
Abstract
Background
Cardiovascular progenitor cells (CPCs) derived from human embryonic stem cells (hESCs) are considered valuable cell sources for investigating cardiovascular physiology in vitro. Meeting the diverse needs of this application requires the large-scale production of CPCs in an in vitro environment. This study aimed to use an effective culture system utilizing signaling factors for the large-scale expansion of hESC-derived CPCs with the potential to differentiate into functional cardiac lineage cells.
Methods and results
Initially, CPCs were generated from hESCs using a 4-day differentiation protocol with a combination of four small molecules (CHIR99021, IWP2, SB-431542, and purmorphamine). These CPCs were then expanded and maintained in a medium containing three factors (bFGF, CHIR, and A83-01), resulting in a > 6,000-fold increase after 8 passages. These CPCs were successfully cryopreserved for an extended period in late passages. The expanded CPCs maintained their gene and protein expression signatures as well as their differentiation capacity through eight passages. Additionally, these CPCs could differentiate into four types of cardiac lineage cells: cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts, demonstrating appropriate functionality. Furthermore, the coculture of these CPC-derived cardiovascular lineage cells in rat tail collagen resulted in cardiac microtissue formation, highlighting the potential of this 3D platform for studying cardiovascular physiology in vitro.
Conclusion
In conclusion, expandable hESC-derived CPCs demonstrated the ability to self-renewal and differentiation into functional cardiovascular lineage cells consistently across passages, which may apply as potential cell sources for in vitro cardiovascular studies.
Graphical Abstract

Similar content being viewed by others


Background
Cardiovascular diseases (CVDs) stay remained at the top of global mortality and morbidity for a long time, with a particularly increasing prevalence in recent decades [1]. Despite advances in the treatment of CVDs, available strategies have not effectively restored the injured myocardium. In addition, the innate regenerative capability of cardiac tissue is limited, which could be associated with the terminally differentiated cardiomyocytes that stay in the G0/G1 phase of the cell cycle lifelong [2]. In this regard, recent studies have focused on cell therapy to restore the cardiomyocyte population of injured myocardium as a novel and promising therapeutic approach [3]. Among all candidate cell sources, cardiac precursor cells as well as their derivatives, such as cardiovascular progenitor cells (CPCs) and CPC-derived cardiomyocytes, have gained growing attention for cardiac cell therapy [4]. In addition to therapeutic applications, CPC-derived cardiomyocytes could be employed for in vitro drug testing, cardiotoxicity assessments, and disease modeling, which provides incredible opportunity and great advancement in the field of cardiovascular research [5, 6].
CPCs are a heterogeneous population of clonogenic, self-renewing, and multipotent cells with the capability of differentiating into various cardiac lineage cells, including cardiomyocytes (CMs), endothelial cells (ECs), cardiac fibroblasts (CFs), and smooth muscle cells (SMCs), both in vitro and in vivo [7, 8]. To date, multiple CPC populations have been identified, which are characterized by the expression of different surface markers and/or transcription factors, such as NKX2.5. The precise mechanisms by which CPCs are differentiated into cardiac and vascular lineages are still being investigated; however, some growth factors and the related signaling pathways are known to be contributed [4, 9,10,11]. Using this knowledge, protocols for in vitro differentiation of human pluripotent stem cells-derived CPCs are developing [4]. However, due to the innate proliferative and multipotent characteristics of CPCs, the ultimate goal is now to achieve an effective culture strategy with the capability for in vitro large-scale expansion of CPCs while preserving the differentiation capacity into various cardiovascular lineages. This could provide an unprecedented opportunity for cardiac regenerative approaches as well as developmental studies [12,13,14]. So far, some research groups have suggested protocols and technologies for the generation of expandable CPCs from pluripotent cells, including human embryonic stem cells (hESCs) and/or human-induced pluripotent stem cells (hiPSCs) [15, 16]. However, this remains a challenging endeavor. For instance, in a study performed by Cao et al., the homogeneous cardiovascular progenitor cells were generated and expanded from human pluripotent stem cells by using a serum-free culture medium supplemented with a combination of three small molecules. These cells showed over 107-fold expansion, retained their molecular characteristics as well as the potential to differentiate into CM, EC, and SMC [16]. Likewise, in the previous study, we developed a culture system for generation of expandable cardiogenic mesodermal cells (early CPCs) with differentiation capacity into CMs, ECs, and SMCs [15]. However, the functionality of CPC-derived cardiac linage cells was not assessed in the previous studies. Hence, the findings suggested an urgent need for the development of efficient differentiation protocols in order to obtain functional cardiovascular lineage cells. Moreover, previous studies have not investigated the differentiation of cardiac fibroblast from CPC, despite the crucial role of these fibroblasts in cardiac disorders. Therefore, this study took the advantage of our previous defined, costly, concise, and universal protocol for the extended-term expansion of hPSC-derived CPCs, while concurrently focusing on the retention of their inherent capacity for differentiation into various functional cardiac lineage cells by establishing highly efficient protocols. By doing so, the expandability and preservation of hESC-derived CPCs were supported up to 8 passages while maintaining their morphological characteristics, molecular profile, and the ability to differentiate into functional CMs, ECs, CFs, and SMCs in vitro across subsequent passages. Furthermore, we used the resulting cells for the formation of a microtissue by co-culturing the CPC-derived CMs, ECs, CFs, and SMCs at passages 0 and 8 (P0 and P8). The constructed cardiac microtissue showed the presence and distribution of cardiac lineage cells as well as excitability. Altogether, our findings may offer a novel strategy for the generation of expandable CPCs with preservation of differentiation capability into functional cardiac linage cells and maintenance in freeze-thaw cycles. This represents a crucial step towards commercialization, advancements in disease modeling and cardiotoxicity testing in in vitro studies.
