T cells is improved by using 5-aza-2-deoxycytidine (DNA methyltransferase inhibitor) before PD-1 blockade.
Research has indicated that the absence of H3K79me2 leads to reduced levels of STAT5 expression and compromised immune response in T cells. Furthermore, the addition of methionine enhances H3K79me2 and STAT5 expression in T cells, resulting in heightened T-cell immunity in patients with CRC [24]. An additional study has also indicated that TET2 (demethylase) loss in CAR T cells increases the longevity and tumor regulation of CAR T cells, which suggests that disrupting TET2 could prevent T-cell exhaustion and promote the formation of T-cell memory [25].
Myeloid cells
Tumor-associated macrophages (TAMs) are the main immune cells in the context of CRC. Epigenetics affect the production, recruitment and exhaustion of TAMs and reprogram TAMs to promote or inhibit tumors. Studies have shown that in a coculture system of macrophages and CRC cells, the use of EZH2i (an inhibitor of EZH2) suppresses the levels of H3K27me3 on the promoters of STAT3, which is a crucial transcription factor responsible for M1 macrophage polarization. The tumor suppressive effect of EZH2i is achieved by transforming M2 macrophages into effector M1 macrophages, thereby regulating macrophages [26]. Researchers found that histone lactylation promotes immune evasion by regulating the immunosuppressive effects of myeloid cells that infiltrate tumors. The function of antitumor T cells is also inhibited by myeloid-derived suppressor cells (MDSCs). Studies have shown that HDAC11 is a key regulator of IL-10 gene expression in MDSCs, which suggests that histone modifications also contribute to regulating various aspects of MDSCs [27, 28].
Treg cells
Treg inhibitory activity requires the expression of FoxP3. The regulation of FoxP3 expression in T cells is controlled by DNA methylation of the CpG island located in the enhancer region. Additionally, researchers have found that the transcriptional program and epigenetic characteristics of Treg cells are diminished when TET2 and TET3 are lost. Conversely, vitamin C (TET activator) alters the epigenetic landscape to closely resemble Treg cells produced in vivo and enhances the expression of Treg signature genes [29, 30]. EZH2 activity plays a key role in the suppression of antitumor immunity by Treg cells. Furthermore, using drugs to inhibit EZH2 resulted in enhanced CD8 + T-cell responses inside tumors, which hindered tumor advancement and correlated with decreased FOXP3 expression in Tregs. Thus, inhibiting EZH2 activity remolds the TME and leads to enhanced tumor immunity [31, 32] (Fig. 2).

Epigenetic regulation shapes the role of the immune microenvironment in CRC
Epigenetic alterations facilitate cancer development by influencing multiple pathways in a wide range of histologies and by affecting the activation, differentiation and function of immune cells. Gal-9: Galectin-9, Tim-3: T cell immunoglobulin and mucin domain-containing protein 3, PD-L1: Programmed death-ligand 1, PD-1: Programmed cell death protein 1, Tn: naive T cells, Tpex: exhausted T cells, Tex: exhausted T cells, IFN-γ: Interferon-γ pathway, MHC-1: Major Histocompatibility Complex class I, CXCL-9/10: C-X-C motif chemokine ligand 9/10, MDSC: Myeloid-derived suppressor cells, Treg: Regulatory T cells.
Targeted epigenetic immunoregulation sensitized immunotherapy
Epigenetic immune drugs (e.g., DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis)) can inhibit the renewal and proliferation of tumor stem cells while promoting the reactivation of tumor suppressor genes, thus leading to cycle arrest or apoptosis [33,34,35,36,37,38,39]. These drugs also activate endogenous retroviruses (ERVs) and cancer testicular antigen (CTA), thus activating the virus analog state [40, 41] and increasing immunogenicity [42, 43]. DMNTi inhibits T-cell exhaustion and increases early memory T cells. HDACis inhibit Treg cells, promote CD4 + T-cell effects [44] and regulate the tumor microenvironment [45, 46]. Apparently, epigenetic immune drugs combined with immune checkpoint inhibitors will enhance T-cell infiltration, suppress MDSCs and M2-type macrophages [44], and reduce TME-mediated immune escape. This suggests that targeted epigenetic immunoregulation will sensitize cells to immunotherapy.
DNA methylation
Epigenetic modification DNA methylation plays a key role in regulating gene expression, including DNA methylation and DNA demethylation. The DNA methylase family consists of five members, namely DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L [47, 48]. DNA demethylases mainly refer to the 10–11 translocation protein (TET) family, including TET1, TET2 and TET3.
