
Abstract
Mitochondria have multiple functions such as supplying energy, regulating the redox status, and producing proteins encoded by an independent genome. They are closely related to the physiology and pathology of many organs and tissues, among which the brain is particularly prominent. The brain demands 20% of the resting metabolic rate and holds highly active mitochondrial activities. Considerable research shows that mitochondria are closely related to brain function, while mitochondrial defects induce or exacerbate pathology in the brain. In this review, we provide comprehensive research advances of mitochondrial biology involved in brain functions, as well as the mitochondria-dependent cellular events in brain physiology and pathology. Furthermore, various perspectives are explored to better identify the mitochondrial roles in neurological diseases and the neurophenotypes of mitochondrial diseases. Finally, mitochondrial therapies are discussed. Mitochondrial-targeting therapeutics are showing great potentials in the treatment of brain diseases.
Similar content being viewed by others

Introduction
The brain operates through a combination of electrical and chemical signals, and this process is highly energy-demanding [1, 2]. Neurons are the basic working units of the brain and are the most power-hungry cell type in the brain. The human brain contains nearly 100 billion neurons [3]. Each neuron is connected to up to 10,000 other neurons, exchanging signals via as many as 1000 trillion synapses [3]. Even in resting states or when neurons are not releasing neurotransmitters to each other, the brain consumes 20% of the body’s overall energy [4].
Mitochondria are essential for brain functions, as they produce adenosine triphosphate (ATP) to be used by brain cells [5]. Insufficient ATP supply will lead to brain cell death [6]. Reactive oxygen species (ROS) are toxic byproducts of ATP generation, produced along with the respiration process [7]. In physiological states, the ROS are maintained at a controllable steady-state level [8] and facilitate normal redox signaling of the brain cells [9, 10]. Excessive production of ROS will oxidize the brain lipids and neurotransmitters to induce enrichment of unsaturated lipids, neurotransmitter auto-oxidation, RNA oxidation, etc. [11,12,13]. Interestingly, mitochondria also possess antioxidant enzymes and endogenous antioxidants to balance cellular oxidation and reduction states [19, 20]. In addition, the membrane dynamics, genetic information storage and the quality control system of mitochondria are closely related to the homeostasis of the brain [14,15,16,17].
Disturbances in immune processes, protein deposition, neurogenesis, and organelle functions are reported as the pathological mechanisms underlying certain brain diseases, and all these aspects have a close relationship with mitochondrial dysfunctions. Neuronal loss in neurodegenerative diseases (NDDs) is often attributed to mitochondrial energy shortage. The neuron types affected in NDDs, such as dopaminergic neurons in Parkinson’s disease (PD) and motor neurons in amyotrophic lateral sclerosis (ALS), often have a complex structure with extensive and highly branched axonal arbors, requiring large amount of ATP for normal functions [18,19,20,21]. In healthy neurons, mitochondria provide energy for neuronal activities, and also modulate neuronal degeneration and death under stress [22,23,24]. Inflammatory cytokines, notably interleukin 6 (IL-6), are regarded as the endogenous biomarker and therapeutic target for depression [25]. The inflammatory cytokines can be induced by mitochondrial DNA (mtDNA) as a danger-associated molecular pattern (DAMP) via the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) signaling pathway [26]. Epilepsy is a pathological state with abnormal electrical signals in the brain, among which the calcium signaling is a crucial point [27]. It has been demonstrated that the mitochondria-mediated calcium buffering is fundamental for neuronal activity set points in epilepsy [28, 29], which may also provide foundations for other brain pathological conditions associated with aberrant network activity. Collectively, mitochondria are involved in both the integrative mechanisms of brain diseases and specific pathologies. In this review, we summarize the roles of mitochondrial biology in brain functions and the mitochondria-dependent cellular events regarding brain pathophysiology. We also discuss the involvement of mitochondrial dysfunctions in the progression of different neurological diseases. On this foundation, mitochondrial-based therapies and advanced technologies are discussed. We highlight that mitochondrial therapy is one of the most prospecting therapeutics for brain pathology.
Mitochondrial biology maintains brain physiology
Mitochondria are double-membrane-bound organelles present in almost all eukaryotic cells with two aqueous compartments, the inter-membrane space (IMS) and the matrix. The IMS houses about 5% of the mitochondrial proteome [30], but is responsible for multifaceted functions including molecular exchange between mitochondrion and cytosol [30, 31], initiation of apoptotic cascades [32], biogenesis of respiratory chain complexes [33], as well as control of mitochondrial structural integrity and morphogenesis [34]. The internal matrix is the main working area of mitochondria, containing hundreds of enzymes for the oxidation of fats and carbohydrates in the tricarboxylic acid (TCA) cycle. The mitochondrial matrix and the inner membrane together constitute the functional compartment for urea cycle, protein synthesis and amino acid metabolism, supporting mitochondria as the bio-synthetic hub [35]. Beyond these metabolic substances, the mitochondrial matrix also possesses an independent genome for protein synthesis inside mitochondria [36]. In addition to the bi-layer membranes and the independent genome, another unique feature of mitochondria is their self-reproduction by binary fission, leading to mitochondrial versatile membrane dynamics, including fusion, fission, and degradation [37]. Below, we discuss the multifaceted contributions of mitochondrial biology to brain physiology (Fig. 1).

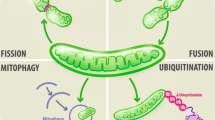
Mitochondrial biology maintains brain physiology. a Mitochondria are the power house and generate ATP through relevant processes of glucose, FA and amino acid metabolism. They tightly support normal brain functions dominated by neuronal activity including synaptic transmission, neuroelectrical activity, and ion exchange. b The mitochondrial ETC is the site of mitochondrial ROS generation. During oxidative metabolism, electrons combine prematurely with oxygen to form O2•−, which is dismutated to H2O2 by SOD2 and then converted to H2O by catalase and GPx. There are also mitochondria-targeted antioxidants essential for controlling ROS homeostasis in the brain, such as PDRX3, PDRX5 and TRX2. c The entire protein-coding capacity of mtDNA is devoted to the synthesis of mitochondrial complexes except complex II. Mutagenesis in mitochondrial genome occurs at a much higher rate than that in the nuclear genome, leading to the collapse of mitochondrial functions, which is closely related to neurological diseases. d Mitochondrial membrane dynamics including mitochondrial fission/fusion, membrane interactions with other organelles and ultra-structural membrane remodeling, renders the multifaceted involvement of mitochondria in cell biology. ATP, adenosine triphosphate; cyto c, cytochrome c; ER, endoplasmic reticulum; ETC, electron transport chain; FAs: fatty acids; GPx, glutathione peroxidases; GSH, glutathione; H2O2, hydrogen peroxide; lyso, lysosome; O2•−, superoxide; PDRX, peroxiredoxin; ROH, organic alcohol; ROS, reactive oxygen species; SOD2, manganese-dependent superoxide dismutase; TCA, tricarboxylic acid; TRX, thioredoxin
Mitochondria feed the brain
As mentioned above, the brain has an extremely high metabolic demand. It utilizes approximately 20% of the body’s total oxygen and glucose consumption with only 2% weight [38]. About 70% of the calculated energy expenditures are used to support neuronal signaling including resting potentials, action potentials, postsynaptic receptor activation, glutamate cycling, and postsynaptic Ca2+ signaling, while the remainder is for non-signaling activities like biomacromolecule turnover, axonal transport, mitochondrial proton leak and actin cytoskeleton remodeling [39] (Fig. 1a). Neurons display most of the energy consumption. They generate ATP predominantly within mitochondria through oxidative phosphorylation (OXPHOS), with a small portion of ATP from aerobic glycolysis in the cytoplasm. Astrocytes are highly glycolytic and they transform glucose into lactate with low-oxygen consumption; the lactate is then delivered to neurons for complete oxidation. This process largely supports the neuronal energetic needs by supplying metabolic substrates [40, 41]. Oligodendrocytes also obtain ATP primarily by aerobic glycolysis. They use lactate for their own energy needs and also supply neighbouring axons with lactate [42]. Microglia are predominantly fueled by OXPHOS but are metabolically reprogrammed to an aerobic glycolysis-predominant phenotype under certain neurological circumstances [43, 44]. The metabolic features of the brain change with age. Throughout human lifespan, the brain glucose utilization peaks at age 4–5 years dominantly in the form of aerobic glycolysis [45]. This high-level aerobic glycolysis in the developmental stage of life is reported to support the maximal lipid biosynthesis for neurite growth [46]. Normal aging induces a global decrease in brain metabolism, with the glucose uptake decrease exceeding the oxygen use reduction, implying loss of brain aerobic glycolysis [47]. Oxidative metabolism of glucose is relatively stable, and persists to support synaptic transmission [48, 49].
It is reported that metabolic shifts in the brain contribute to neurological conditions. Disruption of mitochondrial complex I induces a Warburg-like shift in metabolism that enables neuronal survival, but triggers a progressive loss of nigrostriatal axons resembling human parkinsonism [50]. Though astrocytes possess low mitochondrial OXPHOS activity, this metabolic mode of astrocytes is indispensable for brain lipid homeostasis. Aberrant astrocytic OXPHOS would induce lipid droplet accumulation followed by development of Alzheimer’s disease (AD)-related neurodegeneration [51]. Moreover, hexokinase 2 (HK2) gates the glycolytic flux of microglia. HK2 elevation in microglia under AD and stroke pathology attenuates and promotes the pathology, respectively [44, 52], representing different effects of microglial metabolic change in brain diseases.
Balance between ROS generation and clearance
ROS encompass a collection of molecular species derived from oxygen, including oxygen free radicals, such as superoxide anion radical (O2•−) and hydroxyl radical (•OH), and nonradical oxidants, such as hydrogen peroxide (H2O2) and singlet oxygen (1O2) [8, 53]. The major endogenous sources of ROS are trans-membrane NADPH oxidases (NOXs) and the mitochondrial electron transport chain (ETC) [8]. For ROS generated from ETC (Fig. 1b), the orderly flow of electrons down the mitochondrial ETC to complex IV results in their final deposition into oxygen to form water. However, during this process, the electrons can also react prematurely with oxygen at sites in the ETC (mainly Complexes I and III, rarely Complex II) to form O2•−, which can then be dismutated to H2O2 [8, 54]. Among NOXs, ETC and other enzymatic pathways, the mitochondrial ETC is estimated as the predominant oxidant generator in the C2C12 myoblasts [55]. Nevertheless, the assumption that mitochondria are main producers of cellular ROS is hardly conclusive [53]. The origins of cellular ROS production vary significantly, depending on the category of ROS, the types of cells, tissues or species, and the specific physiological or pathological conditions [56,57,58]. In the brain, mitochondria would generate more ROS under conditions of stress or gene mutations [59,60,61,62]. NOX-generated ROS are prone to be physiological second messengers and regulate the sequential and inter-dependent events of neuronal development: neurogenesis [9], neuronal polarization [63], and maturation of polarized neurons [64, 65]. Despite no unified verdict on which one is the leading ROS generator, a consensus has been reached that controlled ROS generation is critical for physiological brain function through redox-sensitive signaling pathways, while excessive ROS generation leads to oxidative stress and central nervous system (CNS) diseases [8, 66].
The mitochondria are equipped with powerful antioxidant defense systems, which are required for brain redox homeostasis (Fig. 1b). It is estimated that one-third of the cellular antioxidant enzymes (glutathione peroxidase and catalase) reside in mitochondria [53], and the manganese-dependent superoxide dismutase (Mn-SOD or SOD2) is exclusively located in mitochondria [67]. Mitochondria also contain 10%-15% of total cellular glutathione (GSH), the most abundant non-enzymatic antioxidant with a redox-active thiol [68]. There are also other antioxidants localized in mitochondria that are essential for the control of ROS homeostasis in the brain, including peroxiredoxin 3 (PDRX3), which contributes to the majority of hydrogen peroxide reduction in mitochondria and is closely related to glioblastoma therapeutics [69]; PDRX5, which protects against mitochondrial ROS and prevents hippocampal neuron death [70, 71]; and thioredoxin 2, a mitochondria-specific redox protein, homozygous stop mutation of which is reported to induce infantile-onset neurodegeneration in a 16-year-old adolescent [72].
Independent genome, exclusive functions and mutagenesis susceptibility
mtDNA is a double-stranded circular structure (Fig. 1c). Each mitochondrion harbors 10–10,000 copies of mtDNA, in contrast to the nuclear DNA (nDNA) which contains only two copies per cell. The mitochondrial genomes are organized with factors such as mitochondrial transcription factor A (TFAM), into mtDNA-protein structures called nucleoids for gene packaging, transcription, and replication [73, 74]. mtDNA contains 37 genes, encoding 13 essential proteins of the mitochondrial ETC, as well as 22 transfer RNAs and 2 ribosomal RNAs for the function of mitochondrial ribosomes [75]. The entire protein-coding capacity of mtDNA is devoted to the mitochondrial OXPHOS [75], with the additional assistance of nuclear-encoded proteins imported into the mitochondria [76]. Of the mitochondrial ETC, the complex I is the largest component, composed of 46 sub-units with seven (ND-1, -2, -3, -4, 4L, -5 and -6) encoded by mtDNA, while the complex II genes are entirely nuclear. Meanwhile, one out of 11 sub-units of complex III (mt-CYB), three out of 13 sub-units of complex IV (mt-CO1, 2 and 3) and two of 16 sub-units of complex V (mt-ATP6, 8) are encoded by mtDNA [77, 78]. Almost all these mtDNA-encoded components form the core sub-units of complexes (except mt-ATP8 of complex V), and are conserved across all the domains of life [78].
However, mutations in the mitochondrial genome occur at a much higher rate than that in the nDNA [79, 80] due to the following reasons. First, mtDNA replication occurs continuously, depending on the cellular energy demands. The proximity of mtDNA to the sites of OXPHOS renders them more prone to be oxidized by ROS compared with its nuclear counterpart [81]. Second, limited DNA repair capacity could also be a significant factor for mtDNA mutagenesis [73]. Diseases caused by mtDNA mutation involve multiple organ systems and often present with neurological disturbances, which will be discussed in later sections.
Membrane dynamics
Mitochondria are surrounded by a bi-layer membrane system. The outer mitochondrial membrane (OMM) serves as a platform for molecular exchange with sub-cellular compartments. The inner mitochondrial membrane (IMM) delimits the mitochondrial matrix and is further divided into the inner boundary membrane (IBM) and the cristae. The IBM hosts various channel transporters shuttling ions, ATP, ADP and small metabolites. The cristae are invaginations towards the matrix and harbor the machinery required for mitochondrial respiration [82, 83]. The mitochondrial membrane presents a high degree of morphological variability, with constant reshaping to coordinate various cellular functions (Fig. 1d). Mitochondrial fusion is the fusion of two mitochondria mediated by mitofusins (Mfn1/2) at the OMM and by optic atrophy 1 (Opa1) at the IMM [37, 84, 85]. Fission is mediated by the translocation of cytosolic dynamin-related protein 1 (Drp1) to the OMM with the guidance of fission 1 protein (Fis1) and mitochondrial fission factor (Mff) [37, 84, 86]. The two processes play important roles in maintaining functional mitochondria under metabolic or environmental stresses. Fusion could mitigate stress by mixing the contents of partially damaged mitochondria as a form of complementation, while fission enables both mitochondrial biogenesis and the removal of damaged mitochondria [87, 88].
Cristae remodeling is another aspect of mitochondrial dynamics [84, 89, 90]. The shape of cristae is constantly changing based on the metabolic state of mitochondria. The changes include increased abundance, tightening between cristae membranes, and opening of the cristae junctions (CJs, referring to the sites where crista membrane and IBM are joined) [84] (Fig. 1d). Upon respiration activation, the mitochondrion assumes a condensed appearance with the matrix contracted and cristae lumen expanded [91, 92]. Also, the cristae biogenesis increases during energy-demanding conditions [93]. Apart from involvement in OXPHOS, the CJ integrity is important for retaining the bulk of mitochondrial apoptogenic molecule cytochrome c (cyto c) inside the cristae lumen. Disruption of CJ integrity results in the egress of cyto c into the cytoplasm for apoptosis induction [94, 95].