Methods
Generation of human embryonic stem cells-derived cardiovascular progenitor cells (hESC-CPC)
Human embryonic stem cells (hESC), Royan H6 (RH6) line which was established and characterized as previously reported [17], was received from Royan Stem Cell Bank and expanded in adherent culture according to a previously described protocol [18]. In order to prompt hESC for cardiogenic differentiation, the expansion system was changed to static suspension culture, where hESC were expanded as previously described [18]. Briefly, the 5-day-old hESC spheroids were subjected to cardiogenic differentiation using a cocktail of small molecules including CHIR (Stemgent, 04-0004-10), IWP2 (Tocris Bioscience, 3533), SB-431,542 (Cayman, 13031), and purmorphamine (Pur) (Stemgent, 04–0009), according to a previous study [18]. A detailed description of the differentiation method can be found in the supplemental information.
Culture and expansion of hESC-CPC
The 4-day-old differentiated hESC-CPC spheroids were dissociated into single cells and cultured on Matrigel (Sigma-Aldrich, E1270)-coated plates with specific expansion medium (CPCxm) composed of a basal medium and fresh small molecules; bFGF, CHIR, and A83-01 (Stemgent, 04–0014). Furthermore, hESC-CPC was passaged every 72 hs and expanded for 8 passages. Characterization of hESC-CPC was performed at passages 0 (P0), P4, and P8 using flow cytometry and immunofluorescence staining against CPC-specific markers. For a detailed description, see supplemental information. Furthermore, the freeze-thaw procedure of hESC-CPC was assessed, and the resulting cells were characterized, for which details can be found in supplemental information. Population Doubling Time (PDT) and expansion fold assays were evaluated as described previously [15], whose details are described in supplemental information.
Differentiation of hESC-CPC towards cardiovascular lineage cells
Differentiation of hESC-CPC into cardiomyocytes, endothelial and smooth muscle cells
To generate cardiovascular lineage cells, hESC-CPCs (P0 and P8) were seeded on Matrigel-coated plates at a density of 3 × 104 cells/cm2. Induction towards cardiomyocytes (CM) was achieved by using CM differentiation medium (CMm) composed of RPMI1640 (Gibco, 52400-041) supplemented with B27 without insulin (Gibco, A18956-01), 2 mM L-glutamine, 1% NEAA, and 0.1 mM β-mercaptoethanol, as well as growth factor and small molecules including BMP4 (10 ng/ml) (314-BP-010/CF R&D) and IWP2 (5 µM) (Tocris). hESC-CPCs were differentiated into endothelial cells (EC) by using EC differentiation medium (ECm) composed of DMEM/F12 supplemented with 2% B-27 without vitamin A, 2 mM L-glutamine, 1% NEAA, and 0.1 mM β-mercaptoethanol, as well as growth factors VEGF (50 ng/ml) (Royan Biotech, PRP-1109) and bFGF (10 ng/ml). Smooth muscle cell differentiation required the culture of hESC-CPC in SMC differentiation medium (SMCm) composed of DMEM/F12 supplemented with 2% B-27 without vitamin A, 2 mM L-glutamine, 1% NEAA, and 0.1 mM β-mercaptoethanol, as well as growth factors of PDGF-BB (10 ng/ml) (Royan Biotech, RP-1111) and TGF-β (2 ng/ml) (Fitzgerald 30R-AT072). hESC-CPCs were differentiated into cardiac fibroblasts (CF) using a two-step protocol, which will be explained in a separate section.
For EC and SMC differentiation, hESC-CPCs were cultured in ECm and SMCm for 12 days, and the medium was renewed every 2 days. For differentiation to CM, hESC-CPCs were cultured in CMm for 3 days and subsequently in CMm without growth factors and with B27 complete (Thermo Fisher Scientific, 17504044) for 9 days.
Differentiation of hESC-CPC into cardiac fibroblasts
Regarding differentiation of CFs from hESC-derived CPCs (P0 and P8), we used the following protocol, which obtains CFs from epicardial origin. Initially, hESC-CPCs were seeded on Matrigel-coated plates at a density of 3 × 104 cells/cm2 and maintained in a basal differentiation medium (CFbm1) consisting of RPMI1640 supplemented with B27 without insulin, 2 mM L-glutamine, 1% NEAA, and 0.1 mM β-mercaptoethanol for 24 h in order to adapt to 2D culture. After this time, the CFbm1 was renewed and supplemented with a concentration of 3 µM of the small molecule CHIR for 48 h. Subsequently, the medium was replaced by CFbm1, and the culture medium was exchanged every 24 hs. After 48 h, epicardial-like cells were obtained and subjected to CF differentiation. To do so, the medium was replaced by DMEM/F12 supplemented with 2 mM L-glutamine, 1% NEAA, 0.1 mM β-mercaptoethanol, 15% FBS, and 10 ng/ml bFGF for a further 6 days.
Characterization of hESC-CPC and differentiated cardiovascular lineage cells
Gene expression analysis by real-time RT-qPCR
The detailed methods of gene expression analysis are described in supplemental information. Briefly, total RNA was isolated using Micro Reagent Kit (Qiagen, 74004). First strand cDNA synthesis was carried out using 2 µg of total RNA with the Pars tous Reagent Kit (A101161), and real-time PCR was conducted using the Amplicon Kit (A325402) and Real-Time PCR machine (AB Applied Biosystems). Relative gene expression was calculated using the 2-ΔΔct formula.
