
Abstract
Background
Infant gut microbiota is highly malleable, but the long-term longitudinal impact of antibiotic exposure in early life, together with the mode of delivery on infant gut microbiota and resistome, is not extensively studied.
Methods
Two hundred and eight samples from 45 infants collected from birth until 2 years of age over five time points (week 1, 4, 8, 24, year 2) were analysed. Based on shotgun metagenomics, the gut microbial composition and resistome profile were compared in the early life of infants divided into three groups: vaginal delivery/no-antibiotic in the first 4 days of life, C-section/no-antibiotic in the first 4 days of life, and C-section/antibiotic exposed in first 4 days of life. Gentamycin and benzylpenicillin were the most commonly administered antibiotics during this cohort’s first week of life.
Results
Newborn gut microbial composition differed in all three groups, with higher diversity and stable composition seen at 2 years of age, compared to week 1. An increase in microbial diversity from week 1 to week 4 only in the C-section/antibiotic-exposed group reflects the effect of antibiotic use in the first 4 days of life, with a gradual increase thereafter. Overall, a relative abundance of Actinobacteria and Bacteroides was significantly higher in vaginal delivery/no-antibiotic while Proteobacteria was higher in C-section/antibiotic-exposed infants. Strains from species belonging to Bifidobacterium and Bacteroidetes were generally persistent colonisers, with Bifidobacterium breve and Bifidobacterium bifidum species being the major persistent colonisers in all three groups. Bacteroides persistence was dominant in the vaginal delivery/no-antibiotic group, with species Bacteroides ovatus and Phocaeicola vulgatus found to be persistent colonisers in the no-antibiotic groups. Most strains carrying antibiotic-resistance genes belonged to phyla Proteobacteria and Firmicutes, with the C-section/antibiotic-exposed group presenting a higher frequency of antibiotic-resistance genes (ARGs).
Conclusion
These data show that antibiotic exposure has an immediate and persistent effect on the gut microbiome in early life. As such, the two antibiotics used in the study selected for strains (mainly Proteobacteria) which were multiple drug-resistant (MDR), presumably a reflection of their evolutionary lineage of historical exposures—leading to what can be an extensive and diverse resistome.
Video Abstract
Similar content being viewed by others
Background
Early life is a crucial window for the development of the neonate as the foundations for later life are laid. The timing and source of colonisation of the microbiota play an indispensable role in developing optimal health. These initial colonisers perform important functions such as the breakdown of complex compounds, e.g., the role of bifidobacteria in the breakdown of human milk oligosaccharides (HMOs) to harvest energy and synthesise vitamins and metabolites such as vitamins B and K and short-chain fatty acids (SCFAs) [1]. They protect against pathogen colonisation—a phenomenon referred to as colonisation resistance [2], and they are involved in neural development and immune programming as the neurocognitive and immunity maturation occur during this period [3,4,5].
Infants have a simple gut microbial structure comprising fewer bacterial species than adults, stabilising by 3 to 4 years of age towards adult life. This early-life gut microbiota is influenced by several factors that include but are not limited to gestational age [6], mode of delivery (vaginal vs C-section) [7], feeding habits (breastfeeding vs formula feeding) [8], and antibiotic uptake, both by the infant and the mother [9,10,11,12].
The infant gut is considered an antibiotic resistance gene (ARG) reservoir [13, 14]. This high ARG abundance is associated with Gammaproteobacteria, owing to their intrinsic resistance and high colonisation levels in early life [15,16,17,18,19,20]. C-section (CS) born infants are colonised by a high abundance of opportunistic pathogens belonging to taxa such as Escherichia coli, Klebsiella, and Enterobacter, often resulting in higher antibiotic use and ARG carriage in these infants [21, 22]. Antibiotic resistance is a global health concern [23]. Septic infections caused by antibiotic-resistant bacteria are responsible for 214,000 neonatal deaths each year, of which multi-drug resistant (MDR) bacteria cause 30% [24]. The immediate effect after antibiotic treatment is generally an overall decrease in gut microbiota diversity, including commensals, benefiting the colonisation of antimicrobial-resistant (AMR) strains due to selective pressure [25]. Increasing evidence has demonstrated the association between antibiotic use and altered microbiota in early life, the rise in ARGs [11, 26], and its linkage to the development of disease conditions in later life [27].
