
Abstract
Background
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes and nemaline myopathy are two rare genetic conditions. We report the first case reported in world literature with coexistence of both these rare disorders.
Case presentation
A 11-year-old previously healthy Sri Lankan male child, product of a nonconsanguineous marriage with normal development presented with acute onset short lasting recurring episodes of right-sided eye deviation with impaired consciousness. In between episodes he regained consciousness. Family history revealed a similar presentation in the mother at 36 years of age. Examination was significant for short stature and proximal upper and lower limb weakness. His plasma and cerebrospinal fluid lactate were elevated. Magnetic resonance imaging brain had evidence of an acute infarction in the right occipital territory. Sanger sequencing for common mitochondrial variants of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes confirmed this diagnosis. Whole exome sequencing revealed pathogenic compound heterozygous variants in NEB gene implicating in coexisting nemaline myopathy. Acute presentation was managed with supportive care, antiepileptics, and mitochondrial supplementation. Currently he is stable on daily supplementation of arginine and limb-strengthening physiotherapy. He is being monitored closely clinically and with serum lactate level.
Conclusion
Genetic diseases are rare. Coexistence of two genetic conditions is even rarer. Genetic confirmation of diagnosis is imperative for prediction of complications, accurate management, and genetic counseling.
Similar content being viewed by others


Background
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), which is characterized by encephalomyopathy, lactic acidosis, and stroke-like episodes is a maternally inherited mitochondrial disorder that was first described in 1984 [1]. It is a rare disease with a prevalence of 0.2:100,000 was described in Japan [2]. Mutation carrier state is reported to be 16–18:100,000 in Finland [3, 4], while a higher prevalence of 236:100,000 was reported in Australia [5].
The molecular basis of MELAS was first delineated in 1990 with the discovery of the most common pathogenic variant associated with the condition in MT-TL1 gene m.3243A > G [6, 7]. MELAS study group committee in Japan published a set of diagnostic criteria by which the diagnosis is considered definitive with at least two category A criteria (headaches with vomiting, seizures, hemiplegia, cortical blindness, and acute focal lesions in neuroimaging) and two category B criteria [high plasma or cerebrospinal fluid (CSF) lactate, mitochondrial abnormalities in muscle biopsy, and a MELAS related gene mutation] [2].
Nemaline myopathy is a rare genetic muscle disease leading to skeletal muscle weakness with an incidence of 0.02 per 1000 live births [8].
We report a case of definitive diagnosis of MELAS according to above criteria with positive family history and heteroplasmy for the MT-TL1 gene mutation presenting with seizures and focal lesion on MRI and compound heterozygosity for nemaline myopathy genetic variants. This is the first case in literature with co-occurrence of MELAS and nemaline myopathy.
Case presentation
An 11-year-old previously healthy Sri Lankan male child, product of a non-consanguineous marriage with normal development presented with acute onset right-sided eye deviation with impaired consciousness.; six similar episodes had occurred, each lasting for around 3 minutes in duration over a period of 1 hour with regaining of consciousness in between episodes. He did not have fever, abnormal movements, tongue biting, incontinence, limb weakness, or recent history of trauma. Preceding the event, he had strenuous physical activity for 4 hours at school with reduced oral intake as well.
His birth history was complicated as he was born prematurely (POA 29 weeks) in a twin pregnancy (dichorionic diamniotic), in which the other fetus had miscarried in the first trimester. His delivery was via emergency lower segment cesarean section due to antepartum hemorrhage. Birth weight was 900 g and he was ventilated initially invasively and then non-invasively for 6 weeks post-delivery. He developed retinopathy of prematurity and was offered laser therapy. However, up until this admission he was well and was following education in a mainstream school with good performance.
This child’s family history was significant for a similar episode that had developed in his mother at 36 years of age, which was 6 months before his presentation. She developed an acute episode of impaired consciousness associated with abnormal eye deviation that lasted for 15 minutes. Magnetic resonance imaging (MRI) of her brain revealed a right-sided occipital lobe infarction. However. with supportive care she recovered within 2 weeks without any residual neurological deficit. She did not have any obvious vascular risk factors and the etiology for her presentation was still under investigation at the time of this child’s presentation.
