
Abstract
Background
A gap exists in our mechanistic understanding of how genetic and environmental risk factors converge at the molecular level to result in the emergence of autism symptoms. We compared blood-based gene expression signatures in identical twins concordant and discordant for autism spectrum condition (ASC) to differentiate genetic and environmentally driven transcription differences, and establish convergent evidence for biological mechanisms involved in ASC.
Methods
Genome-wide gene expression data were generated using RNA-seq on whole blood samples taken from 16 pairs of monozygotic (MZ) twins and seven twin pair members (39 individuals in total), who had been assessed for ASC and autism traits at age 12. Differential expression (DE) analyses were performed between (a) affected and unaffected subjects (N = 36) and (b) within discordant ASC MZ twin pairs (total N = 11) to identify environmental-driven DE. Gene set enrichment and pathway testing was performed on DE gene lists. Finally, an integrative analysis using DNA methylation data aimed to identify genes with consistent evidence for altered regulation in cis.
Results
In the discordant twin analysis, three genes showed evidence for DE at FDR < 10%: IGHG4, EVI2A and SNORD15B. In the case-control analysis, four DE genes were identified at FDR < 10% including IGHG4, PRR13P5, DEPDC1B, and ZNF501. We find enrichment for DE of genes curated in the SFARI human gene database. Pathways showing evidence of enrichment included those related to immune cell signalling and immune response, transcriptional control and cell cycle/proliferation. Integrative methylomic and transcriptomic analysis identified a number of genes showing suggestive evidence for cis dysregulation.
Limitations
Identical twins stably discordant for ASC are rare, and as such the sample size was limited and constrained to the use of peripheral blood tissue for transcriptomic and methylomic profiling. Given these primary limitations, we focused on transcript-level analysis.
Conclusions
Using a cohort of ASC discordant and concordant MZ twins, we add to the growing body of transcriptomic-based evidence for an immune-based component in the molecular aetiology of ASC. Whilst the sample size was limited, the study demonstrates the utility of the discordant MZ twin design combined with multi-omics integration for maximising the potential to identify disease-associated molecular signals.
Similar content being viewed by others
Background
Autism spectrum condition (ASC) is a neurodevelopmental disorder typified by deficits in social communication and by stereotyped behaviours. Twin, family and DNA-based studies consistently indicate a strong genetic basis for the disorder, with both common and rare variants contributing to overall risk [1,2,3,4,5,6]. While a significant proportion of genetic liability for ASC can now be captured by polygenic profile scores derived from large-scale genome-wide association studies [1, 7], there exists a gap in our ability to build mechanistic models of how genetic and non-genetic risk factors influence early brain development and result in autism symptoms. Complementary functional genomic approaches that profile gene expression and epigenetic regulation could prove valuable in this regard. Because these intermediate molecular traits lie between genotype and phenotype, they can be informative about underlying mechanisms even when the trait is multifactorial and highly genetically heterogeneous, by providing evidence of convergence of risk on common downstream pathways [8]. In addition, gene regulatory and expression signatures may also have utility as biomarkers [9] as they can display individual level variability that is not completely heritable [10,11,12], may persist long after the initial exposure [13] and may be detectable in tissues other than those primarily affected [14, 15].
A number of studies have characterised global gene expression in ASC, typically using microarrays and whole blood or blood-derived cells. A recent mega-analysis of seven primary blood-based microarray datasets extended previous array-based meta-analyses [16, 17] and found support for reduced-expression of transcripts relating to innate and adaptive immunity [18]. Brain-based transcriptomic investigations of ASC using arrays are scarcer and generally limited to examination of post-mortem brain regions across a wide age range [19, 20], some of which have recently been meta-analysed to populate a searchable differential gene expression database for ASC [21].
More recently, gene expression studies have utilised RNA-seq as it can interrogate gene expression in much greater resolution than expression arrays, including low abundance transcripts as well as alternatively spliced, and non-coding regulatory transcripts [22]. RNA-seq has been used to characterise genome-wide patterns of expression for a range of neurodevelopmental and psychiatric conditions including major depressive disorder (MDD) [23], schizophrenia [24], bipolar disorder [25] and Alzheimer’s disease [26]. RNA-seq has also been used to investigate gene expression in a developmental context in healthy controls [27], and also in ASC-affected individuals [19, 28]. The largest published autism RNA-seq study of post-mortem brain tissue found evidence for cortex-specific differential gene expression and alternative splicing events, with enrichment for genes expressed in microglia and astrocytes [29]. A second (smaller) study identified an ASC-associated gene co-expression module that was enriched for microglia cell states and type 1 interferon pathway, and the authors hypothesised a putative link between neuronal activity-dependent gene regulation mediated by innate immune response [28].
To date, the majority of gene expression studies of ASC have utilised case-control designs that are limited in their capacity to investigate the influence of environmental factors, due to the potential for genetic and other sources of confounding. Intermediate molecular traits such as DNA methylation and transcription can be influenced by genetic variation (i.e. mQTLs and eQTLs), in addition to displaying environmentally linked and stochastic variability, complicating attempts to separate genetically driven from environmentally driven effects (and their interactions) [10]. To overcome some of these challenges, a number of authors have recommended a discordant identical twin-based design [30, 31]. Because identical (monozygotic; MZ) twins are matched for age, sex, genetic background, as well as family-wide factors [32], divergence in gene expression patterns or epigenetic marks can indicate causes or consequences of disease that are independent of genetic effects [33]. In ASC, despite the high heritability, MZ concordance rates do not rise above 90% [34], suggesting a role for non-shared effects such as epigenetic, gene expression, other environmental and/or stochastic factors [35, 36]. Epidemiological investigation of what these specific non-shared environmental factors might be is difficult, especially given that the relevant exposures are likely to occur early in development. Investigation of the intermediate molecular phenotype of gene expression might therefore act as a useful proxy for relevant non-shared environmental effects as it is tangibly linked to underlying cellular processes and can reflect the aggregate effects of environmental influences that could persist long after the initial exposure. Identification of environmentally attributable differential gene expression would provide critical insights into the mechanistic developmental origins of ASD, as well as provide a potential biomarker for diagnostics, treatment assessment or intervention studies.
Genome-wide gene expression profiling of discordant MZ twins has previously been performed for a range of cognitive, physical, psychiatric and neurological traits including BMI [37, 38], intelligence [39], bipolar disorder [40] and Alzheimer’s disease [41]. There has however been limited application of this methodology to ASC. A study by Hu et al. examined gene expression in three pairs of MZ ASC discordant and two pairs of MZ ASC concordant twins and identified expression differences in gene networks involved in neurological function, nervous system development and inflammation [42]. Several studies have successfully utilised MZ twins to investigate altered DNA methylation in psychiatric traits [43,44,45,46]. We previously performed genome-wide DNA methylation profiling in the cohort used in the current study that included ASC discordant, ASC concordant, as well as unaffected concordant MZ twin pairs, which investigated differences within discordant pairs and between groups (cases and controls) [47]. While ASC was not associated with global changes in DNA methylation within-discordant pairs, a number of CpG sites were found to be differentially methylated between co-twins, with the top association a CpG site located in the promoter of the NFYC gene.