According to studies, DNA methylation is associated with the tumor microenvironment. DNMT1 suppresses chemokines expression, such as CXCL-9/10, which are essential for CD8 + T-cell migration [19]. Therefore, targeting DNMT1 sensitizes cells to immunotherapy and antitumor activities. GHONEIM et al. [23] discovered that exhaustive formation of T-cell precursors was promoted by de novo DNA methylation mediated by DNMT3A. However, CD8 + T-cell exhaustion was improved after decitabine (DNMTi) treatment. Decitabine combined with PD-1 decreases tumor burden and alters transcription factor expression. Furthermore, the methylation of DNA has an impact on genes related to the cell cycle, thereby enabling cancer cells in their early stages to avoid checkpoint blockade and senescence [49]. TET2 has the ability to facilitate the IFN-γ-JAK-STAT signaling pathway, subsequently regulating chemokines and PD-L1 expression, enhancing lymphocyte infiltration and improving the antitumor immune response [50]. However, when TET2 is absent, it promotes tumor immune escape and produces resistance to anti-PD-L1 treatment. Two studies [41] showed that azacytidine (DNMTi) could induce ERV expression and transcription of LTR and activate the antiviral-related interferon signaling pathway, which could induce cell cycle arrest and apoptosis. Similarly, decitabine (DNMTi) could also upregulate MHC-I molecular expression by reversing DNA methylation, thus enhancing the immunogenicity of tumors. Huang et al. [51] found that decitabine could also enhance immune-related gene expression and T cells infiltration in CRC. Moreover, they observed a significant improvement in the effectiveness of PD-L1 inhibitors for immunotherapy. Furthermore, using the cofactor TET2 can improve TET2 activity, upregulate chemokine expression and increase TIL quantity, thus enhancing the immune response against tumors and enhancing the effectiveness of immunotherapy [52].
Histone methylation
Histone methylation is an important mode in which histone modifications regulate chromatin structure and gene expression. Histone methylation involves two types: protein arginine methyltransferase (PRMT) and histone lysine methyltransferase (HKMT). Histone demethylases are broadly divided into the LSD family and the JMJD family [53].
As a histone lysine methyltransferase, EZH2 contributes to the proliferation of CRC cells and regulates various pathways related to CRC. Many immune cells express EZH2 [54]. EZH2 inhibits Th1 and Th2 differentiation, while EZH2 functions as a coactivator of BCL6 transcription, thereby promoting T follicular helper cell (Tfh) differentiation. EZH2 assists in maintaining the stable functional phenotype of activated Tregs, promotes CD8 + T memory cell generation and silences the differentiation and activity of CD8 + T effector cells [55]. EZH2 enhances tumor cell viability, decreases tumor antigen expression, reduces the release of chemokines by recruited T cells and reduces NKG2D expression, thus generating tolerance to immune cells (Fig. 2). In some studies, EZH2i has been found to potentially revive the immunogenicity of certain tumors and enhance the reaction to ICIs [56]. LSD1, in the histone demethylase family, is the first histone lysine demethylase. Studies have shown that the loss of the H3K4 demethylase LSD1 or KDM5B could enhance T-cell infiltration and PD-1 inhibitor efficacy. Furthermore, LSD1 deficiency also enhances the immunogenicity of tumors and T-cell infiltration in low-immunogenicity tumors [57, 58]. In CRC cells, knockdown of JMJD2D inhibited the Hedgehog signaling pathway, which attenuated CRC cell growth and metastasis [59]. The regression of human tumors has been demonstrated with the use of EZH2i. Research has indicated that EZH2i has the potential to diminish the impact of Tregs within the TME, enhance the manifestation and introduction of antigens, and stimulate the secretion of chemokines (such as CXCL 9 and CXCL10) by tumor-infiltrating DCs [56, 60]. Currently, tazemetostat is approved for use in follicular lymphoma and metastatic or refractory epithelioid sarcoma (Fig. 3).

EZH2i reshapes the TME to enhance the antitumor immune effect
EZH2i: EZH2 inhibitor, CXCL-9/10: C-X-C motif chemokine ligand 9/10, Teff cell: Effector T cells, Treg: Regulatory T cells, MDSC: Myeloid-derived suppressor cells, NK cell: Natural Killer cells.
Histone acetylation
The regulation of histone acetylation involves histone acetyltransferases (HATs) and histone deacetylases (HDACs) [61]. Through reversible mechanisms, these two enzymes modify the lysine side chains of histones in chromatin to control gene expression.
There are three general categories of HATs: GNAT, MYST and p300/CBP subfamilies, which regulate basic cell biological processes. Aberrant HATs are closely related to cancer. P300-mediated acetylation of lysine (K) at position 240 increases the stability of TRIB3 which may reduce T-cell infiltration into tumor tissue by inhibiting signal transduction in CRC and promoting immune rejection [62]. Tumors often exhibit high levels of HDAC expression, which is typically linked to reduced acetylation of histones in chromatin, an inactive chromatin structure for transcription and a decline in gene expression [63, 64]. HDAC inhibitors induce antitumor effects via various mechanisms, including triggering cell cycle arrest and apoptosis, regulating cell autophagy, inhibiting tumor blood vessel formation and regulating the immune system. Kim et al. [65] showed that HDAC is promoted IL-2 and IFN-γ expression in tumor tissues and increased the infiltration of cytotoxic T cells and NK cells while concurrently suppressing the proliferation of Treg cells and M2 macrophage polarization and reducing MDSCs. CG-745 (HDACis) can also inhibit Treg proliferation and regulate the TME, enhancing the activity [44] of PD-1 therapy. Through polarization, TMP195 (an HDACi) increases the proportion of M1 macrophages. Furthermore, TMP195 also enhanced the effects of PD-1 inhibitors PD-1. Therefore, combining TMP195 with PD-1 inhibitors may offer a potential approach for treating CRC [66].