CS and antibiotic use are common medical practices [28, 29], but both disturb the natural microbial colonisation process, impacting the long-term health of the host [30, 31]. However, there are still important gaps in the understanding of how disruption of microbiota by differing modes of delivery and antibiotic use can enhance or reduce the gut resistome as the infant ages. In this study, the combined effects of CS birth mode and antibiotic uptake on the meta-taxonomic diversity, and frequency of ARGs over the first few weeks of life were investigated by comparing it to CS-born and vaginally delivered control groups without antibiotics. Furthermore, the effect of antibiotic exposure in early life on the resistome and functional profile of infant gut microbiota longitudinally up to 2 years of age was investigated. CS delivery and antibiotic exposure in early life were seen to pose a twofold disadvantage, resulting in significantly lower microbial diversity carrying a higher ARG frequency with antibiotic use being the dominant influencing factor. Furthermore, the impact of antibiotic use in early life resonated up to 2 years of age with amplified resistome profile observed in antibiotic-exposed infants.
Methods
Study group participants
In this study, DNA extracted from faecal samples from full-term infants from two study cohorts—INFANTMET and MYNEWGUT—were used to investigate the evolution of the infant gut resistome using shotgun metagenomics throughout the first 24 weeks of life up to 2 years of age. MYNEWGUT (MNG) recruited full-term CS-born infants administered antibiotics in the first 4 days of life. INFANTMET (IM) included infants born at full term via CS and vaginal delivery and were not administered antibiotics in the first week of life [32]. A subset of subjects from these two studies (n = 45) were selected randomly and divided into three groups. Group 1 (MNG + ab/CSab) (n = 10) included infants born by CS and given antibiotics during the first week of life. Group 2 (IM-Ab/CSnoab) (n = 17) contains infants born by CS, not administered antibiotics, and group 3 (IM-Ab/VDnoab) (n = 18) has infants born via vaginal delivery (VD) not administered antibiotics in the first few days of life. The cohorts and control groups were matched by gestational age (> 35 weeks of gestation), birth mode (CS and VD), antibiotic treatment (i.e. benzylpenicillin and gentamicin), diet (mostly breastfed or combination of breast and formula feeding), and time points of sample collection. The demographics for these three infant groups are as follows (Table 1).
DNA extraction
Metagenomic DNA was extracted from fecal samples using the QIAmp Fast DNA Stool Mini kit (Qiagen, UK) with a modified protocol combined with a repeated bead beating method [33]. Briefly, 1 ml of lysis buffer was added to the stool sample in the bead-beating tube. The samples were homogenised with the lysis buffer using a mini beadbeater, incubated at 70 °C for 15 min with mixing at regular intervals, followed by another 15-min incubation at 37 °C, and then centrifuged at 4 °C for 5 min at 16,000 × g. The supernatant was then treated with ammonium acetate to remove impurities and incubated on ice for 5 min. This was followed by another centrifugation step at 16,000 × g at 4 °C for 10 min, and then, the DNA was precipitated by combining with an equal volume of isopropanol and stored overnight at − 20 °C. The next day, the DNA was pelleted by centrifugation at 16,000 × g at 4 °C for 15 min, washed with ethanol and Tris–EDTA, and treated with Proteinase K, RNAse, and purified according to the manufacturer’s instructions (QIAmp Fast DNA Stool Mini kit; Qiagen, UK). The DNA was quantified using Qubit and stored at − 30 °C until further use.