Flow cytometry
The methods of flow cytometry are fully described in supplemental information. In brief, the hESC-CPCs were dissociated into single cells and subsequently fixed in 4% paraformaldehyde (PFA). The cells were permeabilized and blocked in a PBS solution containing 0.1% Triton X-100 and 1% BSA. The cells were incubated overnight with the diluted primary antibody (1:50 for NKX2.5). Then, secondary antibody (1:100 for Goat anti-rabbit, Sant Cruz, Sc-3739) was added to the cells. The stained cells were analyzed with a flow cytometer and flowJo software.
Immunofluorescence staining
A full description of the method can be found in the supplemental information. Briefly, cells were fixed with 4% PFA. For permeabilization and blocking, cells were incubated in PBS containing 0.1% Triton X-100 and 1% BSA. Primary antibodies diluted in the fresh permeabilization/blocking buffer were added to the cells, followed by an overnight incubation. Secondary antibodies diluted in fresh permeabilization/blocking buffer were added to the cells, followed by incubation of the cells with DAPI (Sigma-Aldrich, D8417) (2 µg/ml) for nuclear staining.
Functional analysis
Field potential recording of CPC-derived cardiomyocytes
We evaluated the functional characteristics of CPC-derived CMs by conducting an extracellular field potential (FP) recording using a microelectrode array (MEA) data acquisition system (Multi Channel Systems in Reutlingen, Germany). The MEA plates had a grid of 60 titanium nitride electrodes (30 μm) with a 200 μm inter-electrode distance. Before the experiment, the MEA plates were sterilized, hydrophilized with FBS for 30 min, rinsed with sterile water, and coated with gelatin for 1 h. For the analysis, 4–5 × 105 CPCs were placed in the center of a sterilized MEA plate in a medium containing 20% FBS for 24 h. Subsequently, the MEAs were connected to a head-stage amplifier. The extracellular field potentials were recorded at a sampling rate of 10 kHz, and all the measurements were conducted at 37 °C. Recordings lasted for 100 s at baseline. The FP signals were assessed for rhythmicity. The data were analyzed using Cardio2D+ software.
Tube formation assay of CPC-derived endothelial cells
To prepare for the experiment, a 96-well cell culture plate and P100 (t100 LRS) tips were placed at -80 °C for 10 min. After that, 50 µl of ice-cold Matrigel (Corning, USA) was coated in the wells. The plates were then incubated at 37 °C for 30 min to allow the Matrigel to solidify. CPC-derived ECs (CPC-EC) were seeded on the Matrigel at a density of 5 × 104, and a mixture of ECm plus 10% FBS was added. For control, HUVECs were used at a density of 13 × 103 cells [19]. After 2 hs, images were captured using a CKX41 inverted (OLYMPUS) microscope from five random fields per well. These images were analyzed with the Angiogenesis Analyzer macro for ImageJ [20], and the number of nodes, junctions, and branches were calculated and compared. Each experiment was repeated three times.
Contraction assay of CPC-derived smooth muscle cells
CPC-SMCs were cultured on Matrigel-coated 4-well plates. After 24 h, carbachol (Sigma-Aldrich, C4382) was added to the culture medium at a final concentration of 10 µM. Images were taken 10 min after the treatment [21]. The cell surface area was measured using ImageJ 1.54 h software, and the average reduction in cell area was calculated.
Activation of CPC-derived cardiac fibroblasts
In order to activate CPC-CFs, the cells were incubated in serum-free culture media for 24 h at 5% CO2 and 37 °C, followed by treatment with doxorubicin (DOX) 0.5 µM for 48 h. Both DOX-treated and control CFs were collected and subjected to RT-qPCR analysis for the detection of αSMA expression level, as previously described. The expression levels were then compared between the two groups.
Cardiac microtissue formation by co-culture of CPC-CM, CPC-EC, CPC-SMC and CPC-CF
For microtissue formation, CM, EC, SMC, and CF, which were derived from CPC differentiation, were added to 2.5 mg/ml of collagen derived from rat tail at a ratio of 2:1:1:2 and a final density of 2 × 106 cells/ml and cultured in 24-well plates. The co-culture medium (MTm) consisted of two media in an equal proportion: [1] DMEM F12, 15% FBS, 2 mM L-glutamine, 1% NEAA, 1% penicillin/streptomycin, and 0.1 mM β-mercaptoethanol, and [2] RPMI1640 supplemented with complete B27, 2 mM L-glutamine, 1% NEAA, and 0.1 mM β-mercaptoethanol. The medium was refreshed every day for one week. The microtissue started to form after 1 h and continued to make a 3D-like structure, and the area-to-volume ratio decreased over the next 24 h. The cardiac microtissues were then harvested for histological analysis.
Histological analysis
The detailed methods of histological analysis are described in supplemental information. Briefly, the microtissues were collected 7 days after co-culture for histological assessment. The microtissues were fixed with 4% PFA, incorporated in 2% agar gel, embedded in paraffin blocks, and cut into 5 μm-thick sections. For immunofluorescence staining, deparaffinized and rehydrated sections were undergone antigen retrieval. Subsequently, samples were blocked with 10% BSA, followed by the permeabilization with PBS containing 0.5% Triton X100. Then, the sections were incubated overnight with primary antibodies, which followed by incubation with secondary antibodies and staining with DAPI.
Confocal imaging
To evaluate the cell distribution in the microtissue, rhodamine Rhodamine Phalloidin (Invitrogene, B7474) staining was used and visualized by confocal laser scanning microscopy (CLSM; LSM800, Carl Zeiss, Germany). The detailed descriptions can be found in the supplemental information.
Statistical analysis
Statistical analysis was performed using the GraphPad-Prism software (9.0.2), and all results were presented as mean ± standard deviation (SD) or mean ± standard error of mean (SEM). The data is obtained from a minimum of three independent replicates. Following the assessment of normal distribution, significant differences between groups were calculated using proper statistical tests, including an unpaired t-test or one-way and two-way ANOVA. A statistically significant level was considered as P < 0.05.