Histone lactylation
Lactylation, as a new histone modification, has been reported. In 2020, researchers [67] described a different biological activity of lactate in tumors compared to normal tissue, which contributes to the establishment of specific immune states. Cancer cells secrete lactate into the surrounding environment, which facilitates the advancement of cancer. Research shows that the buildup of lactate in metabolism can serve as a substrate for inducing lactylation modification of histone lysine, which regulates gene expression and participates in the homeostatic regulation of M1 macrophages [68]. Studies have also revealed that alteration controls the immunosuppressive role of tumor-infiltrating myeloid cells (TIMs) to facilitate immune evasion from tumors [69]. Mechanistically, lactate promotes the expression of METTL3 in TIMs through histone H3K18 lactylation. METTL3, as an m6A methyltransferase, plays a crucial role in epigenetic regulation by affecting mRNA stability, splicing, nuclear export, translation and degradation through m6A modification. Specifically, METTL3-mediated m6A modification of Jak1 mRNA interacts with YTHDF1. YTHDF1, as an m6A "reader" protein, recognizes and binds to m6A-modified mRNA, increasing its translation efficiency and consequently enhancing the activation of the JAK1-STAT3 signaling pathway. The activation of the JAK1-STAT3 signaling pathway further initiates the expression of downstream immunosuppressive molecules, thereby resulting in immunosuppression [70, 71] (Table 1).
Epigenetic genes serve as potential biomarkers of immunotherapy
Epigenetic alterations in tumor cells can affect the immune response. First, the epigenetic histone modifications of the PRC2 and SWI/SNF complexes affect the tumor immune response through regulation of IFN-γ signaling [18, 19]. In addition, defective expression of some epigenetic genes will unsilence ERV expression, activate the IFN-γ pathway, increase T-cell infiltration and sensitize cells to immunotherapy [18, 41]. Furthermore, some epigenetic regulatory genes, such as ARID1A and KMT2D, maintain genomic stability. However, when these genes are absent, the genomic stability of tumor cells will decrease. Meanwhile, the tumor mutational load or tumor neoantigen load will increase, thus activating the antitumor immune response (Fig. 4).

Epigenes influence the immune response through endogenous interferon response pathways, immunogenicity and genome stability
IFN-γ: Interferon-γ pathway, Interferon alpha/beta receptor 1/2: Interferon alpha/beta receptor 1/2, JAK-1: Janus kinase 1, JAK-2: Janus kinase 2, STAT1: Signal Transducer and Activator of Transcription 1, CXCL-9/10: C-X-C motif chemokine ligand 9/10, MHC-1: Major Histocompatibility Complex class I, ERV: Endogenous retroviruses, TLRs: Toll-like receptors, dMMR: deficient mismatch repair.
DNA methylation status
DNA methylation biomarkers hold significant potential in early screening, prognosis and predicting treatment responses in colorectal cancer. Currently, early diagnosis of colorectal cancer involves detecting abnormal tumor DNA methylation in both blood and stool samples [72]. The US Food and Drug Administration (FDA) has approved a fecal-based screening test for colorectal cancer, including detection of KRAS gene mutations as well as NDRG4 and BMP3 gene methylation [73]. Additionally, the methylation biomarker SEPT9 in plasma has demonstrated high sensitivity and specificity for early-stage colorectal cancer [74, 75]. Methylation of the VIM (Vimentin) gene in stool can also serve as a screening method [76]. DNA methylation patterns can further predict prognosis in colorectal cancer patients. For instance, the high methylation of the SEPT9 and SFRP2 genes is associated with poorer prognosis [61]. DNA methylation also serves as a predictive biomarker for treatment responses. The methylation status of the MGMT gene can predict how patients respond to treatment with temozolomide [77]. CpG island methylation phenotype (CIMP) can also predict responses to certain chemotherapy drugs such as 5-fluorouracil (5-FU) [78].
The methylation status of DNA in tumor cells or immune cells may become a marker for early screening and efficacy prediction in CRC. Recently, studies have reported DNA methylation markers related to CRC diagnosis, such as the methylation levels of cg13096360 and cg12993163 in feces and blood and the combination of both can be used as diagnostic markers for CRC [79]. Researchers then expanded the sample size to validate the finding that it was also progressively upregulated in colorectal cancers of different stages.
In addition, the DNA methylation-derived signature of CD8 + TILs could function as a marker of the immune response and prognosis in CRC. By analyzing DNA methylation patterns across the entire genome in immune cells [80] and colonic epithelial cells, the researcher identified specific sites (DMPs) with differential methylation in CD8 + T cells and found that most of the genes were enriched for immune-related functions. The authors used these DMPs to construct a CD8 + MeTIL assessment system to assess the correlation between CD8 + TIL and survival outcomes in CRC. In addition, MSI-H tumors were found to have a correlation with low CD8 + MeTIL scores (enriched CD8 + tumor-infiltrating lymphocytes), which in turn were predictive of improved survival among those patients. A previous study published in the Journal Frontiers in Immunology explored the development of a machine learning model to generate forecasts on the effectiveness of immunotherapy by analyzing the methylation profiles of tumor DNA. The authors also used a screened ensemble of methylation features to predict pancancer immunotherapy response, which was validated in an independent immunotherapy cohort [81]. All these findings indicate that DNA methylation could become a potential biomarker for diagnosing CRC and evaluating the effectiveness of immunotherapy.