As far as we are aware, there are no previous studies that have utilised a MZ differences twin design to characterise genome-wide gene expression and DNA methylation differences in the same individuals. Here, we conduct a genome-wide gene expression study using RNA-seq to profile global patterns of gene expression in subjects from a cohort of ASC discordant, ASC concordant and unaffected concordant control MZ twin pairs for whom we have previously characterised DNA methylation [47]. Genome-wide gene expression profiles were generated and two main analyses carried out. Firstly, we performed within-group analysis of the discordant ASC twins with the aim to identify genes and pathways showing altered expression in common in ASC compared to non-ASC co-twins, attributable to non-shared environmental factors. Secondly, we performed a case-control analysis by comparing ASC affected from both discordant and concordant groups with unaffected, age-matched controls to identify genes and pathways commonly disrupted in ASC, which could be attributable to both genetic and/or environmental factors. Finally, we performed data integration with previously published methylation array data to determine if identified gene expression signatures showed any relation to previously identified DNA methylation signatures, and whether such an integrative analysis could lend further support to identified pathways and mechanisms.
Methods
Subjects
Subjects were originally recruited as part of the Twins Early Development Study (TEDS; https://www.teds.ac.uk/), a longitudinal study investigating the cognitive and behavioural development of twins born in England and Wales between January 1994 and December 1996 [48,49,50]. TEDS participants completed various web and telephone-based tests and questionnaires at regular intervals over childhood and adolescence designed to assess various aspects of cognition, language and behaviour (see [50] for further details). Twins were assessed for ASC-related traits and behaviours at ages 8 and 12 using the Childhood Autism Spectrum Test (CAST). The CAST is a 31-item questionnaire that assesses ASC traits [51, 52]. The 31 items are combined additively to give a total CAST score out of 31, with those scoring ≥ 15 categorised as “at risk” of having an ASC. Individuals identified as at risk were also formally assessed at home using the Autism Diagnostic Interview-Revised (ADIR) [53] and the Autism Diagnostic Observation Schedule (ADOS) [54] both considered gold standard diagnostic tools. Twins were selected from the TEDS sample of ~ 10,000 twin pairs based on scores on the CAST total score or whether a clinical diagnosis of ASC had been made, and assigned to three study groups: concordant ASC (MZ pairs where both members of the twin pair had a formal ASC diagnosis; 28 pairs identified, of which 25 male), discordant ASC (MZ pairs where one member of the twin pair had formal ASC diagnosis; 14 pairs identified, 11 male) and low total CAST controls (unaffected MZ pairs where both twins scored less than or equal to the sample mean in total CAST score (≤ 6); 120 pairs identified, of which 45 were male). We additionally required that the concordant MZ twin pairs with low ASC symptoms had birth weights within 1 SD of each other, had no reported medical conditions and were not perinatal outliers. All samples were of white ethic origin. Identified families were contacted and invited to participate in the study, which resulted in a total of 23 MZ twin pairs visiting the SGDP lab to have whole blood samples collected by a trained phlebotomist: six concordant ASC, six discordant ASC and 11 control pairs (total N = 46). Blood samples were successfully obtained for 40 individuals: ten concordant ASC (four pairs, two individual members of a twin pair), 11 discordant ASC (five pairs, one individual), 19 concordant low total CAST (unaffected controls) (eight pairs, three individuals).
RNA isolation, library preparation and sequencing
Whole blood samples were collected and stored using the PAXgene system (PreAnalytiX QIAGEN, Germany). Total RNA was extracted (5 μg for each sample) and the Ribo-Zero Globin kit (Ambion, USA) was used to deplete ribosomal and globin RNAs. Sample concentration and quality was assessed using the RNA 6000 Nano LabChip and Agilent 2100 BioAnalyzer systems (Agilent Technologies, USA). The majority of samples (36/40) had RNA integrity (RIN) values between 7 and 9 (mean = 8.2). The four remaining samples had RIN values < 7 (mean = 4.8), with one sample from the concordant ASC group found to be severely degraded (RIN = 2.6) which was subsequently excluded from further analysis (see Fig. 1). Stranded total RNA libraries were prepared for the remaining 39 samples using the Illumina TruSeq Stranded Total RNA Library Prep Kit v3 (Illumina, USA) following the manufacturer’s protocol for a low-throughput experiment. The final libraries consisted of indexed fragments with median length 326 bp (160 bp inserts). The Agilent BioAnalser system was used to assess the fragment length distribution. The libraries were then pooled and randomised across the lanes of a single flow cell of the Illumina HiSeq 2000 (Illumina, USA) sequencing in 100-bp paired end mode. Sample libraries were randomised across lanes in an approximation of a balanced design (to the extent that was possible with the starting sample characteristics), with twin pairs split across the lanes and each lane containing subjects from each of the three study groups (discordant ASC, concordant ASC and concordant controls). To ensure each lane had the same number of samples and read density, one sample from the concordant ASC group was run in duplicate, so that in total RNA libraries for 40 samples were sequenced (for 39 individuals). The resulting reads were demultiplexed by filtering according to the 6 bp index sequences contained within the adaptors and unique to each sample.

Study group sample characteristics (N = 39)
Data preprocessing, mapping and quantification
The fastq files containing the sequenced reads for each sample were downloaded and initial data preprocessing and quality control were carried out. The FastQC program (v0.11.2) [55] was used to produce general sequencing metrics. The number of reads per library ranged between 38 and 78 M, with a mean of approximately 54 M reads per sample. Plots for per-base sequence quality, per-sequence quality, sequence length distribution, GC content were inspected, and found to be as expected for all samples. The average sequence duplication rate for each library was around 0.21, also in line with expectations [56]. Next, to improve mapping efficiency, the cutadapt tool (v1.4.1, [57]) was used to trim lower quality base pair calls from the ends of sequences, and to remove any reads mapping to Illumina adaptor sequences.
Reads were mapped to the Ensembl hg19 reference genome using Tophat2 (v2.0.12) [58]. To assess the effectiveness of read mapping and the quality of the alignments, RNA-SeQC (v1.1.8) [59] was used to produce summary statistics including total proportion of mapped reads, proportion of reads mapping in pairs, the effectiveness of the stranded protocol, fraction of reads mapping to exonic or intronic regions and coverage along the length of the transcripts. These metrics were compared to guidelines from GEUVADIS [56]. The overall indications were that the sequencing data generated was of a high quality, with on average 78% of total reads mapping in their forward and reverse read pairs, 47% of these reads originating from exonic regions, a mean coverage of 6× for less abundant transcripts (bottom 1000) and 732× for transcripts present at higher levels (top 1000).
The featureCounts program [60] from the subread package (v1.4.6) [61] was next used to summarise and quantify the mapped reads, by counting the number of reads overlapping Ensembl gene annotations. Finally, filtering was performed to remove genes with low counts with required counts of ≥ 1 CPM (counts per million) in at least three samples leaving count data for 17,765 genes.
Post quantification QC
Following quantification, a number of QC steps were performed to assess the quality of the final gene expression profiles and identify any outliers. Firstly, concordance between expression profiles within the twin pairs was examined. As gene expression is at least partially heritable (mean h2 ~ 0.14 to 0.3) [11, 62], twins would be expected to show higher correlation in gene expression profiles than in comparison to unrelated subjects. To confirm this, counts were normalised by total library size and transformed into logCPM (log counts per million), and Pearson’s correlation was calculated. A heatmap was produced to visualise the results, and mean correlation values within pairs and between unrelated subjects were recorded. Next, clustering and data reduction techniques were used to produce visualisations for examining the similarity of the expression profiles and determine if any clustering by technical or biological factors was observed, specifically by case status, study group, sex or flow cell lane. Firstly, hierarchical, unsupervised clustering was performed with the hclust function in R using Euclidean distance between samples based on gene expression profiles. These were plotted as dendrograms (Additional file 1: Figure S1). Multi-dimensional scaling (MDS) plots were also generated for further visualisation of sample similarity based on leading log fold changes between pairs of samples (Additional file 1: Figure S2).