Examination revealed him to be dehydrated with Glasgow Coma Scale (GCS) 14/15 and normally reactive pupils. Vital parameters were stable with a blood pressure of 110/70 mmHg and capillary blood sugar was 115 mg/dl. Further examination after initial stabilization revealed him to be short statured (more than two standard deviations below the mean height for his age and sex according to standardized Sri Lankan growth chart), and there was significant muscle tenderness accompanied with proximal limb weakness [proximal limb power—3/5 according to Medical Research Council (MRC) scale]. The rest of the neurological examination was unremarkable, including fundi.
Investigations revealed his plasma and CSF lactate to be elevated with a value of 6.5 mmol/l (0.5–2.3 mmol/l) and 4.25 mmol/l (0.5–2.3 mmol/l), respectively, with metabolic acidosis in arterial blood gas analysis, which was highly indicative of mitochondrial disease. There was neutrophil leukocytosis with white cell count of 18,000 × 103/μl, but inflammatory markers and bacterial cultures in blood and CSF were negative. Creatine kinase level was significantly elevated to 4213 units/l (normal range 55–170 units/l).
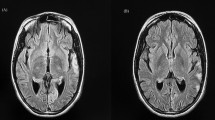
Electroencephalogram (EEG) showed evidence of background slowing suggestive of encephalopathy and right occipito-temporal periodic lateralized epileptiform discharges reflecting focal cortical involvement. MRI brain revealed evidence of acute posterior cerebral territory infarction (Fig. 1). Nerve conduction and electromyogram did not reveal evidence of myopathy. Muscle biopsy was not performed considering the invasiveness of the procedure and low therapeutic benefit, and 2D echocardiogram and hearing assessment were normal.

Magnetic resonance imaging brain of the patient depicting acute right posterior cerebral territory infarction
MELAS was considered in this clinical context supported by investigations and targeted genetic analysis carried out using Sanger sequencing method for common mitochondrial mutations in MT-TL1 and MT-ND5 genes implicating in MELAS. He was found to be harboring a pathogenic variant in MT-TL1 gene m.3243A > G, which confirmed MELAS genetically. He was heteroplasmic for the mutation (Fig. 2).

Electrophorogram of the patient produced via Sanger sequencing depicting m.3243A > G variant in MT-TL1 gene with heteroplasmy
Whole exome sequencing was performed for the genomic DNA extracted from the peripheral venous blood obtained from the affected individual by IlluminaR NovaSeqR 6000 Next Generation Sequencer on exon targets isolated by capture using the SureSelectXT R Human (Mouse) All Exon V6 5190-8864 kit. An in-house bioinformatics pipeline was used for the genetic analysis. Generated paired end sequences were mapped to the published human genome reference sequence build UCSC hg19/GRCh37 using BWA-MEM. Variant calling was performed using Genome Analysis Tool Kit (GATK) after converting generated SAM to BAM files, removing duplicates, and indexing the BAM files using Picard tools. The generated variants were annotated using SNP-EFF, which included nucleotide and amino acid annotations, Clivar, population frequencies (1000 Genomes, Exome sequencing project and internal databases), amino acid conservation scores, and in silico prediction tools. A virtual phenotype-driven gene panel was created and filtered out genetic variants were classified according to the standard American College of Medical Genetics and Genomics (ACMG) guidelines.
Whole exome sequencing was done, which revealed him to be compound heterozygous for pathogenic variants implicated in nemaline myopathy in NEB gene. Two pathogenic variants were detected in the two alleles of NEB gene at c.20089G > A and c.11300A > G. Sanger sequencing revealed his parents to be heterozygous for the nemaline myopathy pathogenic variants he was harboring.
His asymptomatic 8-year-old female sibling and symptomatic mother were screened with Sanger sequencing for MT-TL1 pathogenic variant, which revealed them to be carrying the mutation and they were homoplasmic for the mutation as erll (Fig. 3).