ARID1A
ARID1A mutations were shown to be significantly and positively associated with mutations in the MSI and MMR genes in three GI cancer cohorts [82,83,84,85]. This finding confirms that the absence of ARID1A is linked to the genomic characterization of MSI and impaired MMR in cancer. In addition, the combined data from clinical studies of multiple CRC patients concluded that ARID1A mutant tumors are characterized by higher genomic instability such as high MSI or TMB, heightened PD-L1 expression and increased cytotoxic T lymphocyte infiltration [86]. To confirm the association between ARID1A and immunological characteristics, the TCGA and MD Anderson Cancer Center databases were utilized for DNA sequencing and gene expression analysis of CRC patients. The findings indicated that the MSS and ARID1A mutation cases exhibited the highest rise in the frameshift mutation rate. Compared to ARID1A wild type, the mutant phenotype exhibited statistically significant expression of important checkpoint genes (such as PD-L1 and CTLA4) and gene sets (including antigen presentation, cytotoxic T-cell function and immune checkpoint). Increasing levels of neoantigens in ARID1A mutant cases may contribute to their immunogenicity, which is possibly caused by elevated TMB and code-shift mutations. In the future, there may be an increased likelihood of immunotherapy benefiting patients with ARID1A mutations. Additionally, this suggests that mutations in ARID1A could become a promising biomarker for the application of immunotherapy.
KMT2 family
In tumor cells, the KMT2 family includes KMT2A, KMT2B, KMT2C and KMT2D [87, 88]. The researchers discovered that KMT2 mutant tumors exhibited elevated levels of TMB when comparing the tumor immunogenicity between KMT2 mutant and wild-type samples. KMT2 gene family mutations can improve tumor immunogenicity [89]. By using CRISPR-GEMMs in ICB settings, researchers discovered that KMT2D plays a key role in regulating the ICB response in various types of cancer. They also found that tumors with KMT2 mutations exhibit heightened immune infiltration in humans. Our team discovered that KMT2C/D loss-of-function variants are abundant in many cancer types and found a significant correlation between the abundance of KMT2C/D loss-of-function variants and elevated TMB levels. Notably, KMT2C/D loss-of-function mutations were linked to increased levels of PD-L1 expression and TIL in CRC. Significantly, there was a correlation of statistical significance between KMT2C/D loss-of-function mutations and the response to ICIs in CRC [90]. All these studies suggest that KMT2 mutation may also be a potential biomarker for immunotherapy.
TET1/2
Three homologous proteins were identified in the TET enzyme family, named TET1, TET2 and TET3. Researchers found that TET1 mutation was prevalent in tumors and correlated with clinical response to ICI treatment [91]. By comparing the tumor immunogenicity and antitumor immunity of TET1 mutation type with TET1 wild-type tumors, it was found that both TET1 mutation-type tumors were significantly higher than TET1 wild-type tumors. Further immune signature analysis showed that coinhibition and costimulators of antigen-presenting cells and T cells were also significantly elevated in TET1 mutation-type tumors. Additionally, we compared the distinct expression patterns of immune-related genes in tumors with TET1 mutation-type and TET1 wild-type tumors. The results showed that TET1 mutation-type tumors commonly exhibited elevated levels of various immune stimulators, including chemokines (CXCL9, CXCL10, CCL5). These results suggest that patients treated with ICIs have a better clinical response and prognosis when they have TET1 mutation type. Another study found that knocking down TET2 in colon tumor cells decreased chemokine expression and the TIL count, enhancing tumor immune escape [52]. This suggests that TET1/2 holds the capacity to become a biomarker for forecasting the effectiveness of immunotherapy intervention.
DNMT3A/B
Previous studies have shown that enhancement or upregulation of DNMT3A and DNMT3B can mediate aberrant DNA methylation, which is strongly linked to the development and advancement of numerous malignancies in humans. DNMT3A/B can also be used as potential markers of immunotherapy efficacy. One of the advanced melanoma patients with a DNMT3A mutation developed hyperprogression when treated with immunotherapy, which was 182% compared to the first 2 months of immunotherapy.
Clinical study
Epigenetics plays a key role in regulating the immune response in CRC. Currently, many clinical studies on epigenetic combination immunotherapy are currently underway. The MAYA study [92], a phase II trial conducted at multiple centers, used a single-arm design to assess the effectiveness and safety of immunosensitization approaches. Using temozolomide commenced in eligible patients and was subsequently followed by a combination of low-dose ipilimumab and nivolumab. Patients reaching stage 2 were evaluated for the 8-m PFS rate, which served as the main outcome measure in the study. The study was conducted to assess the effectiveness and security of the immunosensitization strategy. At the end of the study, a total of 12 patients (36%) with > 8 m progression-free survival (PFS) had reached stage 2. The results showed that temozolomide with low-dose ipilimumab and nivolumab induced immunosensitization in pMMR/MSS and MGMT-silenced metastatic colorectal cancer patients. According to another study, the use of epigenetic modulators in MSS CRC enhances responses to pembrolizumab (MK-3475) [93]. This research is a phase 1b trial conducted at a single institution with an open-label design. Enhancing the susceptibility of MSS colorectal tumors to MK-3475 can be achieved by administering an epigenetic agent for either 14 or 21 days. This study enrolled 27 patients. The median progression-free survival (mPFS) and overall survival (OS) were 2.79 months and 9.17 months, respectively. One individual achieved a durable partial response, which lasted for approximately 19 months. The study findings show that the use of 5-azacitidine and romidepsin, in addition to pembrolizumab, demonstrated a favorable safety profile and was well tolerated among the participants. The CAPability-01 study, a randomized, open-label, multicenter, two-arm, phase II study, explored the efficacy and safety of chidamide with sintilimab with or without bevacizumab. The study included a total of 48 patients who had MSS/pMMR. Both groups achieved a 42.6% progression-free survival (PFS) rate at week 18 after treatment, which was the primary endpoint of the study. The findings indicated that the effectiveness of this combination regimen was observed in patients with advanced CRC with pMMR/MSS. Multiple clinical studies of epigenetic drug combination immunotherapies are currently underway, and these studies show promise for population-screened epigenetic combination immunotherapies to achieve desirable results, even in MSS CRC (Table 2).