It is important to note that with the membrane dynamics, the mitochondria are engaged in extensive intracellular interactions with other organelles. The mitochondria-endoplasmic reticulum (ER) contact sites are the best studied type of membrane contact, functioning in calcium signaling and lipid homeostasis [90]. ER and mitochondrial OMM form close contacts through cholesterol-rich micro-domains, called mitochondria-associated ER membranes (MAMs) [96]. The abundant lipid-synthesizing enzymes in the MAMs promote lipid synthesis, especially phosphatidylethanolamine, the main phospholipid of cell membranes [97]. Therefore, orchestrated coupling between ER and mitochondria is critical for calcium signaling and phospholipid balance.
The mitochondrial dynamics plays an important role in brain function and pathology. The fusion/fission dynamics of mitochondria is associated with the fate change of neural stem cells (NSCs) during cortical neurogenesis [28, 98]. Proteins enriched in MAMs are closely related to AD pathogenesis via regulation of lipid homeostasis [99,100,101]. A close link between cristae and brain has been indicated in research on Opa1. Studies have shown that reversal of Opa1-related changes ameliorates neuropathological progression by controlling mitochondrial cristae remodeling [95, 102,103,104]. Nevertheless, more studies on the roles of cristae remodeling and its modulatory molecules in the CNS are anticipated.
Mitochondrial quality control
Mitochondrial quality control is a process of homeostatic regulation of the morphology, quantity, and quality of mitochondria. Besides the membrane dynamics, mitochondrial biogenesis and mitophagy are also critical aspects of quality control [105]. Mitochondrial biogenesis involves the synthesis of mtDNA, proteins, and membranes from preexisting mitochondria through mitochondrial fission [106]. Peroxisome proliferator-activated receptor (PPAR) γ coactivator 1-alpha (PGC-1α) is a key regulator of mitochondrial biogenesis [106, 107]. PGC-1α−/− mice show reduced mitochondrial genes and axonal degeneration in the striatum [108], while PGC-1α over-expression increases dendritic spines and enhances the differentiation of synapses in cultured hippocampal neurons [109]. These phenotypes suggest that the PGC-1α-dependent mitochondrial biogenesis is crucial for neurite growth.
Mitophagy is a form of autophagy that selectively clears damaged mitochondria by lysosome-mediated degradation. One of the most characterized mechanisms of mitophagy is the PINK1-Parkin-mediated pathway. At basal levels, mitophagy occurs to recycle the old and damaged organelles, which may balance energy production with the demands of synaptic transmission in neurons [110]. Under proper stress, mitophagy is promoted for metabolic adjustment to external changes in metabolically enhanced neurons [111], and in differentiated oligodendrocytes [112]. However, both mitophagy defects and its broad activation may result in pathologic conditions. Mutations in PINK1 and parkin genes lead to hereditary forms of parkinsonism [113,114,115,116], while hyper-activation of mitophagy induces tauopathy-linked synaptic pathogenesis [117]. Therefore, mitophagy should be tightly controlled in the CNS.
In the brain, the mitochondrial quality control machinery shows a cell-specific pattern. Neurons are especially susceptible to mitochondrial dysfunctions for their polarized structures; therefore, recovery of stressed mitochondria or turnover of damaged ones is a critical step in the maintenance of neuronal homeostasis, especially for axons. Syntaphilin (SNPH) is a neuron-specific static anchor that immobilizes axonal mitochondria and would be released from stressed mitochondria [118]. The release of SNPH from axonal mitochondria enhances the mitochondrial retrograde transport to the soma for degradation [119]. Another regulatory machinary for mitochondrial homeostasis in axons is the m-AAA protease-dependent pathway. Mitochondrial m-AAA proteases function to remove damaged or unnecessary proteins in the IMM. Mutations in genes encoding sub-units of m-AAA protease are associated with neuronal loss and neurodegeneration in humans [120, 121]. Meanwhile, loss of the m-AAA proteases leads to mitochondrial fragmentation and deficiency in the axonal transport of mitochondria in experimental mice [122, 123]. In addition to the intrinsic mitochondrial quality control systems, neurons also maintain mitochondrial functions or dispose damaged mitochondria via communication with other cells [124, 125]. As to the astrocytes and microglia, they hold higher abilities of balancing out cellular stress via antioxidant systems, metabolic reprogramming, etc. [44, 126]. Therefore, they remain functionally healthy even after the loss of their mitochondrial functions [127], but would release signals to damage neurons [51, 128].
Mitochondria as a multifaceted hub in brain pathophysiology
Mitochondria are involved in multiple signaling pathways or mechanisms, and therefore are active players in brain physiology and pathology (Fig. 2).

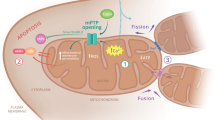
Mitochondria as a multifaceted hub of the brain pathophysiology. a Under cellular stresses, mitochondrial outer membrane permeabilization leads to release of cyto c and ROS, activating the downstream pathways of apoptosis and necroptosis. Ferroptosis is also induced by mitochondrial ETC-promotive lipid peroxide. Pyroptosis is the downstream signal of mitochondrial dysfunctions, and is controlled by mitochondria to initiate apoptosis/necrosis. b Mitochondria contain endogenous inflammatory inducers, including mtDNA, mtRNA, metabolic products and ROS. Mitochondria outer membrane acts as a platform for immune signaling through inflammasome and MAVS activation. MAVS also endows cells with antiviral immunity. c Mitochondria participate in multiple steps of autophagy including autophagy initiation, phagophore elongation, autophagic flux formation and autophagy gene induction. d Mitochondria participate in cellular communication in the brain through membrane contact and cellular organelle transfer. α-syn, α-synuclein; Aβ, β-amyloid; ATP, adenosine triphosphate; cyto c, cytochrome c; ETC, electron transport chain; GSH, glutathione; MAVS, mitochondrial antiviral signaling; ROS, reactive oxygen species; TDP-43: TAR DNA-binding protein 43; TGF-β, transforming growth factor β; VEGF, vascular endothelial growth factor
Manipulators of cell fate
Mitochondria provide power for life; however, they also play roles in cell death [129] (Fig. 2a). Apoptotic cell death is a major form of regulated cell death in which two main signaling pathways are involved: the extrinsic pathway by death receptor and the intrinsic mitochondrial pathway. The mitochondrial pathway is induced by cellular stresses and requires mitochondrial outer membrane permeabilization (MOMP) to release soluble proteins (mainly cyto c, and also SMAC and OMI) from the IMS. The released cyto c initiates activation of the downstream apoptosis executioners, caspase 3 and 7, while SMAC and OMI block the caspase inhibitor to facilitate their activation [129, 130]. BCL-2 family proteins orchestrate the MOMP process and are reported to regulate neuronal death and axonal degeneration [131]. In addition to acting as a central initiator of apoptosis, mitochondria also contribute to other forms of programmed cell death [129]. Mitochondrial ROS facilitate the initiation of necroptosis by promoting the RIPK1/RIPK3-dependent signaling and necrosome formation [132, 133]. Meanwhile, mitochondrial ROS, as well as mtDNA, would evoke pyroptosis, a form of programmed cell death mediated by inflammasome and the downstream gasdermin [134,135,136]. During pyroptosis, mitochondrial dysfunction occurs early, and indispensably controls gasdermin D oligomerization [137] and the pore-forming activity of gasdermin D fragment for plasma membrane rupture [138]. Recently, pyroptosis has been implicated in the pathogenesis of multiple neurological diseases [24, 139,140,141,142,143]. Although neuronal pyroptosis has been widely studied [24, 144], glia are theoretically more likely to experience this type of inflammatory cell death with pivotal significance for disease progression. Astrocytic loss and pyroptosis-related caspase activation coexist in a mouse model of major depressive disorder [145], and inhibiting astrocytic pyroptosis alleviates depressive-like behaviors [145, 146]. Microglial pyroptosis occurs upon ALS symptom onset, and is correlated with neuronal loss [140]. Mitochondria are also the converters between the aforementioned programmed cell death events. Gasdermin E can permeabilize the mitochondria to augment apoptosis [133, 147], and pyroptotic caspase-1 is reported to initiate apoptosis and elicit secondary necrosis by activating the Bid-dependent mitochondrial apoptosis pathway [148]. Ferroptosis is a newly identified modality of regulated cell death, which is triggered by iron-dependent lipid peroxides to attack lipid membranes and tear up cells [149]. A previous study supported a critical role of mitochondria in cysteine-deprivation-induced ferroptosis through accumulation of lipid peroxides [150]. In addition, an antiviral drug for human immunodeficiency virus infection induces mtDNA stress and leads to ferroptotic cell death via autophagy [151]. Conversely, mitochondria hold the abilities to inhibit ferroptosis by mitochondrial GPx4 [152], mitochondrial dihydroorotate dehydrogenase [153], and mitochondria-localized cGAS [154]. Recently, ferroptosis has received increasing attention, especially in brain diseases [155, 156]. On one hand, iron is remarkably important in the biosynthesis of neurotransmitters, mitochondrial respiration, myelin synthesis, and among others. On the other hand, this essential micro-nutrient is engaged in catalyzing the formation of potent oxidants, exposing the nervous tissue to oxidative damage [157]. Brain iron accumulation has been found as a common feature of several NDDs [158,159,160]. Meanwhile, ferroptosis of neural and immune cells is linked to mitochondria and promotes neurodegeneration [161,162,163,164]. Therefore, mitochondria are considered as a central nexus between different modalities of cell death in the CNS.
Immunoregulatory roles of mitochondria
Mitochondria lie at the heart of immunity and neuroinflammation (Fig. 2b). Expelled from stressed mitochondria, mtDNA and its oxidation products can be detected by host pattern-recognition receptors, mainly toll-like receptors, NOD-like receptors and immune interferon-stimulatory DNA receptors, to provoke inflammation via the MYD88-NF-κB signaling, the inflammasome complex and the cGAS-STING pathway, etc. [26, 165]. Meanwhile, many metabolic intermediates of mitochondria such as succinate, itaconate and fumarate are involved in inflammatory processes [166,167,168,169]. Moreover, extracellular ATP even functions as a DAMP by binding to purinergic receptors (mainly P2X7R) expressed on myeloid cells to mediate inflammatory responses, especially inflammasome activation [170, 171]. Mitochondrial ETC also participates in inflammatory events by activating inflammasomes [172]. Further, the OMM anchored by the cardiolipin acts as a platform for inflammasome localization and activation [173]. Damaged/ubiquitinated mitochondria serve as an intracellular scaffold to recruit NF-κB and activate NF-κB signaling [174]. Mitochondrial antiviral signaling (MAVS) protein endows host cells with stronger immunity against viral infection through activation of NF-κB and IRF3 to inhibit viral replication [175]. These mitochondria-related inflammatory events elucidated in the peripheral system also occur in the CNS. Abnormal protein aggregates in the ALS brain promote neuroinflammation via inducing mtDNA release and activating the cGAS-STING pathway [176]. Succinate in the cerebrospinal fluid (CSF) can be a key neuroinflammatory signal in mice with experimental autoimmune encephalomyelitis (EAE) [177]. Sphingolipid metabolism in astrocytes triggers MAVS-associated protein interaction, boosting CNS inflammation in EAE [178]. Aberrant astrocytic OXPHOS initiates brain inflammation by inducing lipid droplet accumulation [51]. It has also been reported that mitochondrial fragmentation in microglia induced by excessive injury causes irreparable neuroinflammation [128]. Suppressed bioenergetics in myeloid cells drives maladaptive pro-inflammatory responses in the ageing brain [179]. These studies collectively emphasize the involvement of mitochondria in neuroinflammation. How do these mitochondria-related immune responses further damage the neuronal cells? Cell–cell interactions may be a mechanism. Microglia with mitochondrial fragmentation release dysfunctional mitochondria to neurons and evoke neuronal damage directly [128]. Activated microglia can also induce A1 type of reactive astrocytes, which then lead to death of neurons by unknown neurotoxic substances (which might be the dysfunctional mitochondria) [128, 180]. Moreover, aberrant astrocytic OXPHOS induces lipid toxicity and astrogliosis in the brain. The lipid-overloaded astrocytes fail to provide neurotrophic and lipid-clearing support for neurons, resulting in neuronal death [51]. In addition to the abovementioned mechanisms, there may be other pathways involved in the mitochondria-related inflammatory response, which deserve explorations in the future.
Mitochondria protect against proteinopathies by autophagy
Macroautophagy is a pathway of degradation of molecules and sub-cellular elements, including aggregates of misfolded proteins, lipid droplets, nucleic acids and defective organelles (like mitochondria, degradation of which is called mitophagy) [181]. As a lysosome-dependent catabolic process, autophagy also requires the assistance of mitochondria [182]. Specifically, newly formed membranes termed phagophores engulf the cargo, leading to the formation of double-membraned autophagosomes that get delivered to lysosomes for degradation [183]. During these processes, mitochondrial proteins interact with autophagy initiators to promote autophagy [184, 185]. Also, the mitochondrial outer membrane provides the anchorage for autophagy proteins, which is required for the elongation of phagophores [186]. Further, autophagic flux and autophagy gene induction require normal mitochondrial respiration, deficiency of which under amino-acid starvation would repress the autophagy process [187]. Additionally, ROS of the mitochondrial origin are also signaling molecules of autophagy, resulting in either survival or cell death under different circumstances [188, 189]. Mitochondrial fission and fusion could both promote autophagy in certain pathological states [190, 191]. Perturbation of the TFAM-dependent mitochondrial biogenesis induces autophagy via cytosolic mtDNA [192].
NDDs all show accumulation of abnormally folded proteins, including β-amyloid (Aβ) and Tau in AD, α-synuclein (α-syn) in PD, TAR DNA-binding protein 43 (TDP-43) in ALS and huntingtin in Huntington’s disease (HD) [193,194,195]. Under neurodegenerative condition, autophagic pathways are particularly important for removing unwanted proteins and damaged organelles caused by these protein deposits (Fig. 2c). It is reported that mutations in autophagy-associated genes are implicated in different NDDs. Mutations in the autophagy receptor P62 have been identified in cases of familial and sporadic ALS and frontotemporal dementia (FTD) [196]. VPS35 is a PD-linked gene that mediates autophagosome formation and elongation, and also ensures mitochondrial stability and function [197,198,199]. Mutations in the autophagy gene WDR45 cause β-propeller protein-associated neurodegeneration [200, 201]. Further, impairments in the mitochondria-dependent autophagy would decrease the proteolytic flux of α-syn and other autophagic substrates, leading to neuronal apoptosis [202]. As such, enhancers/inducers of autophagic pathways (such as latrepirdine, quercetin, trehalose and spermidine) could provide therapeutic benefit for NDDs [203,204,205,206].
Bridge for communication between cells
Brain is a complex system of interactive networks between cells, with heterogeneous patterns of structural connections [207,208,209,210]. There are intimate communications between astrocytes, neurons and microglia, particularly the astrocyte-neuron crosstalk, in the brain (Fig. 2d). Immunofluorescence labeling showed that a single cortical astrocyte enwraps an average of four neuronal somata and up to 300–600 neuronal dendrites, and one hippocampal astrocyte contacts over 100,000 synapses [211]. These structural interactions make the basis for the formation of physiologically functional units. Special focus is given to metabolic crosstalk between astrocytes and neurons. Neurons expend a considerable amount of ATP and generate excessive ROS, while astrocytes provide neurons with metabolic substrates (lactate) and antioxidants (GSH), which are all generated from mitochondria [124, 126, 212]. In addition to energy supply, astrocytic ATP and metabolic products adenosine, glutamate and D-serine act on their receptors on neurons to regulate synaptic transmission, neuronal excitability and axon regeneration, establishing gliotransmitter-dependent neuron-glial networks [213,214,215,216,217]. It was also found that metabolic coupling of fatty acids (FAs) between neurons and astrocytes protect neurons from FA toxicity. Specifically, neurons have a low capacity for FA consumption in mitochondria for energy production. Hyperactive neurons release toxic lipids, which are taken up by neighboring astrocytes through endocytosis. These transferred FAs flow into astrocytic mitochondria for detoxification [218]. Microglia are highly sensitive to the chemical environment of the brain [219]. They identify dying cells by a wide array of signals including the release of mtDNA, ROS, apoptotic signals and metabolites to initiate phagocytosis, especially for neuronal quality control [209]. Beyond the indirect interactions between microglia and neurons though mitochondria-derived factors, microglia also monitor neuronal status via direct junctions through mitochondria-associated membranes [219].