Results
Cardiovascular progenitor cells (CPCs) demonstrated expansion potential in adherent culture
Induction of CPCs from adherently or suspension-expanded hESCs (Supplementary Fig. 1A) was performed by treating the cells with a cocktail of small molecules, including CHIR99021 (CHIR), IWP2, SB-431,542, and Pur, in a static suspension culture, as demonstrated in Fig. 1A.

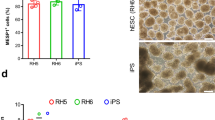
Characterization of cardiac progenitor cells (CPCs) generated from the human embryonic stem cell (hESC). (A) Schematic diagram showing differentiation protocol used for CPC induction from hESCs. hESCs were cultured in suspension as spheroids and differentiated into mesoderm, followed by cardiac progenitor cells differentiation by one-day treatment with 12 µM CHIR99021 (CHIR) and a one-day rest period, then treatment with ISP 5 µM for two-days. The 4-day old differentiated hESC-CPC spheroids were dissociated into single cells and cultured in expansion medium. (B) CPC morphology and immunofluorescence staining of NKX2.5 and Ki67 expressions in P0, 4, 8 CPCs cultured in expansion medium. Cells were counter-stained with DAPI (C) Numeric data of percentages of NKX2.5+ and KI67+ cells in hESC-derived CPCs. Data are presented as mean ± standard error of the mean (SEM). (D & E) Doubling time and expansion fold of CPCs at 0, 4, 8 passages. Data are presented as mean ± standard error of the mean (SEM). (F) Representative graph of total cell counts generated after 8 passages of hESC-derived CPCs
CHIR activates the Wnt/β-catenin signaling pathway by inhibiting GSK3 and is essential for mesendoderm specification by an initial activation of this pathway in hESCs, while a baseline dose of Wnt signaling is critical to direct the cell fate towards cardiac mesoderm [22]. In contrast, IWP2 blocks the secretion of Wnt ligands, leading to inhibition of the Wnt/β-catenin pathway. By modulating Wnt signaling, IWP2 can contribute to the generation of functional cardiomyocytes from hPSCs [23, 24]. SB-431,542 is an antagonist of the TGF-β1 ALK receptors and contributes to CPC specification [25], while Pur is involved in CPC fate by promoting the expression of cardiogenesis genes via activating the Sonic Hedgehog (SHH) signaling pathway, which has a crucial role in heart development [26].
Flow cytometry analysis indicated that day-4 differentiated cells were 88.6% NKX2.5+ (Supplementary Fig. 1B). Also, we demonstrated the upregulation of ADGRL2 and NKX2.5 in day-4 differentiating cells, highlighting the cardiac progenitor nature of these cells (Supplementary Fig. 1C and D). After confirming the CPC’s fate, they were subjected to expansion. Cultured CPCs from P0, P4, and P8 showed mesoderm-like cobblestone morphology in CPCxm medium (Fig. 1B, left column) and co-expression of NKX2.5 and Ki67 in all three passages (Fig. 1B, middle and right columns), which reached more than 80% of the stained cells (Fig. 1C). While the expansion fold of CPCs was > 10 times in all passages (Fig. 1D), the population doubling time (PDT) was ~ 20 h at P0, P4, and P8 (Fig. 1E). As depicted in Fig. 1F, a total of 3 × 109 cells were obtained after 8 passages, starting from an initial cell density of 4 × 105.
CPCs were successfully differentiated into functional cardiovascular lineage cells
CPCs at P0 and P8 were subjected to specific differentiation media containing required growth factors and small molecules for differentiation into CMs, ECs, SMCs, and CFs (Fig. 2A). CPC-derived CMs (CPC-CM) showed elongated morphology compared to cobblestone-like CPCs (Fig. 2B and Supplementary Fig. 2, top row). However, these CPC-CM resembled fetal cardiomyocytes rather than adult mature cells. They expressed cardiac troponin T (cTNT) as relevant in immunostaining and mRNA expression analysis, as well as MLC2v, 12-day post-differentiation. There were no substantial changes in the expression level of the cTNT and MLC2v genes between P0 and P8 CPC-CM (Fig. 2B and C). Twelve-day treatment of CPCs with VEGF (50 ng/ml) and bFGF (10 ng/ml) turned them into cells that generated tube-like structures in the culture (Fig. 2B and Supplementary Fig. 2, CPC-EC, bright field image). The CPC-derived endothelial-like cells (CPC-EC) were positively stained for CD31 protein and showed mRNA expression of KDR and vWF. While vWF mRNA expression did not differ between P0 and P8 CPC-EC, KDR expression showed a 2-fold increase in P8 CPC-EC compared to P0 CPC-EC (Fig. 2B and C). This increase is in favor of endothelial cell derivation. Treatment of P0 and P8 CPCs with PDGF-BB (10 ng/ml) and TGF-b (2 ng/ml) for 12 days resulted in differentiation towards smooth muscle cells, which expressed smooth muscle actin antigen (aSMA+) (Fig. 2B), PDFGRa and MYH14 genes (Fig. 2C). The expandable CPCs were differentiated into cardiac fibroblasts (CFs) by 48 h of treatment with CHIR (3 mM), followed by a 3-day rest and a 10-day treatment with bFGF (10 ng/ml). The differentiated CPC-CFs showed protein expression of the fibroblast cytoskeletal marker VIMENTIN, as well as VIMENTIN and COL1A1 gene expression. Expression of VIMENTIN showed elevated levels in P8 CPC-CFs, while expression of COLA1 did not differ between CFs derived from CPCs in two passages (Fig. 2B and C).