Shotgun sequencing library preparation
The concentration of DNA was determined using Qubit and diluted to 0.2 ng/µl as per the manufacturer’s instructions. Library prep for shotgun sequencing was performed using a Nextera XT kit following the Nextera XT DNA Library Preparation Guide from Illumina. Briefly, genomic DNA (0.2 ng/µl) was tagmented in 20 µl PCR reaction (containing 5 µl of gDNA, 10 µl of tagment DNA buffer, and 5 µl of Amplicon Tagment Mix) volume at 55 °C. The tagmented DNA was then amplified using Illumina index primers, following which the libraries were cleaned using AMPure beads. The quality of the library was then evaluated by running it on the Bioanalyzer using the Agilent High Sensitivity DNA chip. The DNA was quantified using Qubit, normalised, pooled, and outsourced to the Teagasc sequencing facility for sequencing. Samples were sequenced as part of three sequencing runs along with one single sample (I159) ran on a new run making it part of a fourth sequencing run.
Filtering, taxonomic, and functional analysis of metagenomics reads
The quality of raw reads from metagenomic sequencing was assessed using FastQC (v0.11.8) and MultiQC (v1.9). Further, trimming, filtering, and host genome decontamination of the metagenomics reads were done using Trimmomatic (v0.39) and bowtie2 with Homo sapiens reference genome using the default parameters via the KneadData (v0.7.2) program from Huttenhower lab (https://github.com/biobakery/kneaddata). Further HUMAnN3 (v3.0) was used for the functional assignment of the reads, providing microbial gene and pathway abundance information [34], while MetaPhlAn3 was used to provide the taxonomic assignment to the filtered and trimmed reads using default parameters. MetaPhlAn3 output was processed using the microbiomics (v0.0.0.9000) and phyloseq (v1.38.0) package in R (v4.2.1), and relative abundance and mean relative abundance data were used for plotting figures in R. To check for possible batch-effects based on our grouping and sequencing plate variable, we used the R package MBECS v1.4.0 with ComBat as the batch effect correcting algorithm [35]. Briefly, ComBat uses a non-parametric empirical Bayes framework to correct for batch effects. Then, we determined beta-diversity using the Bray–Curtis distance and a PCoA with the corrected batch-effect data. We further calculated the PERMANOVA for all our study variables and compared them with the PERMANOVA based on uncorrected data. We obtained p values and R2 within a 0.0001% range of variation, so we considered that batch effects are negligible.
Metagenomics assembly and contig binning
Post-KneadData filtering, the quality-filtered sequencing reads were first fixed and sorted using bbmap (v38.22). The metagenomes were then assembled using metaSPAdes (v3.14) [36] with default parameters. The scaffolds generated were filtered to obtain resulting scaffolds with a minimum length of 1000 bp (1kbp). Metagenomic bins were generated from the filtered scaffolds using three binning tools (MetaBAT v2.12.1, MaxBin v2.2.622, and CONCOCT v1.0.023) using metaWRAP (v1.3.1) [37] with default parameters. The bins were then refined with Bin_refinement module of metaWRAP with options ‘-c 50—× 10,’ corresponding to the criterion of medium-quality draft metagenome assembled genomes (MAGs). The quality (estimated completeness and contamination) of bins was evaluated with CheckM (v1.0.18) [38], implemented in metaWRAP. This resulted in the formation of 1449 medium-quality MAGs which were used for further analysis.