A more straightforward interplay through mitochondria is the cell-to-cell mitochondrial transfer, in which neurons can release damaged mitochondria to adjacent astrocytes for disposal and recycling [125], and reversely, astrocytes also produce functional mitochondria to support neuronal viability [220]. Similarly, macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain [221], and human brain endothelial cells transfer polarized mitochondria to neurons for protection against ischemia [222]. A general feature of this type of communication is the mitochondrial transfer into neurons for neuronal protection, which is a promising direction for developing neuron-protective methods under brain stresses. Indeed, mitochondrial transplantation is a potential therapeutic strategy. Preclinical studies delivering isolated mitochondria via intraspinal or intracerebral injection to rescue neuronal mitochondrial dysfunction have shown promising effects in acute CNS injuries and neurodegenerative disorders [223, 224].
Mitochondria dysfunctions in neurological diseases and neurophenotypes of mitochondrial diseases
A series of mitochondrial dysfunctions are commonly seen in neurological diseases, including energy hypo-metabolism, decreased oxidation–reduction homeostasis, decline in mitochondrial quality and activity, mitochondrial fragmentation, mtDNA damage, and pro-apoptotic activity [225,226,227,228,229,230,231,232]. In this section, we summarize recent evidence of mitochondrial dysfunction in the ageing brain and in diverse pathological conditions of the CNS including neurodegeneration, mental disorders, brain injuries, motor neuron diseases (MNDs), and brain tumors, with a particular emphasis on omics findings (Fig. 3). We also take an overview of the neurophenotypes of mitochondrial diseases.

Mitochondria in neurological diseases: commonality and specificity. Mitochondrial dysfunctions are commonly seen in neurological diseases, with both commonality and disease specificity from the perspective of mechanism. ASD, autism spectrum disorder; ATP, adenosine triphosphate; BD, bipolar disorder; MDD: major depressive disorder; MNDs, motor neuron diseases; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; TCA, tricarboxylic acid
Ageing
Aging is a normal and inevitable process that predisposes individuals to multiple non-communicable diseases [233, 234]. Although controversy exists over whether aging falls under the category of diseases, efforts can be made to lessen its undesirable impact. Brains of aged people without a diagnosis of neurological disease are reported to show pathological changes including abnormal protein assemblies, neuronal loss and decrease of brain volume [235]. Mitochondrial impairments are also manifested in ageing brains [225]. A recent study analyzing the mammalian RNA-binding proteins (RBPs) [236] showed that Pumilio2 (Pum2) was the only transcript up-regulated in both muscle and brain samples of aged mice and humans. Multi-omics analyses further revealed that this RBP post-transcriptionally targets Mff (the mediator of mitochondrial fission) in old animals. Based on these findings, the authors suggest that modulating the Mff-linked Pum2 level/activity using genetic or pharmacological approaches may restore ageing-related mitochondrial damage, which represents a novel way for anti-aging strategy [236]. Another study presented a single-cell transcriptome atlas of the entire adult Drosophila melanogaster brain sampled across its lifespan, and gene network analysis revealed regulatory states related to OXPHOS [237]. Whether mitochondrial dynamics and respiration are the causes of brain ageing needs to be verified.
Starting from 1930s, researchers discovered that restriction of caloric intake increased lifespan in several species [238,239,240,241], implying that the metabolism process is important for ageing progression. Later, lifespan extension by dietary restriction is found to be mediated by the inhibition of TOR kinase [242, 243], which is a primordial negative regulator of autophagy in organisms from yeast to humans. This points to the critical role of autophagy in the ageing process. Loss-of-function mutations in proteins required for autophagy (TOR, S6K1, Sestrin1, TEFB, Beclin-1) decrease lifespan [243,244,245,246,247], while increased autophagy by pharmacological or genetic methods delays the overall aging and extends longevity [246, 248,249,250,251,252]. The pioneering Mitochondrial Free Radical Theory of Aging emphasizes that mitochondrial ROS is an important factor inducing aging [253]. However, a subsequent study showed that increased ROS production does not always shorten lifespan [254]. Subsequently, the concept that accumulation of mtDNA deletions/mutations may be important in aging came into eyes [255, 256]. mtDNA mutator mice show signs of premature aging including reduced lifespan, reduced fertility, osteoporosis, and hair graying [257]. This notion was in debate later for the observation that the increase of mtDNA mutations was actually far lower than the threshold for mitochondrial changes in normal aging mice [258]. Nevertheless, multiple issues including limitations in research methods and species differences should be taken into account in future studies [259, 260].
Neurodegeneration
Neurodegeneration, referring to the progressive atrophy and loss of function of neurons, occurs in NDDs, including but not limited to AD, PD, ALS, HD, multiple sclerosis (MS), spinal muscular atrophy, FTD, and Creutzfeldt-Jakob disease. Ageing is the primary risk factor for most NDDs [261]. Neurons are targets of disease pathology, although selective neuronal vulnerability is shown in certain neurodegenerative circumstances [262]. Based on the high energy demand of neurons and their definite fate of demise during neurodegeneration, mitochondria appear to be a pathological mechanism of neurodegeneration [38, 262].
AD is the most common NDD, with pathological hallmarks of Aβ plaques and Tau neurofibrillary tangles in the brains of patients. Researchers from St. Jude Children’s Research Hospital profiled the whole proteome and phosphoproteome in frontal cortical samples and CSF of AD patients at different disease stages using deep multi-layer brain proteomics, and found a notable decrease of mitochondrial function in AD [263]. In their later study, further ultra-deep analysis integrating proteomes in cortex, CSF and serum of AD samples revealed that over a half of the differential proteins across three kinds of samples were strikingly mitochondrial proteins and these proteins all showed evident reduction in AD [264]. In contrast to their former study, the current ultra-deep proteomic setting detected larger numbers of differential mitochondrial proteins, which covered multiple aspects of mitochondrial function, especially metabolic processes [264]. Another study characterized tau interactomes and established that tau interacted extensively with proteins involved in mitochondrial bioenergetics. The interaction was beneficial for neuronal bioenergetics, but would decrease with disease severity [265]. Meanwhile, differential gene-expression analyses of the RNA sequencing data in MayoRNAseq dataset also highlighted the down-regulation of mitochondrial respiration and metabolism in AD [266]. A recent omics study also reported reduced mtDNA copy number and mutations of mtDNA in AD [267]. Collectively, mitochondrial energy metabolism is impaired in AD, which is also the leading player in AD progression.
Impairments in energetic pathways also occur in PD. In a study combing omics, biochemical and imaging approaches to reveal the spatiotemporal events associated with Lewy body (LB) formation, researchers found that mitochondrial respiration breakdown and reduced mitochondrial membrane potential are manifested once the LB-like inclusions are formed from α-syn fibrils [268]. Meanwhile, a quantitative proteomics study revealed mitochondrial energetic failure as the earliest event in striatal dopaminergic synapses after α-syn over-expression [269]. In summary, in NDDs represented by AD and PD, collapse in mitochondrial energy metabolism is the earliest and also principal abnormality. From the perspective of protein aggregation in NDDs, autophagy is particularly significant. Impaired autophagy is manifested in most NDDs [270,271,272], while autophagy enhancers counteract neurodegeneration [203, 273].
Psychiatric diseases
The common types of psychiatric disorders include schizophrenia, major depressive disorder (MDD), bipolar disorder (BD), autism spectrum disorder (ASD), addictive behaviors, and obsessive-compulsive disorder. While the majority of patients with mental disorders appear to have widespread mitochondrial dysfunctions, the findings in patients diagnosed with certain psychiatric diseases are less consistent. There are multiple manifestations and mechanisms of mitochondrial dysfunctions in neuropsychiatric disorders, with even phasic characteristics [274,275,276,277]. Defects in bioenergetics in schizophrenia and BD are not only supported by neuroimaging methodologies [278,279,280], but also confirmed by transcriptomics and proteomics in body fluid samples or postmortem brains from patients [281,282,283,284]. However, transcriptomic profiling of the dorso-lateral prefrontal cortex revealed dissimilar mitochondrial alterations between schizophrenia and BD, with 41% of mitochondrial-related genes showing differential expression in the schizophrenia group, whereas 8% in the BD group [285]. This might be caused by the biphasic episodes of this neuropsychiatric disease, as mitochondrial energy metabolism in mania and depressive phases shows opposite and countervailing energy phenotypes [286,287,288]. ASD is highly genetically influenced, with approximately 50% heritability [289]. Whole-genome sequencing (WGS) of ASD revealed that the mtDNA variants accounted for 2% of the ASD genetic variants architecture [290]. Another study investigated the association of mtDNA heteroplasmies (co-existence of mutated and unmutated mtDNA) and content with ASD using ultra-deep sequencing, and showed that they can be used for early risk assessment of future ASD risk in newborns [277]. Although the blood-based mitochondrial respiratory chain function is not a good biomarker for MDD against controls [291], the neuron-derived extracellular vesicles in plasma of MDD subjects contain abnormal levels of proteins involved in mitochondrial dynamics, energy generation, metabolic regulation and anti-oxidant gene responses [292]. In addition, transcriptional profiling of mitochondria-associated genes in post-mortem brains of MDD subjects provided further evidence of oxidative stress in the MDD patients, showing that the ROS-related genes including UCP4 and SOD2 are significantly changed [293]. Some other investigations also support that chronically increased ROS production is the core mechanism in MDD etiology [294,295,296,297], as the stressful life events offer an endocrine basis for the generation of ROS [294, 295].
Brain injuries
Brain injury refers to brain damage caused by an external force, infections, certain diseases, or a lack of oxygen. It is classified into two types depending on the original cause: (1) traumatic brain injury (TBI), like concussion, and (2) non-traumatic brain injury, like stroke, encephalitis and meningitis. The initial stages of TBI are characterized by impaired regulation of cerebral blood flow and metabolism caused by direct tissue damage and hemorrhage. The traumatic site shows an ischemic pattern with inadequate supply of oxygen and glucose, which leads to ATP depletion. The secondary injury develops over time, with release of excitatory neurotransmitters, propagation of damage through energy failure and overload of free radicals [298, 299]. Integrated spatial transcriptomes and metabolome data in injured human brain showed changes in lipid metabolism, energy metabolism, carbohydrate and amino acid metabolism, as well as antioxidant activity [300]. In the same way, stroke also faces these two mitochondrial events, the defective energy metabolism and the imbalanced redox state. An ischemic stroke event occurs when the blood flow to the brain tissue is decreased due to occluded arteries, in which lack of oxygen and nutrients leads to disturbed cellular homeostasis and, eventually cell death. During ischemia–reperfusion, oxygen is restored and ATP is replenished. However, the pro-oxidant enzymatic systems and mitochondria can also employ oxygen as a substrate to generate substantial ROS [231]. For hemorrhagic stroke, the initial bleed leads to an influx of glutamate to the brain parenchyma, which induces Ca2+ overload, membrane depolarization and ROS release. In the second phase of hemorrhagic stroke, ROS is generated in the way similar to the ischemia–reperfusion [301]. Encephalitis and meningitis represent the infection of the brain and the meninges caused by bacteria, virus, fungi and parasites. Mitochondrial oxidative stress underlies the cell death of regulatory T cells in an EAE mouse model [302]. Additionally, genome-wide transcriptomic analysis identified an overt reduction in mtDNA-encoded transcripts in post-mortem brain tissues of herpes simplex virus type-1 (HSV-1) encephalitis (HSE), highlighting mitochondrial damage as a critical event during HSV-1 infection [303]. Collectively, mitochondria play a role in various types of brain injuries due to its multiple functions as an energy factory, a ROS balancer, an innate immune platform, etc.
MNDs
MNDs are a group of neurodegenerative disorders involving both the nervous system and the muscles. ALS is the most common type of MNDs, accounting for 85% of all MND cases [304]. Other types include primary lateral sclerosis, progressive bulbar palsy, progressive muscular atrophy, and spinal-bulbar muscular atrophy [305,306,307,308]. Here, we elaborate on ALS with a focus on motor neurons, to reveal the mitochondrial aspects of the MND pathogenesis. ALS is a genetically heterogeneous disorder, with landmark discoveries of the SOD1 gene mutations in 1993 [309], and C9orf72 in 2011 [310, 311]. As the most abundant enzyme of the SOD family, SOD1 (CuZn-SOD) is primarily a cytosolic enzyme, but it is also reported to localize within the IMS of mitochondria [312, 313]. The mitochondrial SOD1 precludes the exit of mitochondrial superoxide, and as such, protects other cell components from oxidative damage [313]. Further, targeted replacement of SOD1 only in the IMS rescues motor axonopathy of SOD1-deficient mice [314]. C9orf72 is found to be a mitochondria-localized protein, which can be imported into the mitochondrial IMS to regulate OXPHOS by stabilizing mitochondrial complex I assembly [315]. Dominant missense mutations in TARDBP gene (encoding TDP-43) can also cause ALS [316, 317], and the cytoplasmic accumulation of TDP-43 represents a pathological hallmark of ALS [318]. Compelling evidence has revealed that the disease-associated mutations of TARDBP increase TDP-43 mitochondrial localization and cause complex I disassembly [319], while inhibiting the mitochondrial localization of TDP-43 restores mitochondrial bioenergetic malfunctions, neuronal loss, and motor-coordinative and cognitive deficits in TDP-43M337V ALS mice [320]. Apoptosis-inducing factor (AIFM1) localizes in IMS. A study has shown that patients with the Phe210Leu mutation in AIFM1 are afflicted by an inherited axonal polyneuropathy with motor axons being predominantly damaged. This disease-associated mutation in AIFM1 is sufficient to cause misassembly of mitochondrial complexes I and III [321]. Furthermore, a mitochondrial origin for ALS has also been identified in a large family with a late-onset phenotype including MND and cognitive decline, in which a missense mutation in the CHCHD10 gene is detected. CHCHD10 is also a mitochondrial protein located in the IMS and is enriched at CJs [322]. These studies collaboratively show that gene mutations in the IMS-localized proteins lead to axonal atrophy and motor neuron degeneration, and these neurophenotypes are often associated with misassembly of the respiratory complex in mechanism. In addition to the genetic evidence for the association of MNDs with mitochondria, studies using motor neurons derived from induced pluripotent stem cells (iPSCs) from ALS and control subjects also highlighted mitochondrial genes with the most variable expression, especially those involved in the mitochondrial respiratory chain pathway [323]. Probably, motor neurons themselves determine the susceptibility of their mitochondria during MND progression. Motor neurons are highly polarized cells with an extended axon [20, 21], thus facing the special challenge of maintaining mitochondrial integrity and energy homeostasis. Neurons have an intrinsic mechanism responsible for early removal of defective mitochondria from the distal axons via the mitochondrial anchoring protein SNPH [119, 324]. Nevertheless, progressive pathological stress would deplete SNPH and compromise the SNPH-mediated regulation in later disease stages of ALS [119]. These findings all indicate a critical role of mitochondrial respiration in MND pathology, providing insights into the diagnosis and treatment of MNDs in the future.
Brain cancers
Glioma is a common type of tumor in the brain, sometimes in the spinal cord. Among more than 120 different types of brain tumors, about 33% are gliomas. Mitochondria are involved in oncogenesis, from malignant transformation to tumor progression, and even treatment resistance, mainly through the following mechanisms: (1) metabolic flexibility via the interplay between glycolysis and OXPHOS pathways, (2) mitochondrial ROS production, and (3) functional deficits in MOMP and mitochondrial permeability transition (MPT) [325]. Preference of glycolysis over OXPHOS, enhanced ROS generation and abnormalities of mitochondria-mediated apoptotic machinery are frequently observed in various brain malignancies including gliomas [326]. In contrast to tumor cells which display glycolysis metabolic pathways, glioma stem cells rely mainly on the OXPHOS metabolic pathway [327]. Nevertheless, these brain tumor initiating cells harbor fragmented mitochondria, and inhibition of the mitochondrial fission mediator Drp1 leads to decreased oxygen consumption rate and causes metabolic stress in these cells [328].