Differentiation and characterization of cardiomyocyte (CM), endothelial cell (EC), smooth muscle cell (SMC), and cardiac fibroblasts (CF) from CPCs. (A) Schematic diagram of differentiation protocol of CM, EC, SMC, and CF. (B) Morphology and immunofluorescence characterization of CM (with c-TNT), EC (with CD31), SMC (with αSMA), and CF (with VIMENTIN) derived from CPCs at both passages 0 and 8. Scale bar; 200 μm. (C) Characterization of four cardiac lineage cells derived from CPCs at both passages 0 and 8 at gene expression level: c-TNT and MLC2v for CM, KDR and vWF for EC, PDGFRα and MHY14 for SMC, and VIMENTIN and COL1A1 for CF. Data are presented as mean ± standard error of the mean (SEM)
Furthermore, CPC-CMs showed regular field potentials, which resulted from the spontaneous excitability of differentiated cells (Fig. 3A and B). The tube-like structures generated by CPC-ECs resembled those of HUVECs after 2 h of culture, where the number of branches, nodes, and junctions did not differ between CPC(P0)-EC, CPC(P8)-EC, and HUVEC (Fig. 3C and D). Moreover, CPC-SMCs responded to treatment with carbachol (10 mM), an acetylcholine receptor agonist, by a substantial contraction-induced area reduction at CPC(P0)-SMC and a non-significant size decrease in CPC(P8)-SMC (Fig. 3E and F). While differentiated CPC-CF showed spindle-like morphology and a high abundance of VIMENTIN (Fig. 3G), DOX-treated CPC-CF turned to hypertrophied aSMA+ cells, which showed upregulation of ACTA2 in activated derivatives of P8 CPCs (Fig. 3H).

Functional assays for cardiac linaege cells derived from both passages 0 and 8. (A & B) The CPC-derived CMs demonstrated regular field potentials. (C) Tube formation was evaluated in CPC-derived ECs and human umbilical vein endothelial cells (HUVEC) within 2 h. (D) Numerical measurements of number of the branches, nodes, and junctions’ formation of CPC-derived ECs, which was compared with HUVEC, as a control group, representing the comparable ability of tube formation of CPC-ECs. (E) Contractility assay of CPC-derived SMCs using carbachol 10 µM during 10 min. Shortening of SMCs were obviously seen, indicating the ability of contraction of these cells. (F) Numerical assessment also represented a significant reduction in cell area. (G & H) Expression of αSMA in activated CFs with doxorubicin was found at protein level (immunostaining) as well as gene expression level (qRT-PCR). All data are presented as mean ± standard error of the mean (SEM)
Freeze-thaw of CPCs
A cryopreserved ‘off-the-shelf’ CPC product will facilitate its translational use, such as pharmaceutical application or preclinical and clinical trials, as it provides the opportunity to do large-scale and repeated testing. Furthermore, this approach allows preclinical and associated clinical assessments to be performed using the same cryopreserved CPC batch.
Expandable CPCs showed > 80% viability post-freeze-thaw (Fig. 4A and B), as well as upregulation of cardiovascular progenitor-specific genes; NKX2.5 and ISL1, and downregulation of ectoderm- and endoderm-specific genes; SOX17, AFP, and PAX6 (Fig. 4C). The cryopreserved CPCs retained their expandability, as all NKX2.5+ cells were Ki67+ (Fig. 4D and E). Furthermore, they successfully differentiated into cardiac lineage cells (Fig. 4F) and expressed specific markers for CPC-CM, CPC-EC, CPC-SMC, and CPC-CF at the mRNA level (cTNT, vWF, PDGFRa, and VIMENTIN, respectively) (Fig. 4G) and protein level (cTNT, CD31, aSMA, and VIMENTIN, respectively) (Fig. 4H).

Assessment of the cryopreservation of CPCs. (A) Morphology of CPCs after freeze-thaw. (B) Assessing the percentage of viable cells after freeze-thaw that showed more than 80% viablility. (C) Comparison of the gene expression of freeze-thawed CPCs and hESCs. The upregulation of NKX2.5 and ISL1 and downregulation of SOX17, AFP, and PAX6 were observed copmared to hESCs, reflecting the preservation of CPCs transcriptional signature after freeze-thaw. (D & E) Assessing the NKX2.5 and Ki67 positive CPCs in immunofluorescence, showing more than 80% and 90% of cells were positive for NKX2.5 and Ki67, respectively. (F & G & H) Evaluation of morphology, gene expression, and immunofluorescence staining of the CMs, ECs, SMCs, and CFs differentiated from CPCs after freeze-thaw, implying cryopreservation of differentiation capacity of the CPCs. All data are presented as mean ± standard error of the mean (SEM)
Expandable CPC derivatives formed functional cardiac microtissue
Co-culture of 2 × 106 CPC derivatives at a ratio of 2:1:1:2 (CPC-CM: CPC-EC: CPC-SMC: CPC-CF) in 2.5 mg/ml collagen generated a spherical-like tissue after 48 h (Fig. 5A), which showed considerable and homogeneous cellularity all over the microtissue identified by F-actin staining (Fig. 4B). Cell type-specific staining demonstrated the presence of a high population of cTNT+ CPC-CM and VIMENTIN+ CPC-CF within the formed microtissues of P0 and P8 CPC derivatives (Fig. 5C). As CPC-EC and CPC-SMC were used in a lower ratio for microtissue formation, the frequency of aSMA+ and CD31+ cells were lower in the final assembly (Fig. 5D and E). The P0 and P8-derived microtissues exhibited disperse field potentials (Supplementary Fig. 3).