Differential gene expression
The edgeR package [63] was used to perform differential expression (DE) analyses. This uses a generalised linear modelling (GLM) strategy to model systematic and biological sources of variation, and an empirical Bayes method to improve estimates by shrinking gene-wise dispersions towards the common trend. Following the standard edgeR workflow, count data was normalised for total library size, using trimmed mean of M values (TMM), which minimises log FCs between samples for the majority of genes. To test for differential expression attributable to ASC case status, for each gene a negative binomial GLM model was used and a likelihood ratio test (LRT) performed. Multiple testing was controlled at FDR 10%, chosen due to the discovery nature of the study.
Two primary DE analyses were performed. Firstly, a within group analysis of the ASC discordant twins was performed to identify differences in gene expression associated with ASC which could be attributable to non-shared environment. This analysis will be referred to as “discordant group” from this point. The model used specified gene count as the outcome which was regressed on pair—a categorical variable representing twin-pair membership, and case—a binary variable denoting case status. The results were further annotated to indicate whether the direction of effect (log FC) was consistent across pairs. For the second analysis, cases and controls from across the complete sample were compared to identify gene expression differences associated with ASC that could be genetic and/or environmental in origin. We refer to this analysis and the results as “case-control” from here on. The model used was the same as used in the discordant twin analysis above, with gene count the outcome and pair and case status the explanatory variables.
Finally, within-pair gene expression differences were assessed to determine whether these showed consistent differences in magnitude or overall distribution between the three study groups. For each pair separate, edgeR models were fit (comparing one co-twin to the other). Then for each of the study groups, absolute logFCs across all genes and pairs were recorded, and scatter plots produced. Both t tests and Kolmogorov–Smirnov (KS) tests were performed comparing the means and overall distributions for each group (Additional file 1: Figure S5).
Gene set enrichment and pathways
Gene set enrichment testing was performed to investigate whether identified differential expression signals involved known ASC risk genes. The geneSetTest method from limma [64] was used with ranks only, comparing the mean rank of the set of interest with permuted sets. Three different ASC genesets were tested for enrichment. The first of these was from the Simons Foundation Autism Research Initiative (SFARI) gene resource [65, 66], a manually curated reference database of ASC genes from published molecular studies including candidate genes, known common and rare variants and copy number variants linked to ASC. These are divided into different categories based on the strength of evidence. We combined these into two gene sets: category 1 and 2 (high confidence and strong candidate) (n = 91), and categories 3, 4 and 5 (suggestive evidence, minimal evidence and hypothesised) (n = 842). The second gene set was derived from the association results of the iPSYCH-PGC ASD GWAS meta-analysis of over 18,381 individuals with ASD [67], setting a p value threshold of 1 × 10−6 and taking the set of genes that these mapped to (according to Ensembl hg19 annotation) (n = 28). Finally, we used the TADA set of genes from an exome sequencing study of 2270 trios, which identified genes showing evidence for increased burden of de-novo loss of function in autistic subjects (n = 107) [2]. All sets were accessed and downloaded in July 2019.
Pathway enrichment analyses were carried out to identify biological pathways and cellular processes over-represented within the differentially expressed genes. The ROMER method from limma was used. Pathway datasets were from the Molecular Signatures Database (MSigDB) [68], comprising biochemical pathways (from Kyoto Encyclopedia of Genes and Genomes—KEGG [69] and REACTOME [70]), gene families (from Gene Ontology—GO [71]) and various other sources of annotation. Two sets were used, the C2 curated set—containing 4726 canonical metabolic and signalling pathways, and disease gene expression signatures, and the H1 hallmark set—50 gene sets representing well-defined biological states or processes where genes show coordinated expression.
Integration with DNA methylation data
Using an existing Illumina 27 K DNA methylation dataset generated on the same sample [47], an integrative analysis was performed to derive further insights into the functional relevance of DE genes and identify genes with consistent evidence for altered regulation in ASC, representing putative disorder associated cis expression quantitative trait methylation loci (eQTMs). The stored Beta values were first converted to M values as these have distributional properties more suitable for use in standard linear regression [72], and differential methylation analysis performed. The ComBat function from the SVA package [73] was used to identify batch and other unmeasured sources of variation—in this case, none were identified. Next, edgeR [74] was used to fit a linear model for discordant group and case-control comparisons, using the same models described above for the RNA-seq data. Given the limited of coverage of the array, a simple gene-level integration strategy was followed looking for evidence of cis regulatory signals across the datasets. Firstly, to assess the level of concordance, Spearman’s correlation coefficient was calculated between gene-level expression (as normalised counts per million) and gene-level DNA methylation (aggregating CpG probes and taking the median Beta value as representative). Secondly, evidence combination was performed. The SLK method from comb-p [75] was used to obtain spatial correlation adjusted p values for each CpG site, setting the distance parameter to 2000 bp and region filter to 0.05. These were summarised at the gene level by taking the minimum p value. This was then combined with the gene level statistics from the RNA-seq results using empirical Brown’s method [76], which takes into account correlation between measures (using the assay data), to derive a combined p value for genes measured in both assays. The final list of genes was then sorted by combined p value and the top 20 retained.
Male subgroup analysis
Previous studies have identified sex-specific patterns of gene expression associated with ASC [77, 78], with the latter also demonstrating that separate analyses of males and females improved the ability to detect ASC-related differences. Here, because the sample contained only two affected females (both discordant group), we were not adequately powered to perform a sex-stratified analysis. Instead, the main analyses were rerun using male subjects as an explicit control for sex, which in the main analysis was presumed to have been captured by pair status (as a nested effect—since MZ twins are the same sex).
QQ plots were generated and inspected (data not shown) to compare the models in terms of overall distribution of p values. The lists of top differentially expressed genes were also compared to evaluate the impact of excluding female samples on the ranks of FDR significant DE genes and whether this improved our ability to detect ASC-related differences. See Additional file 1: Table S13 and S14.
Blood cell composition and surrogate variable analysis
Issues related to cellular heterogeneity have received much attention in the transcriptomics [79,80,81,82,83] and epigenomics literature [84,85,86,87,88]. Cell composition can act as a confounder, or alternatively represent a variable of interest if disease-associated changes in cell populations or cell subtype-specific expression patterns are disease relevant. In the context of this study, it was not clear how cellular heterogeneity should be dealt with since changes in cell composition could well be ASC relevant, with previous studies finding evidence for increased numbers of activated B and NK cells in ASC affected individuals [89, 90]. As full blood cell counts for our samples were not available, we performed cell subtype estimation as a post hoc analysis, to determine whether the main findings were likely being driven by differences in blood cell composition. The XCell software was used to infer cell counts, which compares the experimental RNA-seq data to expression signatures spanning 64 immune and stromal cell types and calculates an enrichment score [83]. Blood cell adjustment was performed as part of surrogate variable analysis.
Surrogate variable analysis (SVA) [73] was performed firstly to identify any unmodelled technical variation and secondly to reduce the number of parameters required to model inferred blood cell counts—since these could be much greater than the sample size. The two main analyses and the male-only versions were re-run using models including surrogate variable adjustment. Correlation between identified SVs and cell counts were calculated to determine if these were likely to be accounting for these variables. As before, QQ plots (not shown) and relevant summary statistics were generated and inspected to compare the different models, and the ranks of DE genes at FDR 20% between models compared (see Additional file 1: Table S13 and S14).