Electrophorograms produced by Sanger sequencing of the patient’s asymptomatic 8-year-old female sibling (A) and symptomatic mother (B) depicting the same genetic variant m.3243A > G in MT-TL1 gene with homoplasmy
In the acute setting his seizures were managed with intravenous antiepileptic medications including midazolam and levetiracetam, which successfully abated the seizures. He was kept under continuous EEG monitoring which did not show evidence of electrical seizures or non-convulsive status. Considering the possibility of mitochondrial disease, he was started on mitochondrial supplementation coenzyme Q, creatine, carnitine, vitamins E and K, and arginine, which improved his acute condition. Further supportive therapies were undertaken such as optimizing his hydration and nutrition. His lactate level and creatinine kinase levels reduced gradually and are maintained at high normal range. Subsequently he developed similar acute episodes that were managed with intravenous hydration, increased calorie intake, and high-dose arginine.
Though there was no residual neurological deficit, there was a gradual decline in his educational (formally assessed IQ level, 75) and physical performance with moderate degree proximal limb muscle weakness. He was experiencing a persistent low mood with irritability and aggression, however, there were no psychotic symptoms. Currently he is on arginine supplementation daily and being monitored closely clinically and with serum lactate level. His usual stroke-like episodes are heralded by brief seizures with visual symptoms and are aborted by increasing clobazam and arginine doses combined with rest, hydration, and calorie supplementation.
Discussion
MELAS is a disease with multiorgan involvement. Typically, patients present in childhood (65–76%) or before 20 years of age [2, 9, 10]. Presentation before 2 years of age (5–8%) and after 40 years (1–6%) is rare [2, 9,10,11].
The clinical presentation could be quite variable in MELAS. Summary of the clinical manifestations of MELAS are presented in Tables 1, 2.
Apart from the MT-TL1 gene, several other mitochondrial genes could be implicated in MELAS, but they have a low prevalence [11]. Table 3 presents the implicated genes and their percentage attribution to MELAS [11].
The m.3243A > G mutation results in impaired mitochondrial translation that leads to decreased mitochondrial protein synthesis, hence mitochondrial energy production is affected, leading to multiorgan dysfunction [12, 13]. This energy deficiency stimulates mitochondrial proliferation, which causes angiopathy due to proliferation of smooth muscles and endothelial cells in blood vessels, leading to impaired perfusion in the microvasculature. This phenomenon has a significant contribution to stroke-like episodes in MELAS [10, 14, 15]. Nitric oxide (NO) deficiency of multifactorial origin is the other main etiology for the complications observed in MELAS [16,17,18].
The complications encountered by this patient were stroke-like episodes, generalized muscle weakness, and exercise intolerability and lactic acidemia, which are all explainable with these mechanisms. It is believed that these stroke-like episodes occur due to angiopathy and NO deficiency, leading to cerebral perfusion dysfunction [10, 14,15,16,17,18,19,20]. Neuroimaging in these patients reveal ischemic insults not corresponding to classic vascular distribution and are asymmetric, involving predominantly the temporal, parietal, and occipital lobes, with restriction to cortical areas or subcortical white matter [10, 21]. The patient in this case also had similar neuroimaging findings.
Muscle weakness and exercise intolerance could be explained by the deficient energy production due to mitochondrial dysfunction. Lactic acidemia is a result of dysfunctional mitochondria leading to inadequate oxidization of glucose, hence accumulation of pyruvate and shunting of pyruvate to lactate [10] and hypoperfusion due to microangiopathy and NO deficiency.
Extremely variable phenotypes could be seen with m.3243A > G mutation ranging from severe disease in only 10% of individuals to another 10% being asymptomatic carriers. In between these two ends, single-organ to multiorgan disease could exist with variable phenotypes, sometimes overlapping with other mitochondrial syndromes [10, 22, 23].