Coronavirus disease 2019 (COVID-19)-induced neurological manifestations
Acute infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) also leads to neurological features including brain structural changes, acute encephalopathy, paralytic neuromuscular blockade, ischemic strokes and cognitive impairments [329,330,331,332,333]. Evidence shows that the mitochondrial mechanisms might underlie the neurological manifestations of COVID-19. The SARS-CoV-2 invading the brain can be detected in multiple brain regions with a distribution pattern consistent with neurons [329, 334]. SARS-CoV-2 enters host cells through binding of its spike protein to the receptor ACE2, which is also present on neurons [335]. Within host cells, SARS-CoV-2 hijacks double-membrane vesicles derived from mitochondrial membranes to hide and avoid attacks [336]. SARS-CoV-2 is also predicted to have a notable residency signal toward the mitochondrial matrix to alter MAVS function and mitochondrial function [337, 338]. The viral proteins/fragments would also trigger a dramatic reduction in mtDNA content in microglia, as well as activating gliosis and neuroinflammation [329, 339].
Neurophenotypes of mitochondrial diseases
Mitochondrial diseases are caused by mutations in the nDNA and mtDNA that encode mitochondrial proteins or proteins involved in mitochondrial function. This group of diseases are multi-systemic, with substantial involvement of the nervous system. Leber’s hereditary optic neuropathy (LHON) is the first and also one of the most prevalent diseases associated with mtDNA mutations. It is an inheritable neurodegenerative disorder, mainly caused by mutations in mt-ND1, ND4 or ND6. LHON is characterized by blindness due to degeneration of retinal ganglion cells and axons of the optic nerve [340]. Besides the optic nerve, patients with LHON often show brain damage in neuroimaging [341, 342]. Kearn-Saryre-Syndrome (KSS) is caused by a large deletion of mtDNA nucleotides (ranging from 1000 to 10,000) and presents with progressive external ophthalmoplegia, atypical retinal pigmentary degeneration and heart block [343]. Neuroimaging results show cerebral and cerebellar atrophy with focal or diffuse areas of high signal intensity in certain brain regions in KSS patients [344]. Increased tau levels in the CSF are also reported in KSS [345]. In an early case report, patients with MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like syndrome), mainly caused by mutations in mt-TL1, all had lactic acidosis, multiple stroke-like events with secondary neurological deficits, and radiological changes of progressive brain infarction [346]. Other mitochondrial diseases are not listed but reviewed elsewhere [347,348,349]. In addition to the abnormal neuroimaging results and biochemical indications, patients with mitochondrial diseases also experience cognitive problems such as memory impairment, perception deficits, and language deficits [350,351,352], as well as mental health problems like psychosis, chronic confusional states, hallucinations, personality change, or unsteadiness [353,354,355].
Mitochondrial therapies for neurological disorders
Viable mitochondria are critical for the homeostasis of the brain, while mitochondrial malfunctions contribute to the pathogenesis of a variety of neurological conditions. Efficient clearance of damaged mitochondria through mitophagy plays a fundamental role in maintaining mitochondrial and metabolic homeostasis, energy supply, neuronal survival, and health [356]. In neurological diseases, the function of mitophagy is damaged [356], and the impaired mitochondrial functions cannot be compensated for under the condition of an irreversible injury to mitochondria [357,358,359]. Therefore, strategies aimed at supplementation of functional mitochondria have recently gained interest. The earliest trial was in the field of cardiovascular diseases, in which mitochondrial transplantation significantly improved postischemic functional recovery and cellular viability [360]. Later, this concept was extended to the CNS for neuroprotection and neurorecovery [220, 361,362,363]. Here, we summarize the rationale, application and challenges of mitochondrial transplantation for treatment of brain diseases.
Inter-cellular mitochondrial transfer in the brain
Multiple lines of evidence suggest the presence of inter-cellular mitochondrial transfer in the brain, which not only advance our understanding of disease progression, but also provide theoretical foundations for therapeutic strategies of brain diseases [125, 363, 364]. A comprehensive summary of findings is provided in Table 1. It is reported that mitochondria of retinal ganglion cell axons are transferred to adjoining astrocytes for degradation [125]. This autophagy-assisted phagocytosis is named transmitophagy and represents a new way of mitochondrial quality control, which is predicted as a common mechanism in the nervous system [125]. Another mitochondrial quality-control process called mitocytosis has been newly launched in migrating cells like neutrophils. In mitocytosis, damaged mitochondria are released from the rear end of migrating cells to maintain mitochondrial quality and viability, which might share features with transmitophagy [365]. Although mitocytosis has not been reported in the CNS, studies are prospected for its roles in brain pathophysiology, especially within microglia of high mobility. In addition to mitochondrial quality control, mitochondria transfer participates in disease promotion. It is reported that damaged mitochondria released from microglia are sensed by astrocytes to propagate inflammatory signals and provoke neurodegeneration [128]. Nonetheless, the most important aspect of mitochondria transfer lies in its neuro-protective effects. Functional mitochondria of astrocytes are conversely transmitted to neurons, supporting neuronal viability and recovery in stroke [220], or are endocytosed by glioma cell line to inhibit malignant proliferation and enhance glioma radiosensitivity [366]. However, astrocytic mitochondria to glioblastoma can also promote a highly tumorigenic cell phenotype with increased proliferative capacity and self-renewal in a disparate transferring model [367]. Moreover, macrophages, brain endothelial cells and bone marrow mesenchymal stem cells donate their mitochondria to neurons for recovery [221, 222, 224]. Mitochondria transportation almost involves all types of cell in the brain [223, 368]. The aforementioned transferring models provide a theoretical basis for mitochondrial therapy via transplantation.
Mitochondrial transplantation targeting brain diseases
The application of mitochondrial transplantation in neurological disorders is promising (Fig. 4a). For injuries in the CNS including stroke, TBI and spinal cord injury, mitochondrial transfer has been identified as a promising therapeutic strategy [220, 224, 369]. The main strategies for mitochondrial delivery to the brain include intravenous infusion, intra-arterial injection, intraparenchymal and intracerebroventricular transplantation of isolated mitochondria, as well as intravenous delivery of mitochondria-containing extracellular vesicles. Marked effects have been observed, including inhibition of cell apoptosis and oxidative stress, reduction of infarct size and improved neurorecovery [223, 361, 369, 370]. In NDDs including AD, PD and MS, beneficial effects have been reported after intravenous, intracerebroventricular and intranasal administration as well as stereotactic injection of functional mitochondria into the targeted brain regions, or after intracerebroventricular injection of NSCs or mitochondria-containing extracellular vesicles. The beneficial effects include improved mitochondrial function, reduced neuronal loss, decreased gliosis, and significant amelioration of clinical deficits in mouse models [368, 371, 372]. For mental disorders, especially schizophrenia, injection of isolated active normal mitochondria in the prefrontal cortex prevented the occurrence of schizophrenia-like selective attention deficits in a rat model [373]. For brain malignant tumors, starvation-induced endocytosis of exogenous functional mitochondria by glioma cells inhibits their proliferation, promotes death, and enhances radiation sensitivity [366], suggesting the potential application of mitochondrial transplantation for the treatment of glioblastoma and other malignant tumors of the brain.

Advanced mitochondrial therapies for neurological diseases and mitochondrial diseases. a Mitochondrial transplantation via injection of isolated mitochondria, mitochondria-containing vesicles and mitochondria-loaded stem cells is promising for the treatment of brain diseases. b Mitochondrial replacement therapy is conducted by pronuclear transfer or spindle transfer. For pronuclear transfer, a zygote is generated by fertilization and then pronuclei of the zygote containing mutated mtDNA are transferred to the donor’s enucleated zygote. For spindle transfer, the spindle of the oocyte with mtDNA mutation is transferred to the donor’s enucleated oocyte, followed by fertilization. c Mitochondrial genome editing is conducted by editing the nuclease systems using the ZFNs, the TALENs and the CRISPR/Cas9 systems. mtTALENs and mtZFNs are mitochondria-targeted DNA nucleases and promote the degradation of mutant mtDNA for heteroplasmic shifting of mutant mtDNA. Mitochondrial base editing is achievable by DdCBEs, TALED, ZFD and mitoBEs to effectively correct the homoplasmic mtDNA mutation. The mito-Cas9 system enables successful knockin of exogenous DNA into mtDNA, which is promising for manipulating more types of mtDNA base editing. CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-associated Cas9; DdCBE: bacterial cytidine deaminase fused with mitochondrial TALE-linked deaminases; mitoBEs: mtDNA base editors; MRT, mitochondrial replacement therapy; TALEN: transcription activator-like effector nuclease; ZFDs: zinc-finger deaminases; ZFNs: zinc finger nucleases
Challenges and perspectives of mitochondrial transplantation
There are several challenges in mitochondrial transplantation. First, it is challenging to achieve efficient and targeted delivery of functional mitochondria to affected tissues, especially in the CNS with the existence of blood–brain barrier (BBB). Currently, the main mode of mitochondrial administration is the direct injection of isolated mitochondria into the brain lesion [361, 373]. Mesenchymal stem cells (MSCs) loaded with mitochondria also provide therapeutic support for rescuing nerve cells [362, 374], as they have been shown to cross the BBB [375]. Other optimized methods via BBB-penetrating delivery systems may also show therapeutic potentials in the future [376]. Second, retaining the activity or functionality of transferred mitochondria is also a key issue. The integrity of isolated organelles outside the cytoplasm can be fully retained only when they are separated carefully and stored in specific media [377]. In addition, peptide-mediated mitochondrial delivery has been shown as an effective method to sustain the functionality of mitochondria [378]. Third, the immune response triggered by transplanted mitochondria is also a challenge to overcome. Mitochondria derived from different sources, such as allogeneic or xenogeneic sources, may elicit immune reactions and lead to immunological rejection of the transplanted mitochondria or inflammatory responses, as is seen in mitochondrial diseases with congenital genetic mitochondrial dysfunction in all tissues. Strategies to mitigate immune responses, such as immune modulatory approaches or the use of autologous mitochondria, deserve further exploration [379]. Other challenges including ethical concerns, longitudinal effects and consequences are also of important concerns.
Other mitochondrial therapeutic strategies for brain diseases
Chemicals or molecules targeting mitochondria
Some therapeutic targets or small molecules have been developed to (1) alter the mitochondrial signaling pathways or metabolic processes; (2) prevent the organelle-induced damage, including ROS, inflammation and mtDNA; (3) enhance the quality control by clearing damaged mitochondria or altering mitochondrial dynamics; and (4) induce mitochondrial biogenesis [380]. Efficacy has been shown in animal models of brain disease (Table 2).
Pharmacological inhibition of mitochondrial pyruvate carrier by MSDC-0160 ameliorated cerebral glucose metabolism and reduced brain damage in AD patients in a phase 2 clinical trial [381]. Experiments in pre-clinical experimental models of PD showed that MSDC-0160 exerts its effects by targeting energy metabolism [382]. Nicotinamide adenine dinucleotide (NAD+) is a coenzyme/cosubstrate involved in energy metabolism and energy production via participation in pyruvate dehydrogenase, TCA cycle and OXPHOS and activation of sirtuins to comprehensively regulate mitochondrial function [383]. Replenishing the NAD+ pool with molecules such as nicotinamide mononucleotide and nicotinamide riboside shows preventive and therapeutic effects in age-related pathophysiology and disease conditions [384,385,386,387]. SBT-272, a novel molecule targeting the cardiolipin-rich IMM for normal mitochondrial structure, functions to restore mitochondrial structure and respiratory function in motor neurons of the ALS motor cortex [388]. Mitochondria-targeting antioxidants such as MitoQ, and antioxidant peptides like Bendavia (SS31) are protective against mitochondrial damage in brain diseases [389,390,391]. Autophagy is a vital mechanism underlying the effects of metformin to reverse ageing and multiple ageing-related diseases [392,393,394,395]. The natural compound trehalose promotes autophagy to ameliorate neurodegeneration, MNDs and MS [203, 396,397,398,399,400]. Spermidine is an autophagy inducer that extends longevity [250], and ameliorates disease progression in ALS, AD and MS mouse models [206, 401, 402]. Latrepirdine improves neuropathology of AD and PD by stimulating autophagy to reduce neurodegeneration-related protein aggregates in animal models [204, 403,404,405]. Recently, an autophagy-based targeted protein degradation platform has been developed to synthesize chemicals for degrading deposited proteins [406]. By this platform, ATC161 was identified as a promising chemical to treat PD, AD, progressive supranuclear palsy, and ALS [406, 407]. Moreover, UMI-77 induces myeloid leukemia 1-dependent degradation of damaged mitochondria (by mitophagy) and effectively reverses molecular and behavioral phenotypes of AD [408]. Mitochondrial division inhibitor 1 mediates the repression of mitochondrial fragmentation by interfering with the Drp1 assembly at OMM and has shown clinical potential for the treatment of various NDDs and ischemic stroke [409,410,411]. Drp1-derived peptide, P110, has also shown neuroprotective effects in murine models of PD, AD and ALS [128]. PGC-1α and PPAR-γ activators, by stimulating mitochondrial biogenesis, show high significance in the treatment of mitochondrial dysfunction. The FDA-approved drug pioglitazone is an activator of PPAR-γ and its use is associated with a lower risk of dementia [412, 413], and lower incidence of PD and ischemic stroke [414, 415]. Bezafibrate (a pan-PPAR activator), currently used as an antilipemic agent, has been repurposed to correct metabolic defects in mitochondrial myopathies [416, 417], and NDDs [418, 419]. Coniferaldehyde, an agonist of NRF2 that protects mitochondria via targeting mitochondrial biogenesis, also attenuates AD pathology [420]. Despite the promising results in animal models, some mitochondrial therapies entering clinical trials are encountered with failure [421, 422], which may be due to the improper time points for therapy intervention. Particularly, difficulties in noninvasive assessment of mitochondrial function and damage make it hard to determine the optimal time point for the use of mitochondria-targeted compounds. Another main hurdle of mitochondria-targeting therapies for brain diseases is the insufficient brain penetration [422]. To solve this problem, nano-drug delivery systems may enable high bio-availability and targeting of specific regions, representing a promising strategy for mitochondrial drug application in neurological diseases [423]. Moreover, drug conjugation of mitochondrial-targeting moieties such as dequalinium, triphenylphosphonium, and mitochondrial penetrating peptides, can improve the entry of the drugs into the double-membraned mitochondria [424]. Additionally, approved drugs targeting mitochondria including Edaravone, Atovaquone, and Bedaquiline (Table 2), have been repurposed for the treatment of various brain diseases [425,426,427,428,429,430].
Mitochondrial diseases
For the mitochondrial diseases caused by pathogenic variants occurring in the nuclear or mitochondrial genome only symptomatic treatment is available by drugs, metabolic supplements and physical therapy [431,432,433]. Mitochondrial auto-transplantation has a rarely realistic possibility for genetic mitochondrial diseases. Mitochondrial replacement therapy (MRT) (Fig. 4b) by pronuclear transfer and maternal spindle transfer techniques has been developed in human oocytes or embryos [434, 435]. Multiple optimizations have subsequently been conducted on mitochondrial replacement technology. In 2017, Zhang et al. reported the use of MRT to treat a female case of Leigh syndrome carrying mtDNA mutation 8993 T > G. The carrier suffered from a long history of multiple pregnancy losses and offspring deaths due to this disease. She then received MRT treatment and delivered a boy with low neonatal mtDNA mutation [436]. This report raised ethical and legal controversies, as well as scientific questions, guiding the way for refinement of the techniques and highlighting the need for a robust regulatory environment and the importance of cautious clinical implementation in the future.