Conclusions
The immune microenvironment of CRC is significantly influenced by epigenetic regulatory mechanisms. The mutations of some epigenetic genes also reflect the state of the immune response. For example, ARID1A mutations result in an upregulation of PD-L1 expression levels to induce stronger immunogenicity. Therefore, epigenetic gene status can be used as a biomarker for immunotherapy. Currently, epigenetic therapy focuses on two major classes of drugs: HDACis and DNMTis. Studies have found that DNMTis and HDACis remodel the tumor microenvironment, both of which significantly improve the effectiveness of PD-L1 inhibitor immunotherapy in preclinical research [94, 95]. The results suggest that the use of epigenetic drugs in conjunction with immune checkpoint inhibitors holds significant promise in the management of patients with CRC. However, at present, we still lack large-scale clinical cohorts or clinical trials to confirm the synergistic therapeutic efficacy of epigenetic combination immunization. Therefore, we need to conduct more studies in the future.
Perspectives
Epigenetic immunotherapy (epi-immunotherapy) has emerged as a promising treatment strategy. This combined approach enhances the effectiveness of immunotherapy, provides durable antitumor memory and significantly improves prognosis for cancer patients. The CAPability-01 study has successfully explored the treatment of mCRC patients using a combination of chidamide, pembrolizumab ± bevacizumab, offering a hopeful new option. SMARCA4-deficient tumors are highly aggressive with poor prognosis. The combination of HDAC inhibitors (such as vorinostat or romidepsin), DNA methyltransferase inhibitors (such as azacitidine) and PD-1 inhibitors is expected to benefit patients with SMARCA4 deficiency [96, 97]. Currently, guiding colorectal cancer immunotherapy based on DNA methylation and the mutation status of epigenetic genes remains in the academic research stage and has not yet been widely adopted in clinical practice. Future efforts will focus on further clinical validation and prospective studies of these biomarkers to ensure their predictive value and clinical utility. Understanding precision medicine will be key to exploring future combination therapies. For patients with epigenetic gene defects, further investigation into specific gene defect types and potential synergistic lethal mechanisms will be crucial for developing personalized treatment plans. For instance, mutations or loss of ARID1A can affect metabolic vulnerability, immune evasion and DNA repair pathways, presenting opportunities for developing novel combination treatment strategies [98,99,100]. Epi-immunotherapy can significantly enhance treatment outcomes for colorectal cancer patients, thereby improving long-term prognosis. This combined approach aims to improve the current landscape of immune therapy for colorectal cancer and paves the way for new avenues in its treatment.
Availability of data and materials
Not applicable.
Abbreviations
- CRC:
-
Colorectal cancer
- mCRC:
-
metastatic colorectal cancer
- pMMR:
-
Proficient mismatch repair
- dMMR:
-
Defective mismatch repair
- MSI-H:
-
Microsatellite instability-high
- MSS:
-
Microsatellite stability
- ICIs:
-
Immune checkpoint inhibitors
- TMB:
-
Tumor mutation burden
- IFN-γ:
-
Interferon-γ pathway
- PRC2:
-
Polycomb repressive complex 2
- SWI/SNF:
-
SWItch/Sucrose Non-Fermentable
- TAMs:
-
Tumor-associated macrophages
- MDSCs:
-
Myeloid-derived suppressor cells
- ERV:
-
Endogenous retroviruses
- CTA:
-
Cancer testicular antigen
- PRMT:
-
Protein arginine methyltransferase
- HKMT:
-
Histone lysine methyltransferase
- Tfh:
-
T follicular helper cell
- HATs:
-
Histone acetyltransferases
- HDACs:
-
Histone deacetylases
- TIM:
-
Tumor-infiltrating myeloid cells
- PFS:
-
Progression-free survival
- mPFS:
-
Median progression-free survival
- OS:
-
Overall survival
- DNMTi:
-
DNA methylation inhibitors
- HDACi:
-
Histone deacetylase inhibitors
- VIM:
-
Vimentin
- CIMP:
-
CpG island methylator phenotype
- 5-FU:
-
5-Fluorouracil
- FDA:
-
Food and drug administration
- DNMTs:
-
DNA methyltransferases
- TET1-3:
-
Ten-eleven translocation enzymes 1-3
- SMARCA2/4:
-
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2/4
- ARID1A:
-
AT-rich interactive domain-containing protein 1A
- PBRM1:
-
Polybromo 1
- SMARCB1:
-
SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1
- ATRX:
-
Alpha thalassemia/mental retardation syndrome X-linked
- ISWI:
-
Imitation switch
- RSF:
-
Remodeling and spacing factor
- NuRD:
-
Nucleosome remodeling and deacetylase complex
- CHD1-9:
-
Chromodomain-helicase-DNA-binding protein 1-9
- INO80:
-
INOsitol requiring 80
- Ac:
-
Acetylation
- Me:
-
Methylation
- Ub:
-
Ubiquitination
- P:
-
Phosphorylation
- Lac:
-
Lactylation
- Gal-9:
-
Galectin-9
- Tim-3:
-
T cell immunoglobulin and mucin domain-containing protein 3
- PD-L1:
-
Programmed death-ligand 1
- PD-1:
-
Programmed cell death protein 1
- Tn:
-
Naive T cells
- Tpex:
-
T cell precursor exhausted
- Tex:
-
T cell exhaustion
- MHC-1:
-
Major Histocompatibility Complex class I
- CXCL-9/10:
-
C-X-C motif chemokine ligand 9/10
- Treg:
-
Regulatory T cells
- EZH2i:
-
EZH2 inhibitor
- Teff cell:
-
Effector T cells
- NK cell:
-
Natural Killer cells
- JAK-1:
-
Janus kinase 1
- JAK-2:
-
Janus kinase 2
- STAT1:
-
Signal Transducer and Activator of Transcription 1
- TLRs:
-
Toll-like receptors
References
Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Transl Oncol. 2021;14(10): 101174.