Microtissue formation using CM, EC, SMC, and CF in ratio of 2:1:1:2 in collagen derived from rat tail. (A) Macroscopic morphology of the microtissue. (B) Cellularity of the microtissue identified using Rhodamine/Phalloidin staining to show F-actin distribution in the cells, exhibited with Confocal microscope. (C). Detection of CMs, ECs, SMCs, and CFs in the microtissue using immunofluorescence staining: CMs with c-TNT, ECs with CD31, SMCs with αSMA, and CFs with Vimentin
Discussion
In this study, CPCs were initially generated by programming the hESCs with a four-day differentiation protocol in suspension state that utilized small molecules, according to our previously established method [18]. We were able to expand hESC-derived CPCs up to 8 passages while maintaining their morphology, molecular profile, and ability to differentiate into cardiac lineage cells, even after freeze-thawing. Furthermore, we could produce functional cardiomyocytes, endothelial cells, cardiac fibroblasts, and smooth muscle cells and used them to form a cardiac microtissue that showed presence and distribution of cardiac lineage cells as well as excitability. These findings represent an important step towards developing 3D models for disease modeling and cardiotoxicity testing in vitro.
Previously, we introduced the MESP1+ CMCs, which were obtained using a two-day differentiation protocol in suspension state. These cells were expanded approximately 11-fold at early passages, with a slight decrease in expansion rate at later passages. At P10, we obtained 1014 cells with preserved properties such as identity, expandability, and differentiation capacity [15]. However, the current study took advantage of CPCs generated by the same protocol, but harvested at day 4 and being NKX2.5+, as an important molecular marker of CPCs [16, 27]. NKX2.5, one of the earliest cardiac-restricted transcription factors expressed in multipotent CPCs during vertebrate heart development [28, 29], was reported to be expressed in all c-KIT+, SCA-1+, MESP1+, and KDR+ sub-populations [30]. Furthermore, researchers have well documented the crucial role of NKX2.5 as a transcription factor in promoting cardioblast differentiation and proliferation [31]. Interestingly, reactivating the NKX2.5 gene in extracardiac fibroblasts using the CRISPR/Cas9 system could significantly contribute to the successful reprogramming of those into CPCs with renewal and regenerative capacity [32]. In addition, our further analysis revealed the expression of the cardiac mesodermal marker ISL1, which is mainly identified as participating in the differentiation of CPCs into SMCs and ECs [33, 34]. Besides, we demonstrated the expression of ADGRL2, also known as Adhesion G protein-coupled receptor L2 or LPHN2, in CPCs. In line with our results, ADGRL2 was found to be selectively expressed in CPCs during differentiation from hPSCs [35]. This gene expression pattern likely reflects the transition and commitment of mesoderm into CPCs.
Studying the underlying mechanisms of cardiovascular pathologies and pioneering the discovery of novel potential therapeutic agents using in vitro human cardiac cells has gained more attention recently [36,37,38]. Furthermore, due to the complexity of heart tissue, cardiomyocytes alone are insufficient to encompass the entire range of cardiac functionality and pathophysiology [39, 40]. This objective requires the production of human cardiovascular progenitor cells in vitro with self-renewal and differentiation capacity. However, the prolonged expansion of CPCs on a large scale in culture, with the potential to differentiate into functional cardiac lineage cells, has remained challenging [4, 41]. In this study, CPCs were expanded approximately 10-fold, doubling in number within about 22 h. Consistent with the present study, Birket et al. produced NKX2.5+ CPCs from hPSCs using a 6-day differentiation protocol employing growth factors and small molecules such as insulin-like growth factor-1 (IGF-1) and a hedgehog agonist. The clonogenic nature of these CPCs allowed for over 40 population doublings without compromising their characteristics [42]. Recently, Wang et al. developed expandable proliferative NKX2.5+ CPCs from human fibroblasts using a combination of small molecules, including CHIR99021, A83-01, GSK126, Forskolin, CTPB, and AM580.
With respect to cardiac cell type commitment, previous investigations have focused on the differentiation of the CPCs into CMs, ECs, and SMCs for therapeutic purposes [16, 43]. For instance, a 6-day differentiation protocol generated hPSCs-derived NKX2.5 + CPCs, demonstrating their differentiation into functional multi-lineage cardiovascular cells such as pacemaker and ventricular myocytes, ECs, and SMCs [42]. Chemically reprogrammed human fibroblasts produced expandable NKX2.5 + CPCs, which efficiently differentiated into functional CMs, ECs, and SMCs by passage 12 in vitro [10]. Consistent with previous examinations, our CPCs were able to differentiate into CMs displaying electrical activity, ECs capable of tube formation, and SMCs with contractile properties in vitro at both early and late passages, and further progressed into cardiac microtissue by coculture in collagen hydrogel.
Ultimately, in line with our previous study [15], we demonstrated that over 80% of the late-passage CPCs remained viable and retained their characteristics, successfully differentiating into various cardiovascular lineage cells after undergoing freeze-thaw in CPCxm medium. Additionally, we detected no notable expression of SOX17 and AFP endodermal markers and PAX6, an ectodermal marker, in the CPCs. These findings supported our objective of providing an expandable and cryopreservable cardiovascular progenitor cell line.
Nonetheless, there are some limitations in this study, such as: [1] the heterogeneity observed in differentiated CPCs in vitro [2], the incomplete maturation of differentiated cardiovascular lineage cells in vitro [3], insufficient exploration of the functional diversity, maturity, as well as the longevity functionality of the differentiated cardiovascular lineage cells within cardiac microtissues over extended periods.