Genome editing techniques represented by zinc finger nuclease (ZFN) technology, transcription activator-like effector nuclease (TALEN) technology and clustered regularly interspaced short palindromic repeats-associated Cas9 (CRISPR-Cas9) technology have been comprehensively explored. Particularly, the beneficial efficacy of the CRISPR system in clinical practice has opened a new era in treating rare genetic diseases [437], and also shed light on the attempts to edit mitochondrial genome for treating mitochondrial diseases (Fig. 4c). Early studies of mitochondrial gene editing strategies are based on mitochondrial ZFN and TALEN to degrade the damaged mtDNA, which effectively shifts the heteroplasmic level of mtDNA mutation [438, 439]. For homoplasmic pathogenic mtDNA mutations, mitochondrial base editing is an effective way for single-nucleotide conversions. Later, a bacterial cytidine deaminase fused with mito-TALEN was established to induce precise manipulation of mtDNA [440]. Furthermore, other methods for mitochondrial base editing including zinc-finger deaminases [441], TALE-linked deaminases [442], and mtDNA base editors [443], provide a broader scope for mtDNA editing. Nevertheless, engineering the mammalian mtDNA by the CRISPR-Cas technology is hampered by the inability to transport nucleic acids into mitochondria [444]. Recently, a mitochondria-targeting CRISPR-Cas9 system for successful mtDNA editing has been designed, which also represents a promising approach for the treatment of mitochondrial diseases caused by pathogenic mtDNA mutations, especially those with homoplasmic mtDNA mutations [445].
Conclusions
The mitochondrion is a complex organelle, participating in many signaling pathways and cell functions. Mitochondria are involved in the physiological and pathological processes of the brain. A deeper understanding of the basic biology of mitochondria is important for uncovering the mechanisms of brain diseases and facilitates the development of effective therapies. Despite the challenges and obstacles, therapeutic strategies of neurological diseases targeting mitochondria are worth pursuing at long last.
Availability of data and materials
Not applicable.
Abbreviations
- α-syn:
-
α-synuclein
- Aβ:
-
β-amyloid
- AD:
-
Alzheimer’s disease
- AIFM1:
-
Apoptosis-inducing factor
- ALS:
-
Amyotrophic lateral sclerosis
- ASD:
-
Autism spectrum disorder
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood-brain barrier
- BD:
-
Bipolar disorder
- CJs:
-
Cristae junctions
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- DAMP:
-
Danger-associated molecular pattern
- Drp1:
-
Dynamin-related protein 1
- EAE:
-
Experimental autoimmune encephalomyelitis
- ER:
-
Endoplasmic reticulum
- ETC:
-
Electron transport chain
- FAs:
-
Fatty acids
- Fis1:
-
Fission 1
- FTD:
-
Frontotemporal dementia
- GPx:
-
Glutathione peroxidase
- GSH:
-
Glutathione
- H2O2 :
-
Hydrogen peroxide
- HD:
-
Huntington’s disease
- HSE:
-
Herpes simplex virus type-1 encephalitis
- HSV-1:
-
Herpes simplex virus type-1
- IBM:
-
Inner boundary membrane
- IFN:
-
Immune interferon
- IL-6:
-
Interleukin 6
- IMM:
-
Inner mitochondrial membrane
- IMS:
-
Inter-membrane space
- iPSC:
-
Induced pluripotent stem cell
- KSS:
-
Kearn-Saryre-Syndrome
- LB:
-
Lewy body
- LHON:
-
Leber’s hereditary optic neuropathy
- MAMs:
-
Mitochondria-associated ER membranes
- MAVS:
-
Mitochondrial antiviral signaling
- MDD:
-
Major depressive disorder
- Mff:
-
Mitochondrial fission factor
- MNDs:
-
Motor neuron diseases
- MOMP:
-
Mitochondrial outer membrane permeabilization
- MRT:
-
Mitochondrial replacement therapy
- MS:
-
Multiple sclerosis
- MSC:
-
Mesenchymal stem cell
- mtDNA:
-
Mitochondrial DNA
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NDD:
-
Neurodegenerative disease
- nDNA:
-
Nuclear DNA
- NOXs:
-
NADPH oxidases
- NSC:
-
Neural stem cell
- OMM:
-
Outer mitochondrial membrane
- Opa1:
-
Optic atrophy gene 1
- OXPHOS:
-
Oxidative phosphorylation
- PD:
-
Parkinson's disease
- PDRX:
-
Peroxiredoxin
- PGC-1α:
-
Peroxisome proliferator-activated receptor γ coactivator 1-alpha
- PPAR:
-
Peroxisome proliferator-activated receptor
- Pum2:
-
Pumilio2
- RBP:
-
RNA-binding protein
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TALEN:
-
Transcription activator-like effector nuclease
- TBI:
-
Traumatic brain injury
- TCA:
-
Tricarboxylic acid
- TDP-43:
-
TAR DNA-binding protein 43
- TFAM:
-
Mitochondrial transcription factor A
- ZFN:
-
Zinc finger nuclease
References
Pereda AE. Electrical synapses and their functional interactions with chemical synapses. Nat Rev Neurosci. 2014;15:250–63.
Buzsaki G, Kaila K, Raichle M. Inhibition and brain work. Neuron. 2007;56:771–83.
Briscoe J, Marin O. Looking at neurodevelopment through a big data lens. Science. 2020;369(6510):eaaz8627.
Pulido C, Ryan TA. Synaptic vesicle pools are a major hidden resting metabolic burden of nerve terminals. Sci Adv. 2021;7:eabi9027.
Du F, Zhu XH, Zhang Y, Friedman M, Zhang N, Ugurbil K, et al. Tightly coupled brain activity and cerebral ATP metabolic rate. ProcNatl Acad Sci U S A. 2008;105:6409–14.
Alegre-Abarrategui J, Brimblecombe KR, Roberts RF, Velentza-Almpani E, Tilley BS, Bengoa-Vergniory N, et al. Selective vulnerability in alpha-synucleinopathies. Acta Neuropathol. 2019;138:681–704.
Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122:877–902.
Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21:363–83.
Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, et al. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell. 2011;8:59–71.
De Virgiliis F, Hutson TH, Palmisano I, Amachree S, Miao J, Zhou L, et al. Enriched conditioning expands the regenerative ability of sensory neurons after spinal cord injury via neuronal intrinsic redox signaling. Nat Commun. 2020;11:6425.
Morant-Ferrando B, Jimenez-Blasco D, Alonso-Batan P, Agulla J, Lapresa R, Garcia-Rodriguez D, et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat Metab. 2023;5:1290–302.
Graves SM, Xie Z, Stout KA, Zampese E, Burbulla LF, Shih JC, et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat Neurosci. 2020;23:15–20.
Gorg B, Qvartskhava N, Keitel V, Bidmon HJ, Selbach O, Schliess F, et al. Ammonia induces RNA oxidation in cultured astrocytes and brain in vivo. Hepatology. 2008;48:567–79.
Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019;30:630–55.
Khacho M, Harris R, Slack RS. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci. 2019;20:34–48.
Ross JM, Stewart JB, Hagstrom E, Brene S, Mourier A, Coppotelli G, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501:412–5.
Lawrence G, Holley CL, Schroder K. Parkinson’s disease: connecting mitochondria to inflammasomes. Trends Immunol. 2022;43:877–85.
Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–83.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–53.
Hagemann C, Moreno GC, Guetta L, Tyzack G, Chiappini C, Legati A, et al. Axonal length determines distinct homeostatic phenotypes in human iPSC derived motor neurons on a bioengineered platform. Adv Healthc Mater. 2022;11:e2101817.
Sabry J, O’Connor TP, Kirschner MW. Axonal transport of tubulin in Ti1 pioneer neurons in situ. Neuron. 1995;14:1247–56.
DuBoff B, Gotz J, Feany MB. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron. 2012;75:618–32.
Pirooznia SK, Yuan C, Khan MR, Karuppagounder SS, Wang L, Xiong Y, et al. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener. 2020;15:17.
Neel DV, Basu H, Gunner G, Bergstresser MD, Giadone RM, Chung H, et al. Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration. Neuron. 2023;111:1222–40.
Qing H, Desrouleaux R, Israni-Winger K, Mineur YS, Fogelman N, Zhang C, et al. Origin and function of stress-induced IL-6 in murine models. Cell. 2020;182:372–87.
West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17:363–75.
Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, et al. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358:801–7.
Styr B, Gonen N, Zarhin D, Ruggiero A, Atsmon R, Gazit N, et al. Mitochondrial regulation of the hippocampal firing rate set point and seizure susceptibility. Neuron. 2019;102:1009–24.
Nicholls DG. Brain mitochondrial calcium transport: Origins of the set-point concept and its application to physiology and pathology. Neurochem Int. 2017;109:5–12.
Edwards R, Eaglesfield R, Tokatlidis K. The mitochondrial intermembrane space: the most constricted mitochondrial sub-compartment with the largest variety of protein import pathways. Open Biol. 2021;11:210002.
Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, et al. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–69.
Skulachev VP. Cytochrome c in the apoptotic and antioxidant cascades. Febs Lett. 1998;423:275–80.
Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, et al. Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Mol Cell. 2015;58:1001–14.
Aihara T, Nakamura N, Honda S, Hirose S. A novel potential role for gametogenetin-binding protein 1 (GGNBP1) in mitochondrial morphogenesis during spermatogenesis in mice. Biol Reprod. 2009;80:762–70.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54.
Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85:133–60.
Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol-Mech. 2020;15:235–59.
Cunnane SC, Trushina E, Morland C, Prigione A, Casadesus G, Andrews ZB, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2020;19:609–33.
Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019;99:949–1045.
Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity toglucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–9.
Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103.
Saab AS, Tzvetavona ID, Trevisiol A, Baltan S, Dibaj P, Kusch K, et al. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron. 2016;91:119–32.
Pan RY, He L, Zhang J, Liu X, Liao Y, Gao J, et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34:634–48.
Hu Y, Cao K, Wang F, Wu W, Mai W, Qiu L, et al. Dual roles of hexokinase 2 in shaping microglial function by gating glycolytic flux and mitochondrial activity. Nat Metab. 2022;4:1756–74.
Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19:49–57.
Segarra-Mondejar M, Casellas-Diaz S, Ramiro-Pareta M, Muller-Sanchez C, Martorell-Riera A, Hermelo I, et al. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018;37:e97368.
Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017;26:353–60.
Hall CN, Klein-Flugge MC, Howarth C, Attwell D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci. 2012;32:8940–51.
Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–77.
Gonzalez-Rodriguez P, Zampese E, Stout KA, Guzman JN, Ilijic E, Yang B, et al. Disruption of mitochondrial complex I induces progressive Parkinsonism. Nature. 2021;599:650–6.
Mi Y, Qi G, Vitali F, Shang Y, Raikes AC, Wang T, et al. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat Metab. 2023;5:445–65.
Leng L, Yuan Z, Pan R, Su X, Wang H, Xue J, et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to beta-amyloid clearance. Nat Metab. 2022;4:1287–305.
Zhang Y, Wong HS. Are mitochondria the main contributor of reactive oxygen species in cells? J Exp Biol. 2021. https://doi.org/10.1242/jeb.221606.
Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–9.
Wong HS, Benoit B, Brand MD. Mitochondrial and cytosolic sources of hydrogen peroxide in resting C2C12 myoblasts. Free Radic Bio Med. 2019;130:140–50.
He A, Dean JM, Lodhi IJ. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol. 2021;31:656–70.
Cederbaum AI. Molecular mechanisms of the microsomal mixed function oxidases and biological and pathological implications. Redox Biol. 2015;4:60–73.
Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J. 1972;128:617–30.
Hernansanz-Agustin P, Ramos E, Navarro E, Parada E, Sanchez-Lopez N, Pelaez-Aguado L, et al. Mitochondrial complex I deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017;12:1040–51.
Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–5.
Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci U S A. 2012;109:20065–70.
Yardeni T, Cristancho AG, McCoy AJ, Schaefer PM, McManus MJ, Marsh ED, et al. An mtDNA mutant mouse demonstrates that mitochondrial deficiency can result in autism endophenotypes. Proc Natl Acad Sci U S A. 2021;118(6):e2021429118.
Wilson C, Nunez MT, Gonzalez-Billault C. Contribution of NADPH oxidase to the establishment of hippocampal neuronal polarity in culture. J Cell Sci. 2015;128:2989–95.
Munnamalai V, Weaver CJ, Weisheit CE, Venkatraman P, Agim ZS, Quinn MT, et al. Bidirectional interactions between NOX2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J Neurochem. 2014;130:526–40.
Munnamalai V, Suter DM. Reactive oxygen species regulate F-actin dynamics in neuronal growth cones and neurite outgrowth. J Neurochem. 2009;108:644–61.
Wu LJ, Wu G, Akhavan SM, Baker A, Jia Y, Fahey FH, et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci. 2012;15:565–73.
Holley AK, Bakthavatchalu V, Velez-Roman JM, St CD. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci. 2011;12:7114–62.
Ribas V, Garcia-Ruiz C, Fernandez-Checa JC. Glutathione and mitochondria. Front Pharmacol. 2014;5:151.
Huang H, Zhang S, Li Y, Liu Z, Mi L, Cai Y, et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun. 2021;12:3720.
Kropotov A, Usmanova N, Serikov V, Zhivotovsky B, Tomilin N. Mitochondrial targeting of human peroxiredoxin V protein and regulation of PRDX5 gene expression by nuclear transcription factors controlling biogenesis of mitochondria. FEBS J. 2007;274:5804–14.
Lee DG, Kam MK, Lee SR, Lee HJ, Lee DS. Peroxiredoxin 5 deficiency exacerbates iron overload-induced neuronal death via ER-mediated mitochondrial fission in mouse hippocampus. Cell Death Dis. 2020;11:204.
Holzerova E, Danhauser K, Haack TB, Kremer LS, Melcher M, Ingold I, et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain. 2016;139:346–54.
Fontana GA, Gahlon HL. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020;48:11244–58.
Rackham O, Filipovska A. Organization and expression of the mammalian mitochondrial genome. Nat Rev Genet. 2022;23:606–23.
Carelli V, Chan DC. Mitochondrial DNA: impacting central and peripheral nervous systems. Neuron. 2014;84:1126–42.
Kummer E, Ban N. Mechanisms and regulation of protein synthesis in mitochondria. Nat Rev Mol Cell Bio. 2021;22:307–25.
Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–38.
Vercellino I, Sazanov LA. The assembly, regulation and function of the mitochondrial respiratory chain. Nat Rev Mol Cell Biol. 2022;23:141–61.
Song S, Pursell ZF, Copeland WC, Longley MJ, Kunkel TA, Mathews CK. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc Natl Acad Sci U S A. 2005;102:4990–5.
Guo X, Xu W, Zhang W, Pan C, Thalacker-Mercer AE, Zheng H, et al. High-frequency and functional mitochondrial DNA mutations at the single-cell level. Proc Natl Acad Sci U S A. 2023;120:e2093449176.
Lu J, Sharma LK, Bai Y. Implications of mitochondrial DNA mutations and mitochondrial dysfunction in tumorigenesis. Cell Res. 2009;19:802–15.
Wolf DM, Segawa M, Kondadi AK, Anand R, Bailey ST, Reichert AS, et al. Individual cristae within the same mitochondrion displaydifferent membrane potentials and are functionally independent. EMBO J. 2019;38:e101056.
Kondadi AK, Anand R, Reichert AS. Cristae membrane dynamics - a paradigm change. Trends Cell Biol. 2020;30:923–36.
Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. 2016;78:505–31.
Gao S, Hu J. Mitochondrial fusion: the machineries in and out. Trends Cell Biol. 2021;31:62–74.
Kraus F, Roy K, Pucadyil TJ, Ryan MT. Function and regulation of the divisome for mitochondrial fission. Nature. 2021;590:57–66.
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5.
Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten LH, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. 2021;593:435–9.
Quintana-Cabrera R, Scorrano L. Determinants and outcomes of mitochondrial dynamics. Mol Cell. 2023;83:857–76.
Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Bio. 2020;21:204–24.
Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–71.
Hackenbrock CR. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J Cell Biol. 1966;30:269–97.
Latorre-Muro P, O’Malley KE, Bennett CF, Perry EA, Balsa E, Tavares C, et al. A cold-stress-inducible PERK/OGT axis controls TOM70-assisted mitochondrial protein import and cristae formation. Cell Metab. 2021;33:598–614.
Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67.
Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–44.
Prinz WA, Toulmay A, Balla T. The functional universe of membrane contact sites. Nat Rev Mol Cell Bio. 2020;21:7–24.
Petrungaro C, Kornmann B. Lipid exchange at ER-mitochondria contact sites: a puzzle falling into place with quite a few pieces missing. Curr Opin Cell Biol. 2019;57:71–6.