Results
Exploratory analysis
Exploratory analysis was performed to assess the quality of the gene expression profiles generated. Gene expression profiles were compared between individuals; high within-twin correlations were observed (median = 0.98), greater than those observed for unrelated individuals (median = 0.96) (Fig. 2), as expected. Next, sample clustering was performed (Additional file 1: Figure S1a,b). The dendrograms showed male and female subjects segregating into two distinct clusters with no sex outliers, and 15 (of the 16) complete twin pairs clustering together in nodes. MDS plots for the first six dimensions were inspected, which showed that samples mainly clustered by sex, case and experimental group, whereas sequencing lane was not seen to segregate the data (Additional file 1: Figure S2). Inspection of these plots also revealed the presence of three outliers, which were subsequently removed from further analysis. Following these steps, 36 samples were taken forward for differential expression analysis (Additional file 1: Table S1a,b and S2a,b).

Heatmap showing individuals clustered using correlation of genome-wide expression values between samples as distance (also shown as dendrograms). High correlation r values of 0.99 and above are indicated by the dark coloured squares along the diagonal. Twin pairs are found to have an average correlation of 0.98, compared to 0.96 for unrelated subjects. cASC concordant ASC, dASC discordant ASC, cltC concordant low total CAST (control)
Differential expression analysis
The DE genes identified in the within discordant pairs analysis are presented in Table 1 (with extended list in Additional file 1: Table S3). The volcano plot is shown in Fig. 3a, and examination of the QQ plots is suggestive of uncorrected biases, although we were unable to identify any single factor (see Additional file 1: Figures S3a and S4a). Overall, three genes pass FDR 10%: IGHG4 (logFC = 2.15, FDR = 0.0004), EVI2A (logFC = − 0.66, FDR = 0.02) and SNORD15B (logFC = − 0.85, FDR = 0.02). Further down the list of suggestive genes at FDR 20%, a number of ribosomal genes are identified (RPL9, RPL30, RPL41, RPS3A). The majority of the top ranked genes were found to have a consistent direction of effect across all discordant pairs (ten out of 16 genes at FDR < 20%), which were all in the direction of decreased expression in ASC affected compared to unaffected co-twins. Additional file 1: Figure S6 shows the expression levels for DE genes at FDR < 10% for each of the twin pairs, stratified by ASC affected status. The direction of the effect is largely consistent for all three genes, with IGHG4 showing mainly increased expression in ASC cases compared to unaffected co-twin (for four out of five pairs), and EVI2A and SNORD15B showing decreased expression in all affected twin pair members.

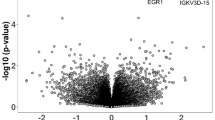
a, b Volcano plots showing relationship between significance and logFC for a discordant and b case control analyses. DE genes passing FDR < 0.1 are indicated by the coloured points and labelled
The DE genes from the case-control analysis are presented in Table 2 (extended results in Additional file 1: Table S4), and the volcano plot is shown in Fig. 3b (further plots provided in S3b and S4b). Four genes pass FDR 10%: IGHG4 (logFC = 2.04, FDR = 0.10), PRR13P5 (logFC = 1.53, FDR = 0.10), DEPDC1B (logFC = − 1.32, FDR = 0.10) and ZNF501 (logFC = − 0.88, FDR = 0.10), all four of which were also highly ranked in the discordant pairs analysis. Annotation of the 19 genes identified as nominally differentially expressed (FDR < 20%) across the discordant twin and case-control analyses revealed two non-coding RNAs (SNORD15B and AL662889.1), and suggested a bias towards RNA binding activity. See Additional file 1: Table S5.
Within-pair comparisons
To assess whether within-pair expression differences varied between discordant ASC, concordant ASC and concordant low total CAST twins, the distributions of within-pair log FCs were compared (Additional file 1: Figure S5). The mean log FCs differed by group: discordant ASC = 0.44, concordant ASC = 0.39, low total CAST = 0.39 and t tests showed this difference was significant when comparing discordant ASC to concordant ASC (p < 2.2 × 10−16) and discordant ASC to concordant low total CAST (p < 2.2 × 10−16). The cumulative distributions of the log FCs also differed by group, with KS tests indicating a significant difference between discordant ASC and concordant low total CAST (p < 2.2 × 10−16), discordant ASC and concordant ASC (8.3 × 10−14) and concordant ASC and concordant low total CAST (p < 2.2 × 10−16). Additional file 1: Figure S7 shows distributions of the expression levels for all four DE genes identified at FDR < 10% in the case-control comparison, stratified by study group and case status. The distribution of expression values can be seen to vary depending on study group, with differences between affected and unaffected co-twins in the discordant ASC appearing on visual inspection to be greater than differences between concordant ASC twin pairs, and concordant low total CAST twin pairs.
Gene set testing
ASC-associated genes
Focused testing of a number of ASC-associated genesets was performed to test for possible enrichment of previously identified ASC-risk genes. The results are presented in Additional file 1: Table S6. There is evidence for enrichment of ASC-associated genes from the SFARI gene set in gene score categories 1 and 2, and categories 3, 4 and 5 in both the discordant and case-control DE gene lists. The iPSYCH-PGC and TADA genesets do not show enrichment in either of the DE gene lists.
Pathways
The lists of DE genes from both analyses were tested for enrichment of MSigDB pathways. The results are presented in Additional file 1: Tables S7–S10. For the discordant analysis, a number of potentially relevant pathways (unadjusted p < 0.05) were identified including those involved in cell signal integration (mTORC1 signalling), regulation of gene expression (MYC targets, E2F targets), immune response (Bohn primary immunodeficiency syndrome) and genomic stability (chromosome maintenance). For the case-control analysis, regulation of gene expression (E2F targets, MYC targets), cell signal integration (mTORC1 signalling, PI3K AKT mTOR signalling) and immune response pathways were similarly identified (Browne HCMV infection, Bohn primary immunodeficiency syndrome).
Integration with DNA methylation data
The gene expression data was integrated with DNA methylation data on the same individuals. The top 20 results are given in Additional file 1: Tables S11 and S12. In the discordant analysis, a number of genes show some combined evidence for differential expression and methylation which is consistent with the findings from DE and pathways analysis implicating immune signalling—DAPP1, and regulation of expression/translation: ZNF501, EIF5A2, HIST1H2AG. In the case-control results, ZNF501, DAPP1 and EIF5A2 were also highly ranked.
Post hoc sensitivity analyses
Sensitivity analyses were performed, including a male-only analysis and a surrogate variable-based modelling approach intended to capture blood cell composition and other unmeasured batch effects. The identified DE genes were found to be largely robust to the inclusion of surrogate variables in the model, and were not influenced by the exclusion of female samples in the male subjects only analysis (see Additional file 1: Tables S13 and Table S14). In the discordant analysis, the top 3 identified DE genes IGHG4, EVI2A and SNORD15B were consistently found to be within the top 0.03% (rank ≤ 5) to ~ 3% (rank ≤ 600) of DE genes for SV adjusted and male-only analysis. For the case-control analysis, similarly IGHG4, DEPDC1B and ZNF501 were also consistently within the top ~ 3%.
Discussion
Differential expression
We performed gene expression profiling by RNA-seq in identical twins discordant for ASC. The overall findings are consistent with the hypothesis that gene expression differences in discordant twins could contribute to (or are a consequence of) phenotypic differences, and we further identify specific gene expression differences associated with ASC affected status. In the discordant group analysis comparing gene expression in ASC affected compared to unaffected co-twins, three DE genes were identified at FDR < 10%: IGHG4, EVI2A and SNORD15B. The case control analysis examining expression differences between ASC affected and control samples identified four DE genes at FDR < 10% including IGHG4, DEPDC1B and ZNF501. Comparison of mean within-pair logFC expression differences revealed that differences were greater in the discordant ASC pairs than in concordant ASC and control pairs. Inspection of expression levels for FDR significant genes identified in the case control analysis suggests that larger differences in expression for trait-associated genes are apparent in the discordant ASC pairs.