Conclusions
In conclusion, this study reported a platform that enables large-scale production of functional cardiovascular lineage cells using expandable CPCs for the purpose of in vitro cardiac microtissue formation as well as other anticipated experimental and preclinical expenditures.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CPCs:
-
Cardiovascular progenitor cells
- hESCs:
-
Human Embryonic Stem Cells
- CVDs:
-
Cardiovascular Diseases
- CMs:
-
Cardiomyocytes
- ECs:
-
Endothelial Cells
- CFs:
-
Cardiac Fibroblasts
- SMCs:
-
Smooth Muscle Cells
- hiPSCs:
-
Human-Induced Pluripotent Stem Cells
- PDT :
-
Population Doubling Time
- MEA:
-
Microelectrode array
- DOX:
-
Doxorubicin
- IGF-1:
-
Insulin-Like Growth Factor-1
References
Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. 2020;76(25):2982–3021.
Leone M, Magadum A, Engel FB. Cardiomyocyte proliferation in cardiac development and regeneration: a guide to methodologies and interpretations. Am J Physiol Heart Circ Physiol. 2015;309(8):H1237–50.
Guo Y, Yu Y, Hu S, Chen Y, Shen ZJCD, Disease. Therapeutic Potential Mesenchymal stem Cells Cardiovasc Dis. 2020;11(5):349.
Barreto S, Hamel L, Schiatti T, Yang Y, George VJC. Cardiac progenitor cells from stem cells: learning from genetics and biomaterials. 2019;8(12):1536.
Burridge PW, Keller G, Gold JD, Wu JCJC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. 2012;10(1):16–28.
Desbordes SC, Studer LJN. Adapting human pluripotent stem cells to high-throughput and high-content screening. 2013;8(1):111–30.
Le T, Chong J. Cardiac progenitor cells for heart repair. Cell Death Discovery. 2016;2(1):1–4.
Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sànchez-Dànes A, et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359(6380):1177–81.
Herrero D, Albericio G, Higuera M, Herranz-López M, García-Brenes MA, Cordero A, et al. The vascular niche for adult cardiac progenitor cells. Antioxidants. 2022;11(5):882.
Wang J, Gu S, Liu F, Chen Z, Xu H, Liu Z, et al. Reprogramming of fibroblasts into expandable cardiovascular progenitor cells via small molecules in xeno-free conditions. Nat Biomedical Eng. 2022;6(4):403–20.
Du J, Zheng L, Gao P, Yang H, Yang W-J, Guo F, et al. A small-molecule cocktail promotes mammalian cardiomyocyte proliferation and heart regeneration. Cell Stem Cell. 2022;29(4):545–58. e13.
He L, Nguyen NB, Ardehali R, Zhou BJC. Heart regeneration by endogenous stem cells and cardiomyocyte proliferation: controversy, fallacy, and progress. 2020;142(3):275–91.
Protze SI, Lee JH, Keller GMJC. Human pluripotent stem cell-derived cardiovascular cells: from developmental biology to therapeutic applications. 2019;25(3):311–27.
Barreto S, Hamel L, Schiatti T, Yang Y, George V. Cardiac progenitor cells from stem cells: learning from Genetics and Biomaterials. Cells. 2019;8(12).
Vahdat S, Pahlavan S, Mahmoudi E, Barekat M, Ansari H, Bakhshandeh B, et al. Expansion of human pluripotent stem cell-derived Early Cardiovascular Progenitor cells by a cocktail of signaling factors. Sci Rep. 2019;9(1):16006.
Cao N, Liang H, Huang J, Wang J, Chen Y, Chen Z, et al. Highly efficient induction and long-term maintenance of multipotent cardiovascular progenitors from human pluripotent stem cells under defined conditions. Cell Res. 2013;23(9):1119–32.
Baharvand H, Ashtiani SK, Taee A, Massumi M, Valojerdi MR, Yazdi PE, et al. Generation of new human embryonic stem cell lines with diploid and triploid karyotypes. Dev Growth Differ. 2006;48(2):117–28.
Fonoudi H, Ansari H, Abbasalizadeh S, Larijani MR, Kiani S, Hashemizadeh S, et al. A Universal and Robust Integrated platform for the scalable production of human cardiomyocytes from pluripotent stem cells. Stem Cells Transl Med. 2015;4(12):1482–94.
Hosseini M, Ayyari M, Meyfour A, Piacente S, Cerulli A, Crawford A, et al. Cardenolide-rich fraction of Pergularia tomentosa as a novel antiangiogenic agent mainly targeting endothelial cell migration. Daru. 2020;28(2):533–43.
Carpentier G, Berndt S, Ferratge S, Rasband W, Cuendet M, Uzan G, et al. Angiogenesis analyzer for ImageJ—A comparative morphometric analysis of endothelial tube formation assay and fibrin bead assay. Sci Rep. 2020;10(1):11568.
Tian X-X, Kang J, Yan C-H, Xu K, Tao J, Yang G-T, et al. Purification and functional assessment of smooth muscle cells derived from mouse embryonic stem cells. J Geriatric Cardiology: JGC. 2013;10(3):272.
Zhao M, Tang Y, Zhou Y, Zhang J. Deciphering role of wnt signalling in cardiac mesoderm and cardiomyocyte differentiation from human iPSCs: four-dimensional control of wnt pathway for hiPSC-CMs differentiation. Sci Rep. 2019;9(1):19389.
Muneer R, Fatima A, Ahmad W, Salim A, Dini L, Khan I. Wnt signaling pathway inhibitor promotes mesenchymal stem cells differentiation into cardiac progenitor cells in vitro and improves cardiomyopathy in vivo. World J Stem Cells. 2023;15(8):821.