Iwata R, Casimir P, Vanderhaeghen P. Mitochondrial dynamics in postmitotic cells regulate neurogenesis. Science. 2020;369:858–62.
Zhao Y, Hu D, Wang R, Sun X, Ropelewski P, Hubler Z, et al. ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat Commun. 2022;13:1121.
Xu P, Chang JC, Zhou X, Wang W, Bamkole M, Wong E, et al. GSAP regulates lipid homeostasis and mitochondrial function associated with Alzheimer’s disease. J Exp Med. 2021;218:e20202446.
Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017;36:3356–71.
Hering T, Kojer K, Birth N, Hallitsch J, Taanman JW, Orth M. Mitochondrial cristae remodelling is associated with disrupted OPA1 oligomerisation in the Huntington’s disease R6/2 fragment model. Exp Neurol. 2017;288:167–75.
Wu W, Zhao D, Shah S, Zhang X, Lai M, Yang D, et al. OPA1 overexpression ameliorates mitochondrial cristae remodeling, mitochondrial dysfunction, and neuronal apoptosis in prion diseases. Cell Death Dis. 2019;10:710.
Lai Y, Lin P, Chen M, Zhang Y, Chen J, Zheng M, et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020;34:101503.
Youle RJ. Mitochondria-Striking a balance between host and endosymbiont. Science. 2019;365(6454):eaaw9855.
Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–22.
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–24.
Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–35.
Cheng A, Wan R, Yang JL, Kamimura N, Son TG, Ouyang X, et al. Involvement of PGC-1alpha in the formation and maintenance of neuronal dendritic spines. Nat Commun. 2012;3:1250.
Goldsmith J, Ordureau A, Harper JW, Holzbaur E. Brain-derived autophagosome profiling reveals the engulfment of nucleoid-enriched mitochondrial fragments by basal autophagy in neurons. Neuron. 2022;110:967–76.
Han S, Zhang M, Jeong YY, Margolis DJ, Cai Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy. 2021;17:4182–201.
Yazdankhah M, Ghosh S, Shang P, Stepicheva N, Hose S, Liu H, et al. BNIP3L-mediated mitophagy is required for mitochondrial remodeling during the differentiation of optic nerve oligodendrocytes. Autophagy. 2021;17:3140–59.
Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile Parkinsonism. Nature. 1998;392:605–8.
Lucking CB, Abbas N, Durr A, Bonifati V, Bonnet AM, de Broucker T, et al. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson’s Disease and the French Parkinson’s Disease Genetics Study Group. Lancet. 1998;352:1355–6.
Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895–900.
Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MM, et al. PARK6-linked parkinsonism occurs in several European families. Ann Neurol. 2002;51:14–8.
Jeong YY, Han S, Jia N, Zhang M, Sheshadri P, Tammineni P, et al. Broad activation of the Parkin pathway induces synaptic mitochondrial deficits in early tauopathy. Brain. 2022;145:305–23.
Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell. 2008;132:137–48.
Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, et al. Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron. 2017;94:595–610.
Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–21.
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83.
Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, et al. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231–42.
Maltecca F, Aghaie A, Schroeder DG, Cassina L, Taylor BA, Phillips SJ, et al. The mitochondrial protease AFG3L2 is essential for axonal development. J Neurosci. 2008;28:2827–36.
Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, et al. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–8.
Baxter PS, Hardingham GE. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic Bio Med. 2016;100:147–52.
Ragupathy H, Vukku M, Barodia SK. Cell-type-specific mitochondrial quality control in the brain: a plausible mechanism of neurodegeneration. Int J Mol Sci. 2023;24:14421.
Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn GN, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci. 2019;22:1635–48.
Bock FJ, Tait S. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Bio. 2020;21:85–100.
Vringer E, Tait S. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023;30:304–12.
Pemberton JM, Pogmore JP, Andrews DW. Neuronal cell life, death, and axonal degeneration as regulated by the BCL-2 family proteins. Cell Death Differ. 2021;28:108–22.
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329.
Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185:3214–31.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85.
Zhang W, Li G, Luo R, Lei J, Song Y, Wang B, et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp Mol Med. 2022;54:129–42.
Prochnicki T, Vasconcelos MB, Robinson KS, Mangan M, De Graaf D, Shkarina K, et al. Mitochondrial damage activates the NLRP10 inflammasome. Nat Immunol. 2023;24:595–603.
Evavold CL, Hafner-Bratkovic I, Devant P, D’Andrea JM, Ngwa EM, Borsic E, et al. Control of gasdermin D oligomerization and pyroptosis by the ragulator-Rag-mTORC1 pathway. Cell. 2021;184:4495–511.
Miao R, Jiang C, Chang WY, Zhang H, An J, Ho F, et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity. 2023;56:2523–41.
He Z, An S, Chen J, Zhang S, Tan C, Yu J, et al. Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target. Proc Natl Acad Sci U S A. 2020;117:23869–78.
Van Schoor E, Ospitalieri S, Moonen S, Tome SO, Ronisz A, Ok O, et al. Increased pyroptosis activation in white matter microglia is associated with neuronal loss in ALS motor cortex. Acta Neuropathol. 2022;144:393–411.
Moonen S, Koper MJ, Van Schoor E, Schaeverbeke JM, Vandenberghe R, von Arnim C, et al. Pyroptosis in Alzheimer’s disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol. 2023;145:175–95.
Poh L, Fann DY, Wong P, Lim HM, Foo SL, Kang SW, et al. AIM2 inflammasome mediates hallmark neuropathological alterations and cognitive impairment in a mouse model of vascular dementia. Mol Psychiatr. 2021;26:4544–60.
He X, Yang W, Zeng Z, Wei Y, Gao J, Zhang B, et al. NLRP3-dependent pyroptosis is required for HIV-1 gp120-induced neuropathology. Cell Mol Immunol. 2020;17:283–99.
de Dios C, Abadin X, Roca-Agujetas V, Jimenez-Martinez M, Morales A, Trullas R, et al. Inflammasome activation under high cholesterol load triggers a protective microglial phenotype while promoting neuronal pyroptosis. Transl Neurodegener. 2023;12:10.
Li S, Sun Y, Song M, Song Y, Fang Y, Zhang Q, et al. NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in astrocyte pathological injury in mouse model of depression. JCI Insight. 2021;6:e146852.
Li F, Jiang SY, Tian T, Li WJ, Xue Y, Du RH, et al. Kir61/K-ATP channel in astrocytes is an essential negative modulator of astrocytic pyroptosis in mouse model of depression. Theranostics. 2022;12:6611–25.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
Tsuchiya K, Nakajima S, Hosojima S, Thi ND, Hattori T, Manh LT, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun. 2019;10:2091.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354–63.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17:948–60.
Friedmann AJ, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Qiu S, Zhong X, Meng X, Li S, Qian X, Lu H, et al. Mitochondria-localized cGAS suppresses ferroptosis to promote cancer progression. Cell Res. 2023;33:299–311.
Ryan SK, Zelic M, Han Y, Teeple E, Chen L, Sadeghi M, et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat Neurosci. 2023;26:12–26.
Bao WD, Pang P, Zhou XT, Hu F, Xiong W, Chen K, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28:1548–62.
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–60.
Ayton S, Portbury S, Kalinowski P, Agarwal P, Diouf I, Schneider JA, et al. Regional brain iron associated with deterioration in Alzheimer’s disease: a large cohort study and theoretical significance. Alzheimers Dement. 2021;17:1244–56.
Thomas G, Leyland LA, Schrag AE, Lees AJ, Acosta-Cabronero J, Weil RS. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2020;91:418–25.
Thomas G, Zarkali A, Ryten M, Shmueli K, Gil-Martinez AL, Leyland LA, et al. Regional brain iron and gene expression provide insights into neurodegeneration in Parkinson’s disease. Brain. 2021;144:1787–98.
Rothammer N, Woo MS, Bauer S, Binkle-Ladisch L, Di Liberto G, Egervari K, et al. G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis. Sci Adv. 2022;8:eabm5500.
Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. 2021;41:101947.
Adeniyi PA, Gong X, MacGregor E, Degener-O’Brien K, McClendon E, Garcia M, et al. Ferroptosis of microglia in aging human white matter injury. Ann Neurol. 2023;94:1048–66.
Luoqian J, Yang W, Ding X, Tuo QZ, Xiang Z, Zheng Z, et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell Mol Immunol. 2022;19:913–24.
Xian H, Karin M. Oxidized mitochondrial DNA: a protective signal gone awry. Trends Immunol. 2023;44:188–200.
Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238–42.
Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–7.
Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–19.
Zecchini V, Paupe V, Herranz-Montoya I, Janssen J, Wortel I, Morris JL, et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature. 2023;615:499–506.
Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–9.
Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. 2017;47:15–31.
Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. 2022;23:692–704.
Pizzuto M, Pelegrin P. Cardiolipin in immune signaling and cell death. Trends Cell Biol. 2020;30:892–903.
Harding O, Holzer E, Riley JF, Martens S, Holzbaur E. Damaged mitochondria recruit the effector NEMO to activate NF-kappaB signaling. Mol Cell. 2023;83:3188–204.
Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–82.
Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 2020;183:636–49.
Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa A, Kwok CK, Leonardi T, et al. Macrophage-derived extracellular succinate licenses neural stem cells to suppress chronic neuroinflammation. Cell Stem Cell. 2018;22:355–68.
Chao CC, Gutierrez-Vazquez C, Rothhammer V, Mayo L, Wheeler MA, Tjon EC, et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 2019;179:1483–98.
Minhas PS, Latif-Hernandez A, McReynolds MR, Durairaj AS, Wang Q, Rubin A, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590:122–8.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Rambold AS, Lippincott-Schwartz J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle. 2011;10:4032–8.
Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29-34.
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51.
Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–75.
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67.
Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. EMBO J. 2011;30:2101–14.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–60.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12:999–1014.
Li Y, Chen H, Yang Q, Wan L, Zhao J, Wu Y, et al. Increased Drp1 promotes autophagy and ESCC progression by mtDNA stress mediated cGAS-STING pathway. J Exp Clin Cancer Res. 2022;41:76.
Li Y, Yang Q, Chen H, Yang X, Han J, Yao X, et al. TFAM downregulation promotes autophagy and ESCC survival through mtDNA stress-mediated STING pathway. Oncogene. 2022;41:3735–46.
Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51.
Jucker M, Walker LC. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci. 2018;21:1341–9.
Saudou F, Humbert S. The biology of huntingtin. Neuron. 2016;89:910–26.
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–62.
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7.
Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun. 2014;5:3828.
Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–56.
Zhao YG, Sun L, Miao G, Ji C, Zhao H, Sun H, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11:881–90.
Wan H, Wang Q, Chen X, Zeng Q, Shao Y, Fang H, et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy. 2020;16:531–47.
Arduino DM, Esteves AR, Cortes L, Silva DF, Patel B, Grazina M, et al. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum Mol Genet. 2012;21:4680–702.
Rusmini P, Cortese K, Crippa V, Cristofani R, Cicardi ME, Ferrari V, et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy. 2019;15:631–51.
Steele JW, Ju S, Lachenmayer ML, Liken J, Stock A, Kim SH, et al. Latrepirdine stimulates autophagy and reduces accumulation ofalpha-synuclein in cells and in mouse brain. Mol Psychiatry. 2013;18:882–8.
Han X, Xu T, Fang Q, Zhang H, Yue L, Hu G, et al. Quercetin hinders microglial activation to alleviate neurotoxicity via the interplay between NLRP3 inflammasome and mitophagy. Redox Biol. 2021;44:102010.
Choi SH, Yousefian-Jazi A, Hyeon SJ, Nguyen P, Chu J, Kim S, et al. Modulation of histone H3K4 dimethylation by spermidine ameliorates motor neuron survival and neuropathology in a mouse model of ALS. J Biomed Sci. 2022;29:106.
Soto JS, Jami-Alahmadi Y, Chacon J, Moye SL, Diaz-Castro B, Wohlschlegel JA, et al. Astrocyte-neuron subproteomes and obsessive-compulsive disorder mechanisms. Nature. 2023;616:764–73.
Wheeler MA, Clark IC, Lee HG, Li Z, Linnerbauer M, Rone JM, et al. Droplet-based forward genetic screening of astrocyte-microglia cross-talk. Science. 2023;379:1023–30.
Cserep C, Posfai B, Denes A. Shaping neuronal fate: functional heterogeneity of direct microglia-neuron interactions. Neuron. 2021;109:222–40.
Medina-Flores F, Hurtado-Alvarado G, Contis-Montes DOA, Lopez-Cervantes SP, Konigsberg M, Deli MA, et al. Sleep loss disrupts pericyte-brain endothelial cell interactions impairing blood-brain barrier function. Brain Behav Immun. 2020;89:118–32.
Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–92.
Calabrese G, Morgan B, Riemer J. Mitochondrial glutathione: regulation and functions. Antioxid Redox Sign. 2017;27:1162–77.
Lezmy J, Arancibia-Carcamo IL, Quintela-Lopez T, Sherman DL, Brophy PJ, Attwell D. Astrocyte Ca(2+)-evoked ATP release regulates myelinated axon excitability and conduction speed. Science. 2021;374:eabh2858.
Ma Z, Stork T, Bergles DE, Freeman MR. Neuromodulators signal through astrocytes to alter neural circuit activity and behaviour. Nature. 2016;539:428–32.
Wang F, Ruppell KT, Zhou S, Qu Y, Gong J, Shang Y, et al. Gliotransmission and adenosine signaling promote axon regeneration. Dev Cell. 2023;58:660–76.
Yang J, Vitery M, Chen J, Osei-Owusu J, Chu J, Qiu Z. Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron. 2019;102:813–27.
Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, et al. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–84.
Ioannou MS, Jackson J, Sheu SH, Chang CL, Weigel AV, Liu H, et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell. 2019;177:1522–35.
Cserep C, Posfai B, Lenart N, Fekete R, Laszlo ZI, Lele Z, et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science. 2020;367:528–37.
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. 2016;535:551–5.
van der Vlist M, Raoof R, Willemen H, Prado J, Versteeg S, Martin GC, et al. Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron. 2022;110:613–26.
D’Souza A, Burch A, Dave KM, Sreeram A, Reynolds MJ, Dobbins DX, et al. Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J Control Release. 2021;338:505–26.
Norat P, Soldozy S, Sokolowski JD, Gorick CM, Kumar JS, Chae Y, et al. Mitochondrial dysfunction in neurological disorders: exploring mitochondrial transplantation. NPJ Regen Med. 2020;5:22.
Li H, Wang C, He T, Zhao T, Chen YY, Shen YL, et al. Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics. 2019;9:2017–35.
Amorim JA, Coppotelli G, Rolo AP, Palmeira CM, Ross JM, Sinclair DA. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol. 2022;18:243–58.
Wang W, Zhao F, Ma X, Perry G, Zhu X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener. 2020;15:30.
Cheng XT, Huang N, Sheng ZH. Programming axonal mitochondrial maintenance and bioenergetics in neurodegeneration and regeneration. Neuron. 2022;110:1899–923.
Daniels TE, Olsen EM, Tyrka AR. Stress and psychiatric disorders: the role of mitochondria. Annu Rev Clin Psycho. 2020;16:165–86.
Filiou MD, Sandi C. Anxiety and brain mitochondria: a bidirectional crosstalk. Trends Neurosci. 2019;42:573–88.
Cheng G, Kong RH, Zhang LM, Zhang JN. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol. 2012;167:699–719.
Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol. 2018;16:263–75.
Mudassar F, Shen H, O’Neill G, Hau E. Targeting tumor hypoxia and mitochondrial metabolism with anti-parasitic drugs to improve radiation response in high-grade gliomas. J Exp Clin Canc Res. 2020;39:208.
Gladyshev TV, Gladyshev VN. A disease or not a disease? Aging as a pathology. Trends Mol Med. 2016;22:995–6.
The LHL. Is ageing a disease? Lancet Health Longev. 2022;3:e448.
Wyss-Coray T. Ageing, neurodegeneration and brain rejuvenation. Nature. 2016;539:180–6.