The top ranked and most highly significant DE gene in both discordant group and case control analyses was IGHG4—Immunoglobulin heavy constant gamma 4, which in both analyses showed evidence for upregulation in ASC affected compared to unaffected individuals (discordant: logFC = 2.15, FDR = 0.0004, case control: logFC = 2.04, FDR = 0.0973). This gene codes for an immunoglobin (or antibody) membrane-bound protein produced by B lymphocytes that recognises specific antigens, and upon binding results in immune cell activation. While IGHG4 does not appear to have previously been linked to ASC, interestingly two previous expression studies using whole blood identified another related and potentially closely interacting immunoglobin IGHG1 as being upregulated [91, 92] and in one study downregulated [42] in ASC. The existing evidence for the association of IGHG1 with ASC, together with the observation that immune-related genes and pathways are some of the most common and highly replicated findings in ASC gene expression studies (discussed further below), lends further support to our identification of IGHG4 as an ASC-associated DE gene.
DEPDC1B was the second ranked coding gene in the case-control analysis (logFC = 1.32, FDR = 0.0973). This showed suggestive evidence for association in the discordant analysis (logFC = − 1.3, FDR = 0.186), but is also noteworthy as it showed consistent effect directionality, being downregulated in ASC affected compared to unaffected for all discordant twin pairs. This gene is a member of the DEP domain coding family, and is involved in intracellular signal transduction and cell growth regulation. While it has not previously been associated with ASC, variants in DEPDC1B have been associated with intelligence and general cognitive ability in two recent large-scale GWA studies [93, 94]. The evidence taken together suggests that DEPDC1B could be an interesting and potentially relevant signal for further follow up study.
Focused testing of ASC-associated gene sets of interest indicated that overall there was enrichment of previously identified ASC loci from SFARI, whilst the iPSYCH-PGC or TADA sets did not show evidence for enrichment. These findings are consistent with those from previous studies that have tested for overlap between established ASC risk loci and ASC DE genes. In a gene mega-analysis of blood-based transcriptomic studies of ASC by Tylee et al. including 626 affected and 447 comparison subjects, the top identified DE genes were not found to be enriched for ASC GWAS associations [18]. Whilst the lack of enrichment for ASC-associated common genetic variants in differentially expressed gene lists might seem surprising, the accumulated evidence would seem to suggest that genetic association and functional approaches are revealing distinct dimensions to the disorder. ASC is frequently characterised as a disorder of the synapse based on genetic findings, whilst in contrast functional findings often implicate other pathways including immune system, inflammation and transcriptional control [95].
Pathway analysis using collections of genes from MSigDB revealed a number of potentially relevant pathways in common between both discordant and case-control analyses. Pathways were identified implicating the immune system (immune cell signalling/signal integration: mTORC1, immune response: Browne HCMV infection, Bohn primary immunodeficiency syndrome), transcriptional control (MYC targets, E2F targets) and chromosome/genomic stability (DNA repair, chromosome maintenance). In support of these findings, a recent comprehensive review of both blood and brain-based transcriptomic studies found that immune system pathways were among the most frequently identified [95]. More specifically, we found replication for the majority of the top pathways: E2F targets, mTOR and MYC pathways identified in a large mega-analysis based on gene expression in blood [18], and immunodeficiency in a meta-analysis using multiple tissues [16].
The integrative analysis revealed a number of genes showing combined evidence for dysregulation in both discordant and case control comparisons that together with the findings from the differential expression and pathways analyses suggest involvement of immune signalling and transcriptional regulation. We highlight DAPP1 (dual adaptor of phosphotyrosine and 3-phosphoinositides) as a recent large-scale CNV study of > 1600 individuals identified a paternally inherited duplication in two unrelated individuals with ASC that disrupts several exons of DAPP1 [96], and large duplications and deletions in the gene are reported in the DECHIPER database—the majority of which list autism, autistic behaviours, global developmental delay and/or intellectual disability as the associated phenotype [97].
In the wider context of ASC research, immune system disruption is one of the most consistent findings, with epidemiological studies showing that families with ASC-affected individuals have a higher rate of autoimmune disorders [98, 99], serological studies finding evidence of increased numbers of activated B and NK cells [90] and elevated levels of pro-inflammatory cytokines [100] in peripheral blood samples and post-mortem studies finding evidence of microglial activation in the dorsolateral prefrontal cortex [101]. In addition, there is also a potential causal link between established environmental risk factors such as prenatal viral infection and paternal age, with immune dysregulation and inflammation and increased risk of developmental disorders. In the case of viral infection, animal studies have linked maternal influenza infection to altered brain development and behaviour in mouse models of ASC and schizophrenia [102, 103]. As for older paternal age, it has been suggested that the observed increased incidence of ASC, schizophrenia and bipolar disorder could be the result of immune dysregulation [104] with increases in pro-inflammatory cytokines IL-1β and IL-6 observed in all three disorders [105, 106]. The accumulated evidence from these diverse studies and the potential link to environmental factors makes a compelling case for prioritising the immune-related genes identified here for further study, where we might begin to investigate links to specific environmental exposures. Future studies should be designed in order to establish the molecular drivers, look at the underlying mechanisms, ascertain whether it is primary or secondary to ASC and whether such immune markers might have utility for prognosis, diagnosis or endophenotype classification.
Strengths and limitations
We used a population-based cohort of 10,000 twins [50] to identify a rare sample of MZ twins discordant for ASC, with the primary aim of identifying biological differences free from genetic confounding. There are a number of drawbacks and limitations with the approach taken. Firstly, as MZ twins stably discordant for ASC are rare, the sample size was constrained. Whilst sample sizes required for 80% power using disease-discordant MZ twins is ~ 15% smaller than case-control designs, based on published EWAS power simulations our N of 40 would not be powered to detect small methylation differences at p < 1 × 10−6 [107, 108]. While replication using larger sample sizes is required, this study gives an indication of the value of the discordant-twin design and integrative approach for identifying modest biological signals.
Next, there is the use of peripheral blood for expression profiling, since the relationship between individual differences in gene expression in blood and brain is not yet fully understood. Whilst considerable similarities between RNA transcription in blood and brain have been reported for subsets of genes, it is not a prerequisite that peripheral gene expression exactly resembles that of the brain—only that it is a reliable marker of disease and is informative about underlying biological mechanisms [109, 110]. Peripheral blood also offers several important advantages over post-mortem brain tissue such as availability (especially at developmentally relevant time points), control of tissue quality and handling and collection of relevant donor characteristics.
There is the issue of age of the participants at sample collections, since these were collected during adolescence. ASC is a disorder of development and so it is not known whether the molecular differences identified would reflect those present in early development, or whether they are primary or secondary to onset. While the current study is not able to address developmental questions related to onset and trajectory, future genomic studies using prospective birth cohorts containing infants at risk of ASC will be highly valuable in this regard.
One assumption of this study is that that MZ twins are genetically identical. MZ twins have been found to display rare genetic differences, for example point mutations, copy number variants, telomere length, uniparental disomy, somatic mosaicism, chromosomal aneuploidies and mutations in mitochondrial DNA (see [32] for a discussion of these issues). Without comprehensive DNA sequence data on the individuals, the possibility that the observed phenotypic discordances are the result of post-twinning de novo mutations cannot be ruled out. However, this is not likely as whole-genome sequencing and SNP microarray-based studies have failed to find many replicable differences between MZ twins [32, 111], and there have been few reports of discordant MZ twins having a DNA sequence mutation causative for a disease [112].