Tsoi C, Deng R, Kwok M, Yan B, Lee C, Li HS, et al. Temporal control of the WNT signaling pathway during Cardiac differentiation impacts upon the maturation state of human pluripotent stem cell derived cardiomyocytes. Front Mol Biosci. 2022;9:714008.
Mahmood A, Harkness L, Schrøder HD, Abdallah BM, Kassem M. Enhanced differentiation of human embryonic stem cells to mesenchymal progenitors by inhibition of TGF-β/activin/nodal signaling using SB‐431542. J Bone Miner Res. 2010;25(6):1216–33.
Jeong M-H, Leem Y-E, Kim H-J, Kang K, Cho H, Kang J-S. A shh coreceptor Cdo is required for efficient cardiomyogenesis of pluripotent stem cells. J Mol Cell Cardiol. 2016;93:57–66.
Iancu CB, Iancu D, Renţea I, Hostiuc S, Dermengiu D, Rusu MC. Molecular signatures of cardiac stem cells. Rom J Morphol Embryol. 2015;56(4):1255–62.
Caprioli A, Koyano-Nakagawa N, Iacovino M, Shi X, Ferdous A, Harvey RP, et al. Nkx2-5 represses Gata1 gene expression and modulates the cellular fate of cardiac progenitors during embryogenesis. Circulation. 2011;123(15):1633–41.
Deutsch M-A, Doppler SA, Li X, Lahm H, Santamaria G, Cuda G, et al. Reactivation of the Nkx2. 5 cardiac enhancer after myocardial infarction does not presage myogenesis. Cardiovascular Res. 2018;114(8):1098–114.
Mehanna RA, Essawy MM, Barkat MA, Awaad AK, Thabet EH, Hamed HA, et al. Cardiac stem cells: current knowledge and future prospects. World J Stem Cells. 2022;14(1):1.
Chen WP, Wu SM. Small molecule regulators of postnatal nkx 2.5 cardiomyoblast proliferation and differentiation. J Cell Mol Med. 2012;16(5):961–5.
Jiang L, Liang J, Huang W, Ma J, Park KH, Wu Z, et al. CRISPR activation of endogenous genes reprograms fibroblasts into cardiovascular progenitor cells for myocardial infarction therapy. Mol Ther. 2022;30(1):54–74.
Ghazizadeh Z, Fattahi F, Mirzaei M, Bayersaikhan D, Lee J, Chae S, et al. Prospective isolation of ISL1 + cardiac progenitors from human ESCs for myocardial infarction therapy. Stem Cell Rep. 2018;10(3):848–59.
Zhou B, Von Gise A, Ma Q, Rivera-Feliciano J, Pu WT. Nkx2-5-and Isl1-expressing cardiac progenitors contribute to proepicardium. Biochem Biophys Res Commun. 2008;375(3):450–3.
Lee C-S, Cho H-J, Lee J-W, Son H, Chai J, Kim H-S. Adhesion GPCR latrophilin-2 specifies cardiac lineage commitment through CDK5, src, and P38MAPK. Stem Cell Rep. 2021;16(4):868–82.
van Doorn EC, Amesz JH, Sadeghi AH, de Groot NM, Manintveld OC, Taverne YJ. Preclinical models of Cardiac Disease: a comprehensive overview for clinical scientists. Cardiovasc Eng Technol. 2024:1–18.
Parrotta EI, Lucchino V, Scaramuzzino L, Scalise S, Cuda G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: progress, promises and challenges. Int J Mol Sci. 2020;21(12):4354.
Hartman ME, Dai D-F, Laflamme MA. Human pluripotent stem cells: prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv Drug Deliv Rev. 2016;96:3–17.
Aujla PK, Kassiri Z. Diverse origins and activation of fibroblasts in cardiac fibrosis. Cell Signal. 2021;78:109869.
Chen Q, Zhang H, Liu Y, Adams S, Eilken H, Stehling M, et al. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat Commun. 2016;7(1):12422.
Chen IY, Wu JC. Finding expandable induced cardiovascular progenitor cells. Circul Res. 2016;119(1):16–20.
Birket MJ, Ribeiro MC, Verkerk AO, Ward D, Leitoguinho AR, Den Hartogh SC, et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat Biotechnol. 2015;33(9):970–9.
Zhang Y, Cao N, Huang Y, Spencer CI, Fu J-d, Yu C, et al. Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell. 2016;18(3):368–81.
Acknowledgements
The authors declare that artificial intelligence is not used in generation of this study.
Funding
This study was supported by a grant from the Royan Institute.
Author information
Authors and Affiliations
Contributions
SR; design, methodology, and writing the first draft. MR; methodology, writing the first draft. MS methodology, CF differentiation, and characterization. MK; methodology microtissue formation. RH; methodology for functional studies of cardiovascular linage cells. HK; methodology microtissue formation. SV; advisor. SP; supervising, revising the manuscript. HB; supervising, revising manuscript and funding.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The ethical approval was not required for the current study as investigated and issued by the Royan Institute Ethics Committee under the license number IR.ACECR.ROYAN.REC.1399.128 (February 14th, 2021). Because the hESC line which was used in the current study (RH6), was derived in the project entitled “Generation of new human embryonic stem cell lines with diploid and triploid karyotypes”. The project was performed following the approval of Royan Institute Ethics Committee and after obtaining informed consent from the couple undergoing in vitro fertilization treatment (2006, full information can be found in https://hpscreg.eu/cell-line/RIe006-A) [17].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rezaeiani, S., Rezaee, M., Shafaghi, M. et al. Expandable hESC-derived cardiovascular progenitor cells generate functional cardiac lineage cells for microtissue construction. Stem Cell Res Ther 15, 298 (2024). https://doi.org/10.1186/s13287-024-03919-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-024-03919-6