D’Amico D, Mottis A, Potenza F, Sorrentino V, Li H, Romani M, et al. The RNA-binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging. Mol Cell. 2019;73:775–87.
Davie K, Janssens J, Koldere D, De Waegeneer M, Pech U, Kreft L, et al. A single-cell transcriptome atlas of the aging drosophila brain. Cell. 2018;174:982–98.
McCay CM, Maynard LA, Sperling G, Barnes LL. Retarded growth, life span, ultimate body size and age changes in the albino rat after feeding diets restricted in calories. Nutr Rev. 1975;33:241–3.
Mitchell SJ, Bernier M, Mattison JA, Aon MA, Kaiser TA, Anson RM, et al. Daily fasting improves health and survival in male mice independent of diet composition and calories. Cell Metab. 2019;29:221–8.
Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, et al. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun. 2017;8:14063.
Spadaro O, Youm Y, Shchukina I, Ryu S, Sidorov S, Ravussin A, et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science. 2022;375:671–7.
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–90.
Kaeberlein M, Powers RR, Steffen KK, Westman EA, Hu D, Dang N, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–6.
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–4.
Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science. 2010;327:1223–8.
Fernandez AF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–40.
Lapierre LR, De Magalhaes FC, McQuary PR, Chu CC, Visvikis O, Chang JT, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4:2267.
Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–84.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5.
Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14.
Knuppertz L, Hamann A, Pampaloni F, Stelzer E, Osiewacz HD. Identification of autophagy as a longevity-assurance mechanism in the aging model Podospora anserina. Autophagy. 2014;10:822–34.
Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature. 2015;521:525–8.
Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300.
Doonan R, McElwee JJ, Matthijssens F, Walker GA, Houthoofd K, Back P, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Gene Dev. 2008;22:3236–41.
Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18:6927–33.
Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–5.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–23.
Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, et al. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–3.
Redman LM, Smith SR, Burton JH, Martin CK, Il’Yasova D, Ravussin E. Metabolic slowing and reduced oxidative damage with sustained caloric restriction support the rate of living and oxidative damage theories of aging. Cell Metab. 2018;27:805–15.
Wolf AM. MtDNA mutations and aging-not a closed case after all? Signal Transduct Target Therapy. 2021;6:56.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81.
Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018;21:1350–8.
Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, et al. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron. 2020;105:975–91.
Wang H, Dey KK, Chen PC, Li Y, Niu M, Cho JH, et al. Integrated analysis of ultra-deep proteomes in cortex, cerebrospinal fluid and serum reveals a mitochondrial signature in Alzheimer’s disease. Mol Neurodegener. 2020;15:43.
Tracy TE, Madero-Perez J, Swaney DL, Chang TS, Moritz M, Konrad C, et al. Tau interactome maps synaptic and mitochondrial processes associated with neurodegeneration. Cell. 2022;185:712–28.
Crawford K, Leonenko G, Baker E, Grozeva D, Lan-Leung B, Holmans P, et al. Golgi apparatus, endoplasmic reticulum and mitochondrial function implicated in Alzheimer’s disease through polygenic risk and RNA sequencing. Mol Psychiatr. 2023;28:1327–36.
Klein HU, Trumpff C, Yang HS, Lee AJ, Picard M, Bennett DA, et al. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol Neurodegener. 2021;16:75.
Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, et al. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A. 2020;117:4971–82.
Merino-Galan L, Jimenez-Urbieta H, Zamarbide M, Rodriguez-Chinchilla T, Belloso-Iguerategui A, Santamaria E, et al. Striatal synaptic bioenergetic and autophagic decline in premotor experimental parkinsonism. Brain. 2022;145:2092–107.
Reddy PH, Yin X, Manczak M, Kumar S, Pradeepkiran JA, Vijayan M, et al. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum Mol Genet. 2018;27:2502–16.
Manczak M, Kandimalla R, Yin X, Reddy PH. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2018;27:1332–42.
Ji YJ, Ugolino J, Brady NR, Hamacher-Brady A, Wang J. Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy. 2017;13:1254–5.
Paul S, Dansithong W, Figueroa KP, Gandelman M, Scoles DR, Pulst SM. Staufen1 in human neurodegeneration. Ann Neurol. 2021;89:1114–28.
Sullivan CR, O’Donovan SM, McCullumsmith RE, Ramsey A. Defects in bioenergetic coupling in schizophrenia. Biol Psychiat. 2018;83:739–50.
Berk M, Turner A, Malhi GS, Ng CH, Cotton SM, Dodd S, et al. A randomised controlled trial of a mitochondrial therapeutic target for bipolar depression: mitochondrial agents, N-acetylcysteine, and placebo. BMC Med. 2019;17:18.
Chung JK, Lee SY, Park M, Joo EJ, Kim SA. Investigation of mitochondrial DNA copy number in patients with major depressive disorder. Psychiatr Res. 2019;282:112616.
Wang Y, Guo X, Hong X, Wang G, Pearson C, Zuckerman B, et al. Association of mitochondrial DNA content, heteroplasmies and inter-generational transmission with autism. Nat Commun. 2022;13:3790.
Ongur D, Prescot AP, Jensen JE, Cohen BM, Renshaw PF. Creatine abnormalities in schizophrenia and bipolar disorder. Psychiat Res. 2009;172:44–8.
Dogan AE, Yuksel C, Du F, Chouinard VA, Ongur D. Brain lactate and pH in schizophrenia and bipolar disorder: a systematic review of findings from magnetic resonance studies. Neuropsychopharmacology. 2018;43:1681–90.
Song X, Chen X, Yuksel C, Yuan J, Pizzagalli DA, Forester B, et al. Bioenergetics and abnormal functional connectivity in psychoticdisorders. Mol Psychiatry. 2021;26:2483–92.
Enwright IJ, Huo Z, Arion D, Corradi JP, Tseng G, Lewis DA. Transcriptome alterations of prefrontal cortical parvalbumin neurons in schizophrenia. Mol Psychiatry. 2018;23:1606–13.
Martins-de-Souza D, Gattaz WF, Schmitt A, Novello JC, Marangoni S, Turck CW, et al. Proteome analysis of schizophrenia patients Wernicke’s area reveals an energy metabolism dysregulation. BMC Psychiatry. 2009;9:17.
Stacey D, Schubert KO, Clark SR, Amare AT, Milanesi E, Maj C, et al. A gene co-expression module implicating the mitochondrial electron transport chain is associated with long-term response to lithium treatment in bipolar affective disorder. Transl Psychiatry. 2018;8:183.
Pacifico R, Davis RL. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol Psychiatry. 2017;22:441–9.
Glausier JR, Enwright JR, Lewis DA. Diagnosis- and cell type-specific mitochondrial functional pathway signatures in schizophrenia and bipolar disorder. Am J Psychiatry. 2020;177:1140–50.
Machado-Vieira R, Luckenbaugh DA, Ballard ED, Henter ID, Tohen M, Suppes T, et al. Increased activity or energy as a primary criterion for the diagnosis of bipolar mania in DSM-5: findings from the STEP-BD study. Am J Psychiatry. 2017;174:70–6.
Cheniaux E, Filgueiras A, Silva RA, Silveira LA, Nunes AL, Landeira-Fernandez J. Increased energy/activity, not mood changes, is the core feature of mania. J Affect Disord. 2014;152–154:256–61.
Malhi GS, Fritz K, Allwang C, Burston N, Cocks C, Harper M, et al. Agitation for recognition by DSM-5 mixed features specifier signals fatigue? Aust N Z J Psychiatry. 2015;49:499–501.
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311:1770–7.
Trost B, Thiruvahindrapuram B, Chan A, Engchuan W, Higginbotham EJ, Howe JL, et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell. 2022;185:4409–27.
Fernstrom J, Mellon SH, McGill MA, Picard M, Reus VI, Hough CM, et al. Blood-based mitochondrial respiratory chain function in major depression. Transl Psychiatry. 2021;11:593.
Goetzl EJ, Wolkowitz OM, Srihari VH, Reus VI, Goetzl L, Kapogiannis D, et al. Abnormal levels of mitochondrial proteins in plasma neuronal extracellular vesicles in major depressive disorder. Mol Psychiatry. 2021;26:7355–62.
Wang Q, Dwivedi Y. Transcriptional profiling of mitochondria associated genes in prefrontal cortex of subjects with major depressive disorder. World J Biol Psychiatry. 2017;18:592–603.
Bouvier E, Brouillard F, Molet J, Claverie D, Cabungcal JH, Cresto N, et al. Nrf2-dependent persistent oxidative stress results in stress-induced vulnerability to depression. Mol Psychiatry. 2017;22:1701–13.
Brivio P, Audano M, Gallo MT, Gruca P, Lason M, Litwa E, et al. Metabolomic signature and mitochondrial dynamics outline the difference between vulnerability and resilience to chronic stress. Transl Psychiatry. 2022;12:87.
Hursitoglu O, Kurutas EB, Strawbridge R, Oner E, Gungor M, Tuman TC, et al. Serum NOX1 and Raftlin as new potential biomarkers of major depressive disorder: a study in treatment-naive first episode patients. Prog Neuro-Psychopharmacol. 2023;121:110670.
Karabatsiakis A, Schonfeldt-Lecuona C. Depression, mitochondrial bioenergetics, and electroconvulsive therapy: a new approach towards personalized medicine in psychiatric treatment - a short review and current perspective. Transl Psychiatry. 2020;10:226.
Stocchetti N, Carbonara M, Citerio G, Ercole A, Skrifvars MB, Smielewski P, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16:452–64.
Hinzman JM, Wilson JA, Mazzeo AT, Bullock MR, Hartings JA. Excitotoxicity and metabolic crisis are associated with spreading depolarizations in severe traumatic brain injury patients. J Neurotrauma. 2016;33:1775–83.
Zheng P, Zhang N, Ren D, Yu C, Zhao B, Zhang Y. Integrated spatial transcriptome and metabolism study reveals metabolic heterogeneity in human injured brain. Cell Rep Med. 2023;4:101057.
Chen W, Guo C, Feng H, Chen Y. Mitochondria: novel mechanisms and therapeutic targets for secondary brain injury after intracerebral hemorrhage. Front Aging Neurosci. 2020;12:615451.
Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. 2020;32:591–604.
Wnek M, Ressel L, Ricci E, Rodriguez-Martinez C, Guerrero JC, Ismail Z, et al. Herpes simplex encephalitis is linked with selective mitochondrial damage; a post-mortem and in vitro study. Acta Neuropathol. 2016;132:433–51.
Uccelli R, Binazzi A, Altavista P, Belli S, Comba P, Mastrantonio M, et al. Geographic distribution of amyotrophic lateral sclerosis through motor neuron disease mortality data. Eur J Epidemiol. 2007;22:781–90.
Turner MR, Barohn RJ, Corcia P, Fink JK, Harms MB, Kiernan MC, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020;91:373–7.
Benjamins D. Progressive bulbar palsy of childhood in siblings. Ann Neurol. 1980;8:203.
Cervenakova L, Protas II, Hirano A, Votiakov VI, Nedzved MK, Kolomiets ND, et al. Progressive muscular atrophy variant of familial amyotrophic lateral sclerosis (PMA/ALS). J Neurol Sci. 2000;177:124–30.
Mercuri E, Sumner CJ, Muntoni F, Darras BT, Finkel RS. Spinal muscular atrophy. Nat Rev Dis Primers. 2022;8:52.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–68.
Weisiger RA, Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–6.
Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu, Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–9.
Fischer LR, Igoudjil A, Magrane J, Li Y, Hansen JM, Manfredi G, et al. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011;134:196–209.
Wang T, Liu H, Itoh K, Oh S, Zhao L, Murata D, et al. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021;33:531–46.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande VC, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22:869–78.
Wang W, Arakawa H, Wang L, Okolo O, Siedlak SL, Jiang Y, et al. Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Ther. 2017;25:127–39.
Hu B, Wang M, Castoro R, Simmons M, Dortch R, Yawn R, et al. A novel missense mutation in AIFM1 results in axonal polyneuropathy and misassembly of OXPHOS complexes. Eur J Neurol. 2017;24:1499–506.
Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137:2329–45.
Workman MJ, Lim RG, Wu J, Frank A, Ornelas L, Panther L, et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron. 2023;111:1191–204.
Zhou B, Yu P, Lin MY, Sun T, Chen Y, Sheng ZH. Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. J Cell Biol. 2016;214:103–19.
Porporato PE, Filigheddu N, Pedro J, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–80.
Guntuku L, Naidu VG, Yerra VG. Mitochondrial dysfunction in gliomas: pharmacotherapeutic potential of natural compounds. Curr Neuropharmacol. 2016;14:567–83.
Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108:16062–7.
Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–10.
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv Q, et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Therapy. 2021;6:337.
Uginet M, Breville G, Assal F, Lovblad KO, Vargas MI, Pugin J, et al. COVID-19 encephalopathy: clinical and neurobiological features. J Med Virol. 2021;93:4374–81.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. New Engl J Med. 2020;382:2268–70.
Liu YH, Wang YR, Wang QH, Chen Y, Chen X, Li Y, et al. Post-infection cognitive impairments in a cohort of elderly patients with COVID-19. Mol Neurodegener. 2021;16:48.
Douaud G, Lee S, Alfaro-Almagro F, Arthofer C, Wang C, McCarthy P, et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature. 2022;604:697–707.
Stein SR, Ramelli SC, Grazioli A, Chung JY, Singh M, Yinda CK, et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature. 2022;612:758–63.
Xu J, Lazartigues E. Expression of ACE2 in human neurons supports the neuro-invasive potential of COVID-19 virus. Cell Mol Neurobiol. 2022;42:305–9.
Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol-Cell Physiol. 2020;319:C258–67.
Wu KE, Fazal FM, Parker KR, Zou J, Chang HY. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020;11:102–8.
Ganji R, Reddy PH. Impact of COVID-19 on mitochondrial-based immunity in aging and age-related diseases. Front Aging Neurosci. 2020;12:614650.
Pliss A, Kuzmin AN, Prasad PN, Mahajan SD. Mitochondrial dysfunction: a prelude to neuropathogenesis of SARS-CoV-2. ACS Chem Neurosci. 2022;13:308–12.
Lenaers G, Beaulieu C, Charif M, Gerber S, Kaplan J, Rozet JM. Autosomal Recessive Leber Hereditary Optic Neuropathy, A New Neuro-Ophthalmo-Genetic Paradigm. Brain. 2023;146:3156–61.
Inglese M, Rovaris M, Bianchi S, La Mantia L, Mancardi GL, Ghezzi A, et al. Magnetic resonance imaging, magnetisation transfer imaging, and diffusion weighted imaging correlates of optic nerve, brain, and cervical cord damage in Leber’s hereditary optic neuropathy. J Neurol Neurosurg Psychiatry. 2001;70:444–9.
Kovacs GG, Hoftberger R, Majtenyi K, Horvath R, Barsi P, Komoly S, et al. Neuropathology of white matter disease in Leber’s hereditary optic neuropathy. Brain. 2005;128:35–41.
Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. New Engl J Med. 1989;320:1293–9.
Leutner C, Layer G, Zierz S, Solymosi L, Dewes W, Reiser M. Cerebral MR in ophthalmoplegia plus. Am J Neuroradiol. 1994;15:681–7.
Salvador CL, Oppeboen M, Vassli AO, Pfeiffer H, Varhaug KN, Elgstoen K, et al. Increased sphingomyelin and free sialic acid in cerebrospinal fluid of Kearns-Sayre syndrome: new findings using untargeted metabolomics. Pediatr Neurol. 2023;143:68–76.
Koo B, Becker LE, Chuang S, Merante F, Robinson BH, MacGregor D, et al. Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS): clinical, radiological, pathological, and genetic observations. Ann Neurol. 1993;34:25–32.
McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol. 2002;1:343–51.
McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–40.
Ng YS, Bindoff LA, Gorman GS, Klopstock T, Kornblum C, Mancuso M, et al. Mitochondrial disease in adults: recent advances and future promise. Lancet Neurol. 2021;20:573–84.
Amemiya S, Hamamoto M, Goto Y, Komaki H, Nishino I, Nonaka I, et al. Psychosis and progressing dementia: presenting features of a mitochondriopathy. Neurology. 2000;55:600–1.