Currently there is no specific treatment for MELAS and it is being managed symptomatically in a multidisciplinary approach since it is a multiorgan disease. On the basis of limited clinical trials, several supplementations, including antioxidants and cofactors, are being used in MELAS [24]. This patient’s acute presentation was managed with intravenous arginine and he was on oral arginine daily, which is being supported by unblinded studies to be beneficial in acute management and prevention of stroke-like episodes [20, 25]. Increased availability of nitric oxide leading to intracerebral vasodilatation and blood flow is the proposed mechanism of therapeutic effect of arginine in managing stroke-like episodes [16, 26].
Coenzyme Q provides a protective antioxidant effect, which is beneficial in muscle weakness, fatigability, and reducing lactate level [27]. Creatine is an essential phosphate donor for adenosine triphosphate (ATP) regeneration in muscle and brain. Therefore, it increases the strength of high-intensity anaerobic and aerobic activities in individuals with mitochondrial diseases [28]. Carnitine is needed for long-chain fatty acid transportation to the mitochondria, where it undergoes β-oxidation. Secondary carnitine deficiency has been reported in patients with MELAS. Therefore, carnitine supplementation can potentially enhance β-oxidation and replenish the intracellular pools of coenzyme A [29].
This patient has genetically proven nemaline myopathy. It is a genetically heterogeneous condition with nebulin (NEB) gene variants accounting for 50% of cases [30]. However, lack of evidence on a muscle biopsy supporting nemaline myopathy is a limitation in this case. Creatine kinase level was highly elevated (25 times the upper limit of normal) in this patient in the acute setting, which is not a usual finding in MELAS. Therefore, this could have been contributed to coexisting nemaline myopathy. Respiratory muscle weakness is a well-reported complication of nemaline myopathy [30]. Therefore, diagnosis of this genetic condition is extremely important in anticipation and management of future complications of this patient.
The genetic confirmation of both the conditions in this patient ended the diagnostic odyssey not only for him, but also for his whole family. This enabled accurate management and proper genetic counseling for them.
There were no reported cases of both these rare genetic conditions, MELAS and nemaline myopathy, occurring in the same patient to the best of our knowledge. This case illustrates the importance of genetic testing and consideration of rare conditions in the appropriate clinical context.
Conclusion
MELAS is a mitochondrial disease with heterogeneous presentation. Currently there is no specific disease-modifying therapy. However, several therapeutic modalities have shown to be effective in improving symptoms and preventing stroke-like episodes. Rare, unexpected dual pathologies can coexist in the same patient, altering the clinical presentation. Though there is no treatment to offer for nemaline myopathy currently, its diagnosis is important for prediction of future complications, accurate supportive management, and proper genetic counseling.
Data availability
The data generated and analyzed are not publicly available but are available from the corresponding author on reasonable request.
References
Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16(4):481–8.
Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, Kakuma T, Koga Y, Taro Matsuoka for MELAS Study Group in Japan. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta BBA Gen Subj. 2012;1820(5):619–24.
Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Kärppä M, Majamaa-Voltti KA, Rusanen H, Sorri M, Peuhkurinen KJ, Hassinen IE. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Human Genet. 1998;63(2):447–54.
Uusimaa J, Moilanen JS, Vainionpää L, Tapanainen P, Lindholm P, Nuutinen M, Löppönen T, Mäki-Torkko E, Rantala H, Majamaa K. Prevalence, segregation, and phenotype of the mitochondrial DNA 3243A> G mutation in children. Ann Neurol. 2007;62(3):278–87.
Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, Mitchell P, Sue CM. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7(3):230–3.
Goto YI, Nonaka I, Horai S. A mutation in the tRNALeu (UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–3.
Kobayashi Y, Momoi MY, Tominaga K, Momoi T, Nihei K, Yanagisawa M, Kagawa Y, Ohta S. A point mutation in the mitochondrial tRNALeu (UUR) gene in MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes). Biochem Biophys Res Commun. 1990;173(3):816–22.
Agrawal PB, Strickland CD, Midgett C, Morales A, Newburger DE, Poulos MA, Tomczak KK, Ryan MM, Iannaccone ST, Crawford TO, Laing NG. Heterogeneity of nemaline myopathy cases with skeletal muscle α-actin gene mutations. Ann Neurol. 2004;56(1):86–96.