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–9.
Diaz LA Jr, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022;23(5):659–70.
Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019;20(6):849–61.
Wang F, Zhao Q, Wang YN, Jin Y, He MM, Liu ZX, et al. Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5(10):1504–6.
O’Malley DM, Bariani GM, Cassier PA, Marabelle A, Hansen AR, De Jesus AA, et al. Pembrolizumab in patients with microsatellite instability-high advanced endometrial cancer: results from the KEYNOTE-158 study. J Clin Oncol. 2022;40(7):752–61.
Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19(11):776–800.
Bian S, Hou Y, Zhou X, Li X, Yong J, Wang Y, et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science. 2018;362(6418):1060–3.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–20.
Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18(2):90–101.
Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305.
Makaremi S, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Sgambato A, Ghorbaninezhad F, et al. Immune checkpoint inhibitors in colorectal cancer: challenges and future prospects. Biomedicines. 2021;9(9):1075.
Chen Q, Peng K, Mo P, Yu C. Histone demethylase JMJD2D: a novel player in colorectal and hepatocellular cancers. Cancers (Basel). 2022;14(12):2841.
Liao TT, Lin CC, Jiang JK, Yang SH, Teng HW, Yang MH. Harnessing stemness and PD-L1 expression by AT-rich interaction domain-containing protein 3B in colorectal cancer. Theranostics. 2020;10(14):6095–112.
Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA methylation and repressive histones in the promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and galectin-9 genes in human colorectal cancer. Clin Epigenetics. 2018;10(1):104.
Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. 2016;34:539–73.
Du W, Frankel TL, Green M, Zou W. IFNγ signaling integrity in colorectal cancer immunity and immunotherapy. Cell Mol Immunol. 2022;19(1):23–32.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–53.
Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res. 2016;76(2):275–82.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Belk JA, Yao W, Ly N, Freitas KA, Chen YT, Shi Q, et al. Genome-wide CRISPR screens of T cell exhaustion identify chromatin remodeling factors that limit T cell persistence. Cancer Cell. 2022;40(7):768-86.e7.
Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170(1):142-57.e19.
Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature. 2020;585(7824):277–82.
Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. 2022;19(12):775–90.
Li C, Song J, Guo Z, Gong Y, Zhang T, Huang J, et al. EZH2 inhibitors suppress colorectal cancer by regulating macrophage polarization in the tumor microenvironment. Front Immunol. 2022;13: 857808.
Sahakian E, Powers JJ, Chen J, Deng SL, Cheng F, Distler A, et al. Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63(2):579–85.
Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenetics. 2020;12(1):13.
Yue X, Samaniego-Castruita D, González-Avalos E, Li X, Barwick BG, Rao A. Whole-genome analysis of TET dioxygenase function in regulatory T cells. EMBO Rep. 2021;22(8): e52716.
Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda KI, Akanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol. 2017;29(8):365–75.
Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep. 2018;23(11):3262–74.
Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20(1):171.
Nishioka C, Ikezoe T, Yang J, Udaka K, Yokoyama A. Simultaneous inhibition of DNA methyltransferase and histone deacetylase induces p53-independent apoptosis via down-regulation of Mcl-1 in acute myelogenous leukemia cells. Leuk Res. 2011;35(7):932–9.
Hsi LC, Xi X, Wu Y, Lippman SM. The methyltransferase inhibitor 5-aza-2-deoxycytidine induces apoptosis via induction of 15-lipoxygenase-1 in colorectal cancer cells. Mol Cancer Ther. 2005;4(11):1740–6.
Yang D, Torres CM, Bardhan K, Zimmerman M, McGaha TL, Liu K. Decitabine and vorinostat cooperate to sensitize colon carcinoma cells to Fas ligand-induced apoptosis in vitro and tumor suppression in vivo. J Immunol. 2012;188(9):4441–9.
Brodská B, Otevřelová P, Holoubek A. Decitabine-induced apoptosis is derived by Puma and Noxa induction in chronic myeloid leukemia cell line as well as in PBL and is potentiated by SAHA. Mol Cell Biochem. 2011;350(1–2):71–80.