Salsano E, Giovagnoli AR, Morandi L, Maccagnano C, Lamantea E, Marchesi C, et al. Mitochondrial dementia: a sporadic case of progressive cognitive and behavioral decline with hearing loss due to the rare m.3291T>C MELAS mutation. J Neurol Sci. 2011;300:165–8.
Kraya T, Neumann L, Paelecke-Habermann Y, Deschauer M, Stoevesandt D, Zierz S, et al. Cognitive impairment, clinical severity and MRI changes in MELAS syndrome. Mitochondrion. 2019;44:53–7.
Inagaki T, Ishino H, Seno H, Ohguni S, Tanaka J, Kato Y. Psychiatric symptoms in a patient with diabetes mellitus associated with point mutation in mitochondrial DNA. Biol Psychiat. 1997;42:1067–9.
Mancuso M, Ricci G, Choub A, Filosto M, DiMauro S, Davidzon G, et al. Autosomal dominant psychiatric disorders and mitochondrial DNA multiple deletions: report of a family. J Affect Disorders. 2008;106:173–7.
Satogami K, Takahashi S, Kose A, Shinosaki K. Schizophrenia-like symptoms in a patient with Leigh syndrome. Asian J Psychiatry. 2017;25:249–50.
Lou G, Palikaras K, Lautrup S, Scheibye-Knudsen M, Tavernarakis N, Fang EF. Mitophagy and neuroprotection. Trends Mol Med. 2020;26:8–20.
Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–29.
Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120:419–29.
Thomas RR, Keeney PM, Bennett JP. Impaired complex-I mitochondrial biogenesis in Parkinson disease frontal cortex. J Parkinson Dis. 2012;2:67–76.
McCully JD, Cowan DB, Pacak CA, Toumpoulis IK, Dayalan H, Levitsky S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am J Physiol Heart Circ Physiol. 2009;296:H94–105.
Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, et al. Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 2016;25:913–27.
Boukelmoune N, Chiu GS, Kavelaars A, Heijnen CJ. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol Commun. 2018;6:139.
English K, Shepherd A, Uzor NE, Trinh R, Kavelaars A, Heijnen CJ. Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol Commun. 2020;8:36.
Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directionaltransfer of alpha-synuclein and mitochondria. Cell Death Dis. 2023;14:329.
Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. 2021;184:2896–910.
Sun C, Liu X, Wang B, Wang Z, Liu Y, Di C, et al. Endocytosis-mediated mitochondrial transplantation: transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics. 2019;9:3595–607.
Watson DC, Bayik D, Storevik S, Moreino SS, Sprowls SA, Han J, et al. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat Cancer. 2023;4:648–64.
Peruzzotti-Jametti L, Bernstock JD, Willis CM, Manferrari G, Rogall R, Fernandez-Vizarra E, et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021;19:e3001166.
Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH. Extracellular mitochondria for therapy and diagnosis in acute central nervous system injury. JAMA Neurol. 2018;75:119–22.
Dave KM, Stolz DB, Venna VR, Quaicoe VA, Maniskas ME, Reynolds MJ, et al. Mitochondria-containing extracellular vesicles (EV) reduce mouse brain infarct sizes and EV/HSP27 protect ischemic brain endothelial cultures. J Control Release. 2023;354:368–93.
Nitzan K, Benhamron S, Valitsky M, Kesner EE, Lichtenstein M, Ben-Zvi A, et al. Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer’s Disease mice. J Alzheimers Dis. 2019;72:587–604.
Cheng XY, Biswas S, Li J, Mao CJ, Chechneva O, Chen J, et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl Neurodegener. 2020;9:13.
Robicsek O, Ene HM, Karry R, Ytzhaki O, Asor E, McPhie D, et al. Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophrenia Bull. 2018;44:432–42.
Pourmohammadi-Bejarpasi Z, Roushandeh AM, Saberi A, Rostami MK, Toosi S, Jahanian-Najafabadi A, et al. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res Bull. 2020;165:70–80.
Aleynik A, Gernavage KM, Mourad Y, Sherman LS, Liu K, Gubenko YA, et al. Stem cell delivery of therapies for brain disorders. Clin Transl Med. 2014;3:24.
Xie J, Shen Z, Anraku Y, Kataoka K, Chen X. Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials. 2019;224:119491.
Lightowlers RN, Chrzanowska-Lightowlers ZM, Russell OM. Mitochondrial transplantation-a possible therapeutic for mitochondrialdysfunction?: Mitochondrial transfer is a potential cure for many diseases but proof of efficacy and safety is still lacking. EMBO Rep. 2020;21:e50964.
Chang JC, Wu SL, Liu KH, Chen YH, Chuang CS, Cheng FC, et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res. 2016;170:40–56.
Zhang TG, Miao CY. Mitochondrial transplantation as a promising therapy for mitochondrial diseases. Acta Pharm Sin B. 2023;13:1028–35.
Murphy MP, Hartley RC. Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov. 2018;17:865–86.
Shah RC, Matthews DC, Andrews RD, Capuano AW, Fleischman DA, VanderLugt JT, et al. An evaluation of MSDC-0160, a prototype mTOT modulating insulin sensitizer, in patients with mild Alzheimer’s disease. Curr Alzheimer Res. 2014;11:564–73.
Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci Transl Med. 2016;8:368ra174.
Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53.
Ma XR, Zhu X, Xiao Y, Gu HM, Zheng SS, Li L, et al. Restoring nuclear entry of Sirtuin 2 in oligodendrocyte progenitor cells promotes remyelination during ageing. Nat Commun. 2022;13:1225.
Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, et al. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci U S A. 2021;118(37):e2011226118.
Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016;24:795–806.
Brakedal B, Dolle C, Riemer F, Ma Y, Nido GS, Skeie GO, et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022;34:396–407.
Gautam M, Genc B, Helmold B, Ahrens A, Kuka J, Makrecka-Kuka M, et al. SBT-272 improves TDP-43 pathology in ALS upper motor neurons by modulating mitochondrial integrity, motility, and function. Neurobiol Dis. 2023;178:106022.
Ding XW, Robinson M, Li R, Aldhowayan H, Geetha T, Babu JR. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in Diabetes Mellitus and Alzheimer’s disease. Pharmacol Res. 2021;171:105783.
Olesen MA, Torres AK, Jara C, Murphy MP, Tapia-Rojas C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020;34:101558.
Yin X, Manczak M, Reddy PH. Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington’s disease. Hum Mol Genet. 2016;25:1739–53.
Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020;32:15–30.
Xu X, Sun Y, Cen X, Shan B, Zhao Q, Xie T, et al. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell. 2021;12:769–87.
Zu T, Guo S, Bardhi O, Ryskamp DA, Li J, Khoramian TS, et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc Natl Acad Sci USA. 2020;117:18591–9.
Mor DE, Sohrabi S, Kaletsky R, Keyes W, Tartici A, Kalia V, et al. Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci USA. 2020;117:26438–47.
Schaeffer V, Goedert M. Stimulation of autophagy is neuroprotective in a mouse model of human tauopathy. Autophagy. 2012;8:1686–7.
Holler CJ, Taylor G, McEachin ZT, Deng Q, Watkins WJ, Hudson K, et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 2016;11:46.
Lotfi P, Tse DY, Di Ronza A, Seymour ML, Martano G, Cooper JD, et al. Trehalose reduces retinal degeneration, neuroinflammation and storage burden caused by a lysosomal hydrolase deficiency. Autophagy. 2018;14:1419–34.
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9:1308–20.
Haidar M, Loix M, Vanherle S, Dierckx T, Vangansewinkel T, Gervois P, et al. Targeting lipophagy in macrophages improves repair in multiple sclerosis. Autophagy. 2022;18:2697–710.
Yang Q, Zheng C, Cao J, Cao G, Shou P, Lin L, et al. Spermidine alleviates experimental autoimmune encephalomyelitis through inducing inhibitory macrophages. Cell Death Differ. 2016;23:1850–61.
Freitag K, Sterczyk N, Wendlinger S, Obermayer B, Schulz J, Farztdinov V, et al. Spermidine reduces neuroinflammation and soluble amyloid beta in an Alzheimer’s disease mouse model. J Neuroinflammation. 2022;19:172.
Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol Psychiatry. 2013;18:889–97.
Steele JW, Gandy S. Latrepirdine (Dimebon(R)), a potential Alzheimer therapeutic, regulates autophagy and neuropathology in an Alzheimer mouse model. Autophagy. 2013;9:617–8.
Steele JW, Kim SH, Cirrito JR, Verges DK, Restivo JL, Westaway D, et al. Acute dosing of latrepirdine (Dimebon), a possible Alzheimer therapeutic, elevates extracellular amyloid-beta levels in vitro and in vivo. Mol Neurodegener. 2009;4:51.
Lee J, Sung KW, Bae EJ, Yoon D, Kim D, Lee JS, et al. Targeted degradation of ⍺-synuclein aggregates in Parkinson’s disease using the AUTOTAC technology. Mol Neurodegener. 2023;18:41.
Lee J, Yoon D, Sung KW, Bae EJ, Park DH, Suh YH, et al. Targeted degradation of SNCA/alpha-synuclein aggregates in neurodegeneration using the AUTOTAC chemical platform. Autophagy. 2024;20:463–5.
Cen X, Chen Y, Xu X, Wu R, He F, Zhao Q, et al. Pharmacological targeting of MCL-1 promotes mitophagy and improves disease pathologies in an Alzheimer’s disease mouse model. Nat Commun. 2020;11:5731.
Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, et al. Inhibition of Drp1 ameliorates synaptic depression, abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J Neurosci. 2017;37:5099–110.
Li YH, Xu F, Thome R, Guo MF, Sun ML, Song GB, et al. Mdivi-1, a mitochondrial fission inhibitor, modulates T helper cells and suppresses the development of experimental autoimmune encephalomyelitis. J Neuroinflammation. 2019;16:149.
Cui M, Ding H, Chen F, Zhao Y, Yang Q, Dong Q. Mdivi-1 protects against ischemic brain injury via elevating extracellular adenosine in a cAMP/CREB-CD39-dependent manner. Mol Neurobiol. 2016;53:240–53.
Ha J, Choi DW, Kim KJ, Kim KY, Nam CM, Kim E. Pioglitazone use and reduced risk of dementia in patients with diabetes mellitus with a history of ischemic stroke. Neurology. 2023;100:e1799–811.
Masciopinto F, Di Pietro N, Corona C, Bomba M, Pipino C, Curcio M, et al. Effects of long-term treatment with pioglitazone on cognition and glucose metabolism of PS1-KI, 3xTg-AD, and wild-type mice. Cell Death Dis. 2012;3:e448.
Chang YH, Yen SJ, Chang YH, Wu WJ, Lin KD. Pioglitazone and statins lower incidence of Parkinson disease in patients with diabetes mellitus. Eur J Neurol. 2021;28:430–7.
Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. 2016;374:1321–31.
Steele H, Gomez-Duran A, Pyle A, Hopton S, Newman J, Stefanetti RJ, et al. Metabolic effects of bezafibrate in mitochondrial disease. Embo Mol Med. 2020;12:e11589.
Viscomi C, Bottani E, Civiletto G, Cerutti R, Moggio M, Fagiolari G, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011;14:80–90.
Chandra A, Sharma A, Calingasan NY, White JM, Shurubor Y, Yang XW, et al. Enhanced mitochondrial biogenesis ameliorates disease phenotype in a full-length mouse model of Huntington’s disease. Hum Mol Genet. 2016;25:2269–82.
Chung HL, Ye Q, Park YJ, Zuo Z, Mok JW, Kanca O, et al. Very-long-chain fatty acids induce glial-derived sphingosine-1-phosphate synthesis, secretion, and neuroinflammation. Cell Metab. 2023;35:855–74.
Dong Y, Stewart T, Bai L, Li X, Xu T, Iliff J, et al. Coniferaldehyde attenuates Alzheimer’s pathology via activation of Nrf2 and its targets. Theranostics. 2020;10:179–200.
Ranen NG, Peyser CE, Coyle JT, Bylsma FW, Sherr M, Day L, et al. A controlled trial of idebenone in Huntington’s disease. Mov Disord. 1996;11:549–54.
Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–4.
Singh AP, Biswas A, Shukla A, Maiti P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct Target Therapy. 2019;4:33.
Wang Z, Guo W, Kuang X, Hou S, Liu H. Nanopreparations for mitochondria targeting drug delivery system: current strategies and future prospective. Asian J Pharm Sci. 2017;12:498–508.
Bao Q, Hu P, Xu Y, Cheng T, Wei C, Pan L, et al. Simultaneous blood-brain barrier crossing and protection for stroke treatmentbased on edaravone-loaded ceria nanoparticles. ACS Nano. 2018;12:6794–805.
Katlama C, Mouthon B, Gourdon D, Lapierre D, Rousseau F. Atovaquone as long-term suppressive therapy for toxoplasmic encephalitis in patients with AIDS and multiple drug intolerance. Atovaquone Expanded Access Group. Aids. 1996;10:1107–12.
Torres RA, Weinberg W, Stansell J, Leoung G, Kovacs J, Rogers M, et al. Atovaquone for salvage treatment and suppression oftoxoplasmic encephalitis in patients with AIDS. Atovaquone/Toxoplasmic Encephalitis Study Group. Clin Infect Dis. 1997;24:422–9.
Umbrasas D, Arandarcikaite O, Grigaleviciute R, Stakauskas R, Borutaite V. Neuroprotective effect of a novel ATP-synthase inhibitor bedaquiline in cerebral ischemia-reperfusion injury. Int J Mol Sci. 2021;22:9717.
Huang Y, Ma M, Zhu X, Li M, Guo M, Liu P, et al. Effectiveness of idebenone nanorod formulations in the treatment of Alzheimer’s disease. J Control Release. 2021;336:169–80.
Yan J, Sun W, Shen M, Zhang Y, Jiang M, Liu A, et al. Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice. Cell Death Discov. 2022;8:28.
Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–86.
Distelmaier F, Haack TB, Wortmann SB, Mayr JA, Prokisch H. Treatable mitochondrial diseases: cofactor metabolism and beyond. Brain. 2017;140:e11.
Jeppesen TD. Aerobic exercise training in patients with mtDNA-related mitochondrial myopathy. Front Physiol. 2020;11:349.
Paull D, Emmanuele V, Weiss KA, Treff N, Stewart L, Hua H, et al. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature. 2013;493:632–7.
Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–31.
Zhang J, Liu H, Luo S, Lu Z, Chavez-Badiola A, Liu Z, et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34:361–8.
Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, et al. CRISPR-cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493–502.
Gammage PA, Viscomi C, Simard ML, Costa A, Gaude E, Powell CA, et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med. 2018;24:1691–5.
Bacman SR, Kauppila J, Pereira CV, Nissanka N, Miranda M, Pinto M, et al. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat Med. 2018;24:1696–700.
Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–7.
Lim K, Cho SI, Kim JS. Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat Commun. 2022;13:366.
Cho SI, Lee S, Mok YG, Lim K, Lee J, Lee JM, et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell. 2022;185:1764–76.
Yi Z, Zhang X, Tang W, Yu Y, Wei X, Zhang X, et al. Strand-selective base editing of human mitochondrial DNA using mitoBEs. Nat Biotechnol. 2024;42(3):498–509.
Gammage PA, Moraes CT, Minczuk M. Mitochondrial genome engineering: the revolution may not be CRISPR-Ized. Trends Genet. 2018;34:101–10.
Bi R, Li Y, Xu M, Zheng Q, Zhang DF, Li X, et al. Direct evidence of CRISPR-Cas9-mediated mitochondrial genome editing. Innovation (Camb). 2022;3:100329.
Acknowledgements
Not applicable.
Funding
This review was supported by the grants from the National Key R&D Program of China (2021ZD0202901 and 2021ZD0202903) and the National Natural Science Foundation of China (81991523 and 82204370).
Author information
Authors and Affiliations
Contributions
NS designed this review and wrote the manuscript; SM, XW and NS drafted the figures and tables; GH and ML designed this review and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Song, N., Mei, S., Wang, X. et al. Focusing on mitochondria in the brain: from biology to therapeutics. Transl Neurodegener 13, 23 (2024). https://doi.org/10.1186/s40035-024-00409-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00409-w