Hirano M, Pavlakis SG. Topical review: mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994;9(1):4–13.
Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008ct;1142(1):133–58.
El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [Updated 2018 Nov 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022.
King MP, Koga Y, Davidson M, Schon EA. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA (Leu (UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12(2):480–90.
Chomyn A, Enriquez JA, Micol V, Fernandez-Silva P, Attardi G. The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu (UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J Biol Chem. 2000;275(25):19198–209.
Hasegawa H, Matsuoka T, Goto YI, Nonaka I. Strongly succinate dehydrogenase–reactive blood vessels in muscles from patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Ann Neurol. 1991;29(6):601–5.
Goto YI, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M, Nonaka I. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3):545.
El-Hattab AW, Hsu JW, Emrick LT, Wong LJ, Craigen WJ, Jahoor F, Scaglia F. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab. 2012;105(4):607–14.
El-Hattab AW, Emrick LT, Craigen WJ, Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol Genet Metab. 2012;107(3):247–52.
El-Hattab AW, Emrick LT, Chanprasert S, Craigen WJ, Scaglia F. Mitochondria: role of citrulline and arginine supplementation in MELAS syndrome. Int J Biochem Cell Biol. 2014;1(48):85–91.
Naini A, Kaufmann P, Shanske S, Engelstad K, Darryl C, Schon EA. Hypocitrullinemia in patients with MELAS: an insight into the “MELAS paradox.” J Neurol Sci. 2005;15(229):187–93.
Koga YM, Akita YM, Nishioka J, Yatsuga S, Povalko N, Tanabe YM, Fujimoto S, Matsuishi T. l-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64(4):710–2.
Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, DiMauro S, Rowland LP. MELAS: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2(2):125–35.
Koga Y, Akita Y, Takane N, Sato Y, Kato H. Heterogeneous presentation in A3243G mutation in the mitochondrial tRNALeu (UUR) gene. Arch Dis Child. 2000;82(5):407–11.
Fabrizi GM, Cardaioli E, Grieco G, Cavallaro T, Malandrini A, Manneschi L, Dotti MT, Federico A, Guazzi G. The A to G transition at nt 3243 of the mitochondrial tRNALeu (UUR) may cause an MERRF syndrome. J Neurol Neurosurg Psychiatry. 1996;61(1):47–51.
Scaglia F, Northrop JL. The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome. CNS Drugs. 2006;20(6):443–64.
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Katayama K, Matsuishi T. MELAS and l-arginine therapy. Mitochondrion. 2007;7(1–2):133–9.
Koga Y, Akita Y, Junko N, Yatsuga S, Povalko N, Fukiyama R, Ishii M, Matsuishi T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology. 2006;66(11):1766–9.
Chen RS, Chin-Chang H, Chu NS. Coenzyme Q10 treatment in mitochondrial encephalomyopathies. Eur Neurol. 1997;37(4):212.
Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20(12):1502–9.
Scaglia F, Northrop JL. The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: a review of treatment options. CNS Drugs. 2006;20:443–64.
Yin X, Pu CQ, Wang Q, Liu JX, Mao YL. Clinical and pathological features of patients with nemaline myopathy. Mol Med Rep. 2014;10(1):175–82.
Acknowledgements
None.
Funding
The authors declare that no funds, grants, or other support were received during preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by KG. Genetic screening, analysis and article revisions were performed by SP. Genetic screening and analysis were supervised by VD. The first draft of the manuscript was written by KG and critically appraised and revised by PR. All authors commented, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for the study was obtained from the Ethics Review Committee, Faculty of Medicine, University of Colombo, Sri Lanka [EC/19/127]. Informed written consent was obtained from the patient, his legal guardian, and other participants who underwent genetic testing for the participation in the study.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gunawardena, K., Praveenan, S., Dissanayake, V.H.W. et al. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes with coexisting nemaline myopathy: a case report. J Med Case Reports 18, 420 (2024). https://doi.org/10.1186/s13256-024-04723-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13256-024-04723-9