Venturelli S, Berger A, Weiland T, Essmann F, Waibel M, Nuebling T, et al. Differential induction of apoptosis and senescence by the DNA methyltransferase inhibitors 5-azacytidine and 5-aza-2’-deoxycytidine in solid tumor cells. Mol Cancer Ther. 2013;12(10):2226–36.
Newbold A, Falkenberg KJ, Prince HM, Johnstone RW. How do tumor cells respond to HDAC inhibition? Febs j. 2016;283(22):4032–46.
Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2017;169(2):361.
Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162(5):961–73.
Kassiotis G. Endogenous retroviruses and the development of cancer. J Immunol. 2014;192(4):1343–9.
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61.
Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, et al. HDAC inhibitor, CG-745, enhances the anti-cancer effect of anti-PD-1 immune checkpoint inhibitor by modulation of the immune microenvironment. J Cancer. 2020;11(14):4059–72.
Lodewijk I, Nunes SP, Henrique R, Jerónimo C, Dueñas M, Paramio JM. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin Epigenetics. 2021;13(1):63.
Saleh R, Toor SM, Sasidharan Nair V, Elkord E. Role of epigenetic modifications in inhibitory immune checkpoints in cancer development and progression. Front Immunol. 2020;11:1469.
Hermann A, Gowher H, Jeltsch A. Biochemistry and biology of mammalian DNA methyltransferases. Cell Mol Life Sci. 2004;61(19–20):2571–87.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92.
Fang JY, Chen YX, Lu J, Lu R, Yang L, Zhu HY, et al. Epigenetic modification regulates both expression of tumor-associated genes and cell cycle progressing in human colon cancer cell lines: Colo-320 and SW1116. Cell Res. 2004;14(3):217–26.
Alspach E, Lussier DM, Schreiber RD. Interferon γ and its important roles in promoting and inhibiting spontaneous and therapeutic cancer immunity. Cold Spring Harb Perspect Biol. 2019;11(3):a028480.
Huang KC, Chiang SF, Chen WT, Chen TW, Hu CH, Yang PC, et al. Decitabine augments chemotherapy-induced PD-L1 upregulation for PD-L1 blockade in colorectal cancer. Cancers (Basel). 2020;12(2):462.
Xu YP, Lv L, Liu Y, Smith MD, Li WC, Tan XM, et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J Clin Invest. 2019;129(10):4316–31.
Wang N, Ma T, Yu B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct Target Ther. 2023;8(1):69.
Wang X, Brea LT, Yu J. Immune modulatory functions of EZH2 in the tumor microenvironment: implications in cancer immunotherapy. Am J Clin Exp Urol. 2019;7(2):85–91.
Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark Res. 2018;6:10.
Kim HJ, Cantor H, Cosmopoulos K. Overcoming immune checkpoint blockade resistance via EZH2 inhibition. Trends Immunol. 2020;41(10):948–63.
Zhang Z, Zhang C, Luo Y, Zhang G, Wu P, Sun N, et al. RNA N(6) -methyladenosine modification in the lethal teamwork of cancer stem cells and the tumor immune microenvironment: current landscape and therapeutic potential. Clin Transl Med. 2021;11(9):e525.
Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. 2018;174(3):549-63.e19.
Zhuo M, Chen W, Shang S, Guo P, Peng K, Li M, et al. Inflammation-induced JMJD2D promotes colitis recovery and colon tumorigenesis by activating Hedgehog signaling. Oncogene. 2020;39(16):3336–53.
Micevic G, Bosenberg MW, Yan Q. The crossroads of cancer epigenetics and immune checkpoint therapy. Clin Cancer Res. 2023;29(7):1173–82.
Jung G, Hernández-Illán E, Moreira L, Balaguer F, Goel A. Epigenetics of colorectal cancer: biomarker and therapeutic potential. Nat Rev Gastroenterol Hepatol. 2020;17(2):111–30.
Shang S, Yang YW, Chen F, Yu L, Shen SH, Li K, et al. TRIB3 reduces CD8(+) T cell infiltration and induces immune evasion by repressing the STAT1-CXCL10 axis in colorectal cancer. Sci Transl Med. 2022;14(626):eabf0992.
Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3(3):224–9.
Kim JH, Workman JL. Histone acetylation in heterochromatin assembly. Genes Dev. 2010;24(8):738–40.
Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114(Pt 13):2363–73.
Han Y, Sun J, Yang Y, Liu Y, Lou J, Pan H, et al. TMP195 exerts antitumor effects on colorectal cancer by promoting M1 macrophages polarization. Int J Biol Sci. 2022;18(15):5653–66.
Certo M, Tsai CH, Pucino V, Ho PC, Mauro C. Lactate modulation of immune responses in inflammatory versus tumour microenvironments. Nat Rev Immunol. 2021;21(3):151–61.
Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80.
Xiong J, He J, Zhu J, Pan J, Liao W, Ye H, et al. Lactylation-driven METTL3-mediated RNA m(6)A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell. 2022;82(9):1660-77.e10.
Qin Y, Wang CJ, Ye HL, Ye GX, Wang S, Pan DB, et al. WWP2 overexpression inhibits the antitumor effects of doxorubicin in hepatocellular carcinoma. Cell Biol Int. 2022;46(10):1682–92.