The integrative analysis performed combining RNA-seq and DNA methylation data on the same individuals was limited in its scope, due to use of low-resolution methylation data from the Illumina 27 K array and absence of genomic data on the same individuals. Therefore, a basic integrative analysis was performed summarising DNA methylation signals at the gene-level, and limiting to identification of cis regulatory signals—since the sites covered on the array would be expected to mainly fall in to this category. We opted for evidence in combination and did not perform causal inference, due to the lack of DNA sequence data which is typically used to help establish directionality between the intermediate traits and phenotype. Our top-ranked (FDR < 10%) DE genes do not show evidence of differential methylation in our 27k dataset, but we cannot rule out the role of other cis regulatory mechanisms such as histone tail modifications, non-coding RNAs or trans effects. That said, it is encouraging that genes previously associated with ASC showed combined evidence for dysregulation across our gene expression and DNA methylation datasets, and given the limitations of the strategy used, we consider this primarily a useful demonstration of the potential utility of combining a twin-based study design with multi-omics datasets. In future studies, should genotype data and higher resolution methylation data on the same individuals become available, a more comprehensive, genome-wide, site-level eQTM scan could be performed in combination with causal analysis methods to determine the likely relationship between genotype, expression, methylation, and trait [113].
Finally, the current study focused on gene-level expression but the data generated could be utilised to investigate other transcriptomic phenomena such as alternative splicing, allele-specific expression and expression of non-coding RNAs (e.g. lncRNA). Alternative splicing in particular is thought to play a critical role in neuronal development, with some evidence that it could be relevant to ASC pathology [114, 115]. There are also examples of allele-specific gene expression linked to ASC [116, 117].
Conclusions
We characterised gene expression in a cohort of discordant and concordant ASC twin pairs using blood RNA-seq. The results from the discordant twin analysis revealed enrichment for immune system, transcriptional control and cell cycle regulation pathways in the identified DE genes, which may represent downstream risk pathways where non-shared environmental or stochastic factors converge. We further incorporated DNA methylation profiles on the same individuals and performed a combined analysis to identify putative ASC associated cis regulatory signals. While the integrative strategy was limited in its scope, we believe it is a useful demonstration of the potential utility and power of the MZ twins design combined with multi-omics integration for maximising the potential to identify disease-associated signals. We hope the RNA-seq dataset we have generated here will serve as a valuable resource for future investigations into the gene expression patterns underlying ASC (including examination of alternative splicing, allele-specific expression and expression of non-coding RNAs), and work focused on untangling genetic and environmental risk pathways.
Availability of data and materials
Processed read count data can be made available upon request subject to approval by the appropriate ethical boards.
References
Grove J, Ripke S, Als TD, Mattheisen M, Walters R, Won H, et al. Common risk variants identified in autism spectrum disorder. bioRxiv. 2017;33:42. https://doi.org/10.1101/224774.
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–15. https://doi.org/10.1038/nature13772.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46:881–5. https://doi.org/10.1038/ng.3039.
Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21. https://doi.org/10.1038/nature13908.
Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47:582–8. https://doi.org/10.1038/ng.3303.
Pettersson E, Lichtenstein P, Larsson H, Song J, Agrawal A, Børglum AD, et al. Genetic influences on eight psychiatric disorders based on family data of 4 408 646 full and half-siblings, and genetic data of 333 748 cases and controls. Psychol Med. 2018:1–8. https://doi.org/10.1017/S0033291718002039.
Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21. https://doi.org/10.1186/s13229-017-0137-9.
Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16:441–58. https://doi.org/10.1038/nrg3934.
Ladd-Acosta C, Fallin MD. The role of epigenetics in genetic and environmental epidemiology. Epigenomics. 2016;8:271–83. https://doi.org/10.2217/epi.15.102.
Hannon E, Knox O, Sugden K, Burrage J, Wong CCY, Belsky DW, et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018;14:e1007544. https://doi.org/10.1371/journal.pgen.1007544.
Wright FA, Sullivan PF, Brooks AI, Zou F, Sun W, Xia K, et al. Heritability and genomics of gene expression in peripheral blood. Nat Genet. 2014;46:430–7. https://doi.org/10.1038/ng.2951.
Li S, Wong EM, Dugué P-A, McRae AF, Kim E, Joo J-HE, et al. Genome-wide average DNA methylation is determined in utero. Int J Epidemiol. 2018;47:908–16. https://doi.org/10.1093/ije/dyy028.
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105:17046–9. https://doi.org/10.1073/pnas.0806560105.
Cai C, Langfelder P, Fuller TF, Oldham MC, Luo R, van den Berg LH, et al. Is human blood a good surrogate for brain tissue in transcriptional studies? BMC Genomics. 2010;11:589. https://doi.org/10.1186/1471-2164-11-589.
van Baak TE, Coarfa C, Dugué PA, Fiorito G, Laritsky E, Baker MS, et al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Biol. 2018;19:2. https://doi.org/10.1186/s13059-017-1374-0.
Ning LF, Yu YQ, Guo Ji ET, Kou CG, Wu YH, Shi JP, et al. Meta-analysis of differentially expressed genes in autism based on gene expression data. Genet Mol Res. 2015;14:2146–55.
Ch’ng C, Kwok W, Rogic S, Pavlidis P. Meta-analysis of gene expression in autism spectrum disorder. Autism Res. 2015;8:593–608. https://doi.org/10.1002/aur.1475.
Tylee DS, Hess JL, Quinn TP, Barve R, Huang H, Zhang-James Y, et al. Blood transcriptomic comparison of individuals with and without autism spectrum disorder: a combined-samples mega-analysis. Am J Med Genet Part B Neuropsychiatr Genet. 2017;174:181–201. https://doi.org/10.1002/ajmg.b.32511.
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–4. https://doi.org/10.1038/nature10110.
Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain transcriptional and epigenetic associations with autism. PLoS One. 2012;7:e44736. https://doi.org/10.1371/journal.pone.0044736.
Zhang S, Deng L, Jia Q, Huang S, Gu J, Zhou F, et al. dbMDEGA: a database for meta-analysis of differentially expressed genes in autism spectrum disorder. BMC Bioinformatics. 2017;18:494. https://doi.org/10.1186/s12859-017-1915-2.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. https://doi.org/10.1038/nrg2484.
Mostafavi S, Battle A, Zhu X, Potash JB, Weissman MM, Shi J, et al. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol Psychiatry. 2014;19:1267–74. https://doi.org/10.1038/mp.2013.161.
Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19:1442–53. https://doi.org/10.1038/nn.4399.
Pacifico R, Davis RL. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol Psychiatry. 2017;22:441–9. https://doi.org/10.1038/mp.2016.94.
Twine NA, Janitz K, Wilkins MR, Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PLoS One. 2011;6:e16266. https://doi.org/10.1371/journal.pone.0016266.
Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206.
Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, et al. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun. 2014;5:1–8. https://doi.org/10.1038/ncomms6748.
Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing and regional gene expression patterns in autism. Nature. 2016;540:423–7. https://doi.org/10.1038/nature20612.
Zwijnenburg PJG, Meijers-Heijboer H, Boomsma DI. Identical but not the same: the value of discordant monozygotic twins in genetic research. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:1134–49. https://doi.org/10.1002/ajmg.b.31091.
Bölte S, Willfors C, Berggren S, Norberg J, Poltrago L, Mevel K, et al. The roots of autism and ADHD twin study in Sweden (RATSS). Twin Res Hum Genet. 2014;17:164–76. https://doi.org/10.1017/thg.2014.12.
van Dongen J, Slagboom PE, Draisma HHM, Martin NG, Boomsma DI. The continuing value of twin studies in the omics era. Nat Rev Genet. 2012;13:640–53. https://doi.org/10.1038/nrg3243.
Kim K, Lee K, Bang H, Kim JY, Choi JK. Intersection of genetics and epigenetics in monozygotic twin genomes. Methods. 2016;102:50–6. https://doi.org/10.1016/j.ymeth.2015.10.020.
Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet Part B Neuropsychiatr Genet. 2011;156:255–74. https://doi.org/10.1002/ajmg.b.31159.
Plomin R. Commentary: why are children in the same family so different? Non-shared environment three decades later. Int J Epidemiol. 2011;40:582–92. https://doi.org/10.1093/ije/dyq144.
Ronald A, Happé F, Dworzynski K, Bolton P, Plomin R. Exploring the relation between prenatal and neonatal complications and later autistic-like features in a representative community sample of twins. Child Dev. 2010;81:166–82.
van Dongen J, Willemsen G, Heijmans BT, Neuteboom J, Kluft C, Jansen R, et al. Longitudinal weight differences, gene expression and blood biomarkers in BMI-discordant identical twins. Int J Obes. 2015;39:899–909. https://doi.org/10.1038/ijo.2015.24.
Pietiläinen KH, Ismail K, Järvinen E, Heinonen S, Tummers M, Bollepalli S, et al. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int J Obes. 2016;40:654–61. https://doi.org/10.1038/ijo.2015.221.
Yu C-C, Furukawa M, Kobayashi K, Shikishima C, Cha P-C, Sese J, et al. Genome-wide DNA methylation and gene expression analyses of monozygotic twins discordant for intelligence levels. PLoS One. 2012;7:e47081. https://doi.org/10.1371/journal.pone.0047081.
Matigian N, Windus L, Smith H, Filippich C, Pantelis C, McGrath J, et al. Expression profiling in monozygotic twins discordant for bipolar disorder reveals dysregulation of the WNT signalling pathway. Mol Psychiatry. 2007;12:815–25. https://doi.org/10.1038/sj.mp.4001998.
D’Addario C, Candia SB, Arosio B, Di Bartolomeo M, Abbate C, Casè A, et al. Transcriptional and epigenetic phenomena in peripheral blood cells of monozygotic twins discordant for alzheimer’s disease, a case report. J Neurol Sci. 2017;372:211–6.
Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. 2006;7:118. https://doi.org/10.1186/1471-2164-7-118.
Castellani CA, Laufer BI, Melka MG, Diehl EJ, O’Reilly RL, Singh SM. DNA methylation differences in monozygotic twin pairs discordant for schizophrenia identifies psychosis related genes and networks. BMC Med Genet. 2015;8:17. https://doi.org/10.1186/s12920-015-0093-1.
Castellani CA, Melka MG, Gui JL, O’Reilly RL, Singh SM. Integration of DNA sequence and DNA methylation changes in monozygotic twin pairs discordant for schizophrenia. Schizophr Res. 2015;169:433–40. https://doi.org/10.1016/j.schres.2015.09.021.
Fisher HL, Murphy TM, Arseneault L, Caspi A, Moffitt TE, Viana J, et al. Methylomic analysis of monozygotic twins discordant for childhood psychotic symptoms. Epigenetics. 2015;10:1014–23. https://doi.org/10.1080/15592294.2015.1099797.
Dempster EL, Wong CCY, Lester KJ, Burrage J, Gregory AM, Mill J, et al. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry. 2014;76:977–83. https://doi.org/10.1016/j.biopsych.2014.04.013.
Wong CCY, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503. https://doi.org/10.1038/mp.2013.41.
Trouton A, Spinath FM, Plomin R. Twins early development study (TEDS ): a multivariate, longitudinal genetic investigation of language , cognition and behavior problems in childhood. Twin Res. 1996;5:444–8.
Trouton A, Spinath FM, Plomin R. Twins early development study (TEDS): a multivariate, longitudinal genetic investigation of language, cognition and behavior problems in childhood. Twin Res. 2002;5:444–8. https://doi.org/10.1375/twin.5.5.444.
Haworth CMA, Davis OSP, Plomin R. Twins early development study (TEDS): a genetically sensitive investigation of cognitive and behavioral development from childhood to young adulthood. Twin Res Hum Genet. 2013;16:117–25. https://doi.org/10.1017/thg.2012.91.
Scott FJ, Baron-Cohen S, Bolton P, Brayne C. The CAST (childhood Asperger syndrome test). Autism. 2002;6:9–31. https://doi.org/10.1177/1362361302006001003.
Williams J, Scott F, Stott C, Allison C, Bolton P, Baron-Cohen S, et al. The CAST (childhood Asperger syndrome test). Autism. 2005;9:45–68. https://doi.org/10.1177/1362361305049029.
Lord C, Rutter M, Le Couteur A. Autism diagnostic interview-revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–85.
Lord C, Risi S, Lambrecht L, Cook EH, Leventhal BL, DiLavore PC, et al. Autism diagnostic observation schedule (ADOS). J Autism Dev Disord. 2000;30:205–23.
Andrews S. FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
‘t Hoen PAC, Friedländer MR, Almlöf J, Sammeth M, Pulyakhina I, Anvar SY, et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31:1015–22. https://doi.org/10.1038/nbt.2702.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17(1):10–12.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. https://doi.org/10.1186/gb-2013-14-4-r36.
DeLuca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire M-D, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–2. https://doi.org/10.1093/bioinformatics/bts196.
Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. https://doi.org/10.1093/bioinformatics/btt656.
Liao Y, Smyth GK, Shi W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. https://doi.org/10.1093/nar/gkt214.
Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KCC, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–7. https://doi.org/10.1038/ng2109.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. https://doi.org/10.1093/bioinformatics/btp616.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Fischbach GD, Lord C. The Simons simplex collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–5. https://doi.org/10.1016/j.neuron.2010.10.006.
Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. 2013;4:36. https://doi.org/10.1186/2040-2392-4-36.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–40. https://doi.org/10.1093/bioinformatics/btr260.
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. https://doi.org/10.1093/nar/gkr988.
Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–7. https://doi.org/10.1093/nar/gkt1102.
Gene Ontology Consortium. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:258D–261. https://doi.org/10.1093/nar/gkh036.
Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. https://doi.org/10.1186/1471-2105-11-587.
Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:e161. https://doi.org/10.1371/journal.pgen.0030161.
Delhomme N, Padioleau I, Furlong EE, Steinmetz LM. easyRNASeq: a bioconductor package for processing RNA-Seq data. Bioinformatics. 2012;28:2532–3. https://doi.org/10.1093/bioinformatics/bts477.
Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–8. https://doi.org/10.1093/bioinformatics/bts545.
Poole W, Gibbs DL, Shmulevich I, Bernard B, Knijnenburg TA. Combining dependent P- values with an empirical adaptation of Brown’s method. Bioinformatics. 2016;32:i430–6. https://doi.org/10.1093/bioinformatics/btw438.
Werling DM, Parikshak NN, Geschwind DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun. 2016;7:10717. https://doi.org/10.1038/ncomms10717.
Tylee DS, Espinoza AJ, Hess JL, Tahir MA, McCoy SY, Rim JK, et al. RNA sequencing of transformed lymphoblastoid cells from siblings discordant for autism spectrum disorders reveals transcriptomic and functional alterations: evidence for sex-specific effects. Autism Res. 2017;10:439–55. https://doi.org/10.1002/aur.1679.
Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, et al. Cell type–specific gene expression differences in complex tissues. Nat Methods. 2010;7:287–9. https://doi.org/10.1038/nmeth.1439.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. https://doi.org/10.1038/nmeth.3337.
Wijnands KPJ, Obermann-Borst SA, Steegers-Theunissen RPM. Early life lipid profile and metabolic programming in very young children. Nutr Metab Cardiovasc Dis. 2015;25:608–14. https://doi.org/10.1016/j.numecd.2015.02.010.