Sheng H, Li Z, Su S, Sun W, Zhang X, Li L, et al. YTH domain family 2 promotes lung cancer cell growth by facilitating 6-phosphogluconate dehydrogenase mRNA translation. Carcinogenesis. 2020;41(5):541–50.
Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370(14):1287–97.
Ahlquist DA, Taylor WR, Mahoney DW, Zou H, Domanico M, Thibodeau SN, et al. The stool DNA test is more accurate than the plasma septin 9 test in detecting colorectal neoplasia. Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc. 2012;10(3):272-7.e1.
Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014;63(2):317–25.
Potter NT, Hurban P, White MN, Whitlock KD, Lofton-Day CE, Tetzner R, et al. Validation of a real-time PCR-based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin Chem. 2014;60(9):1183–91.
Chen WD, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L, et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst. 2005;97(15):1124–32.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, et al. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122(5):1376–87.
Shen Y, Wang D, Yuan T, Fang H, Zhu C, Qin J, et al. Novel DNA methylation biomarkers in stool and blood for early detection of colorectal cancer and precancerous lesions. Clin Epigenet. 2023;15(1):26.
Zou Q, Wang X, Ren D, Hu B, Tang G, Zhang Y, et al. DNA methylation-based signature of CD8+ tumor-infiltrating lymphocytes enables evaluation of immune response and prognosis in colorectal cancer. J Immunother Cancer. 2021;9(9):e002671.
Xu B, Lu M, Yan L, Ge M, Ren Y, Wang R, et al. A pan-cancer analysis of predictive methylation signatures of response to cancer immunotherapy. Front Immunol. 2021;12: 796647.
Allo G, Bernardini MQ, Wu RC, Shih Ie M, Kalloger S, Pollett A, et al. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Mod Pathol. 2014;27(2):255–61.
Bosse T, ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26(11):1525–35.
Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–23.
Li L, Li M, Jiang Z, Wang X. ARID1A Mutations Are Associated with Increased Immune Activity in Gastrointestinal Cancer. Cells. 2019;8(7):678.
Tokunaga R, Xiu J, Goldberg RM, Philip PA, Seeber A, Battaglin F, et al. The impact of ARID1A mutation on molecular characteristics in colorectal cancer. Eur J Cancer. 2020;140:119–29.
Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15(6):334–46.
Fagan RJ, Dingwall AK. COMPASS ascending: emerging clues regarding the roles of MLL3/KMT2C and MLL2/KMT2D proteins in cancer. Cancer Lett. 2019;458:56–65.
Zhang P, Huang Y. Genomic alterations in KMT2 family predict outcome of immune checkpoint therapy in multiple cancers. J Hematol Oncol. 2021;14(1):39.
Liu R, Niu Y, Liu C, Zhang X, Zhang J, Shi M, et al. Association of KMT2C/D loss-of-function variants with response to immune checkpoint blockades in colorectal cancer. Cancer Sci. 2023;114(4):1229–39.
Wu HX, Chen YX, Wang ZX, Zhao Q, He MM, Wang YN, et al. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J Immunother Cancer. 2019;7(1):264.
Morano F, Raimondi A, Pagani F, Lonardi S, Salvatore L, Cremolini C, et al. Temozolomide followed by combination with low-dose ipilimumab and nivolumab in patients with microsatellite-stable, O(6)-Methylguanine-DNA methyltransferase-silenced metastatic colorectal cancer: the MAYA trial. J Clin Oncol. 2022;40(14):1562–73.
Baretti M, Murphy AG, Zahurak M, Gianino N, Parkinson R, Walker R, et al. A study of using epigenetic modulators to enhance response to pembrolizumab (MK-3475) in microsatellite stable advanced colorectal cancer. Clin Epigenet. 2023;15(1):74.
Goyal A, Bauer J, Hey J, Papageorgiou DN, Stepanova E, Daskalakis M, et al. DNMT and HDAC inhibition induces immunogenic neoantigens from human endogenous retroviral element-derived transcripts. Nat Commun. 2023;14(1):6731.
Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. 2019;16(4):401–9.
Li X, Tian S, Shi H, Ta N, Ni X, Bai C, et al. The golden key to open mystery boxes of SMARCA4-deficient undifferentiated thoracic tumor: focusing immunotherapy, tumor microenvironment and epigenetic regulation. Cancer Gene Ther. 2024;31(5):687–97.
Shinno Y, Ohe Y. Thoracic SMARCA4-deficient undifferentiated tumor: current knowledge and future perspectives. Jpn J Clin Oncol. 2024;54(3):265–70.
Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21(3):231–8.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24(5):556–62.
Williamson CT, Miller R, Pemberton HN, Jones SE, Campbell J, Konde A, et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat Commun. 2016;7:13837.
Acknowledgements
Figures were created with Figdraw.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82373372 and 82102858) and the Natural Science Funding of Heilongjiang (No. YQ2022H017).
Author information
Authors and Affiliations
Contributions
TD and XG wrote the manuscript. LC, YR and HZ conducted investigation. ZC and YL were responsible for resources. CL and YZ designed and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dang, T., Guan, X., Cui, L. et al. Epigenetics and immunotherapy in colorectal cancer: progress and promise. Clin Epigenet 16, 123 (2024). https://doi.org/10.1186/s13148-024-01740-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-024-01740-9