Wang L, Oh WK, Zhu J. Disease-specific classification using deconvoluted whole blood gene expression. Sci Rep. 2016;6:32976. https://doi.org/10.1038/srep32976.
Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. https://doi.org/10.1186/s13059-017-1349-1.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:1–9. https://doi.org/10.1186/gb-2014-15-2-r31.
Holbrook JD, Huang R-C, Barton SJ, Saffery R, Lillycrop KA. Is cellular heterogeneity merely a confounder to be removed from epigenome-wide association studies? Epigenomics. 2017;9:1143–50. https://doi.org/10.2217/epi-2017-0032.
Houseman EA, Kim S, Kelsey KT, Wiencke JK. DNA methylation in whole blood: uses and challenges. Curr Environ Heal Reports. 2015;2:145–54. https://doi.org/10.1007/s40572-015-0050-3.
Houseman E, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. https://doi.org/10.1186/1471-2105-13-86.
Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics. 2016;17:259. https://doi.org/10.1186/s12859-016-1140-4.
Enstrom AM, Lit L, Onore CE, Gregg JP, Hansen RL, Pessah IN, et al. Altered gene expression and function of peripheral blood natural killer cells in children with autism. Brain Behav Immun. 2009;23:124–33. https://doi.org/10.1016/j.bbi.2008.08.001.
Ashwood P, Corbett BA, Kantor A, Schulman H, Van de Water J, Amaral DG. In search of cellular immunophenotypes in the blood of children with autism. PLoS One. 2011;6:e19299. https://doi.org/10.1371/journal.pone.0019299.
Gregg JP, Lit L, Baron CA, Hertz-Picciotto I, Walker W, Davis RA, et al. Gene expression changes in children with autism. Genomics. 2008;91:22–9.
Chien W-H, Gau SS-F, Chen C-H, Tsai W-C, Wu Y-Y, Chen P-H, et al. Increased gene expression of FOXP1 in patients with autism spectrum disorders. Mol Autism. 2013;4:23. https://doi.org/10.1186/2040-2392-4-23.
Hill WD, Marioni RE, Maghzian O, Ritchie SJ, Hagenaars SP, McIntosh AM, et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol Psychiatry. 2018; https://doi.org/10.1038/s41380-017-0001-5.
Davies G, Lam M, Harris SE, Trampush JW, Luciano M, Hill WD, et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat Commun. 2018;9:2098. https://doi.org/10.1038/s41467-018-04362-x.
Ansel A, Rosenzweig JP, Zisman PD, Melamed M, Gesundheit B. Variation in gene expression in autism spectrum disorders: an extensive review of transcriptomic studies. Front Neurosci. 2017;10:601. https://doi.org/10.3389/fnins.2016.00601.
Prasad A, Merico D, Thiruvahindrapuram B, Wei J, Lionel AC, Sato D, et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3. 2012;2:1665–85. https://doi.org/10.1534/g3.112.004689.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84:524–33. https://doi.org/10.1016/j.ajhg.2009.03.010.
Atladóttir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW, et al. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics. 2009;124:687–94. https://doi.org/10.1542/peds.2008-2445.
Keil A, Daniels JL, Forssen U, Hultman C, Cnattingius S, Söderberg KC, et al. Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology. 2010;21:805–8. https://doi.org/10.1097/EDE.0b013e3181f26e3f.
Gupta S, Aggarwal S, Rashanravan B, Lee T. Th1- and Th2-like cytokines in CD4+ and CD8+ T cells in autism. J Neuroimmunol. 1998;85:106–9. https://doi.org/10.1016/S0165-5728(98)00021-6.
Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68:368–76. https://doi.org/10.1016/j.biopsych.2010.05.024.
Fatemi SH, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, et al. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol. 2002;22:25–33.
Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi:23/1/297 [pii]
Alter MD, Kharkar R, Ramsey KE, Craig DW, Melmed RD, Grebe TA, et al. Autism and increased paternal age related changes in global levels of gene expression regulation. PLoS One. 2011;6:e16715. https://doi.org/10.1371/journal.pone.0016715.
Drexhage RC, Knijff EM, Padmos RC, van der Heul-Nieuwenhuijzen L, Beumer W, Versnel MA, et al. The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev Neurother. 2010;10:59–76. https://doi.org/10.1586/ern.09.144.
Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol. 2001;120:170–9. https://doi.org/10.1016/S0165-5728(01)00421-0.
Tsai PC, Bell JT. Power and sample size estimation for epigenome-wide association scans to detect differential DNA methylation. Int J Epidemiol. 2015;44:1429–41.
Saffari A, Silver MJ, Zavattari P, Moi L, Columbano A, Meaburn EL, et al. Estimation of a significance threshold for epigenome-wide association studies. Genet Epidemiol. 2018;42(1):20–33.
Tylee DS, Kawaguchi DM, Glatt SJ. On the outside, looking in: a review and evaluation of the comparability of blood and brain “-omes.”. Am J Med Genet Part B Neuropsychiatr Genet. 2013;162B:595–603.
Hayashi-Takagi A, Vawter MP, Iwamoto K. Peripheral biomarkers revisited: integrative profiling of peripheral samples for psychiatric research. Biol Psychiatry. 2014;75:920–8.
Abdellaoui A, Ehli EA, Hottenga J-J, Weber Z, Mbarek H, Willemsen G, et al. CNV concordance in 1,097 MZ twin pairs. Twin Res Hum Genet. 2015;18:1–12. https://doi.org/10.1017/thg.2014.86.
Kato T, Iwamoto K, Kakiuchi C, Kuratomi G, Okazaki Y. Genetic or epigenetic difference causing discordance between monozygotic twins as a clue to molecular basis of mental disorders. Mol Psychiatry. 2005;10:622–30. https://doi.org/10.1038/sj.mp.4001662.
Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, GuhaThakurta D, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37:710–7. https://doi.org/10.1038/ng1589.
Talebizadeh Z, Aldenderfer R, Wen CX. A proof-of-concept study. Psychiatr Genet. 2014;24:1–9. https://doi.org/10.1097/YPG.0b013e3283635526.
Talebizadeh Z. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J Med Genet. 2005;43:e21. https://doi.org/10.1136/jmg.2005.036897.
Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the Neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–4. https://doi.org/10.1016/j.ajhg.2007.09.015.
Veltman MWM, Craig EE, Bolton PF. Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet. 2005;15:243–54. https://doi.org/10.1097/00041444-200512000-00006.
Acknowledgements
N/A
Funding
We gratefully acknowledge the ongoing contribution of the participants in the Twins Early Development Study (TEDS) and their families. TEDS is supported by a program grant to RP from the UK Medical Research Council [MR/M021475/1 and previously G0901245]. The study was funded by a research grant from the Royal Society awarded to ELM. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
JM, AR and ELM conceived of and designed the study; LS, JM and CCYW generated and analysed the methylation dataset; MA and EN generated the RNA-seq dataset; ELM and FD advised on the analysis and interpretation of the data; AS performed all analyses described and drafted the manuscript. All authors contributed to revisions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for TEDS was provided by the Institute of Psychiatry, Psychology and Neuroscience (IoPPN) ethics committee (reference number 05/Q0706/228). Ethical approval for the study described was provided by the Psychiatry, Nursing & Midwifery Research Ethics sub-committee (PNM/08/9-58). Parental consent and child assent was obtained for all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplementary results tables and figures. (DOCX 1562 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Saffari, A., Arno, M., Nasser, E. et al. RNA sequencing of identical twins discordant for autism reveals blood-based signatures implicating immune and transcriptional dysregulation. Molecular Autism 10, 38 (2019). https://doi.org/10.1186/s13229-019-0285-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13229-019-0285-1