Taxonomic and functional annotation of MAGs
The resulting MAGs were then provided taxonomic annotation with GTDB-Tk (v1.5.0) using ‘classify_wf’ workflow with default settings [39]. The phylogenetic tree for the 1449 MAGs was generated using PhyloPhlAn (v3.0.2) with options ‘-diversity low’ and ‘-fast’ [40]. The protein-coding genes from MAGs were predicted using Prokka (v1.14) using default parameters. dbCAN (v3.0.1) [41] (run_dbcan.py) was used to assign carbohydrate-active enzymes (CAZymes) to all the MAGs using default parameters. Only certain CAZymes as reported to be involved in HMO and FOS metabolism were filtered and examined in downstream analysis. inStrain [42] was used for the identification of identical strains from MAGs, briefly, MAGs were dereplicated using drep (v3.2.0) resulting in 598 unique MAGs which were then mapped on filtered and trimmed shotgun reads using the inStrain profile command using Bowtie2 (v2.3.4). The inStrain profiles are then processed with inStrain compare, where the profiles are compared and those with 99.99% population level—average nucleotide identity (popANI) are termed to be the same strain, belonging to the same cluster and thus considered belonging to the same sample. The resulting strains were then classified as early or late colonisers, if they appeared during the first 8 weeks (early colonisers, t ≤ 8 weeks) of life or later (late colonisers, t > 8 weeks). Early colonisers were further classified as persistent colonisers and non-persistent colonisers. The definition for persistent colonisers was adapted from Lou et al. [43] and persistent colonisers were defined as early colonisers that persisted for 24 weeks (6 months) or more (t ≥ 24 weeks); otherwise, the strain was termed as non-persistent coloniser (t < 24 weeks).
To look for possible contaminants, we examined strains (i.e. same strain with popANI > 99.99%) detected in the same inStrain cluster (set of genomes having popANI > 99.99%) coming from different individuals (supplementary Table S1). If the samples where these strains were detected as identical came from different sequencing plates, we did not classify them as contaminants. These could be individuals living in the same household or siblings (e.g. I08 and I18). Only three samples (M15, M20, M25) from the same sequencing plate and corresponding to different (n = 3) individuals were detected as containing the same strains (n = 2). These strains were classified as possible contaminants and removed from the analysis. We also looked at possible contaminants using the blanks included in the sequencing runs. No contamination was found using this method.
Resistome and virulence analysis
MAGs generated were then processed through ABRicate (https://github.com/tseemann/abricate) using default settings with CARD (Comprehensive Antibiotic Resistance Database) [44], VFDB (Virulence Factor Database) [45], and plasmid finder [46] databases. Results from CARD and VFDB were used further unless otherwise specified. RGI-bwt and CARD (v3.1.4) [47] were used to predict ARGs in the clean and filtered metagenomics reads using default parameters. The RGI-bwt read mapping results at the gene level were normalised by the read counts per sample, converted to copies per million (cpm) of ARGs per sample, and used for downstream analysis. ARG names from ABRicate and RGI output were curated to antibiotic class names based on phenotype from CARD. Any gene that conferred resistance to three or more antibiotic classes was termed MDR in downstream analysis. However, when the antibiotic classes of interest in the study (classes of antibiotics administered to infants in the first week of life in the CSab group—Aminoglycoside and or Penams) appeared, we spelled them out along with MDR for better visualisation purposes.
Differential abundance analysis
Analysis of which taxa or ARG classes drive the difference between the three groups in the present study was done using MaAsLin2 [48] and Songbird [49]. The MaAsLin2 model was fit with overall data, and at week 1 and week 4, treating the group as a fixed effect with each of the three study groups (VDnoab, CSnoab, CSab) used as a reference with min_prevalance = 0.1 and min_abundance = 0. For differential abundance analysis of longitudinal data, Songbird was performed using default parameters to study the bacterial species most associated with each group and driving the differences in the groups (Formula: C(Group, Treatment('VDnoab')) and C(Group, Treatment('CSnoab'))). Only the top 10 associations (both positive and negative top 10) were used for further analysis.
Statistical analysis and data representation
Downstream analysis of the data was done in R (v4.1.0) using packages including Vegan (v2.5.7) [50], Phyloseq (v1.38.0) [51], ggplot2 (v3.3.5) [52], microbiome utilities (v1.0.16), and ggpubR (v0.4.0). Statistical analysis was performed in R using Permanova to check the proportion of explained variance and significance of each variable using pairwiseAdonis (v0.4). Wilcoxon sum tests, paired t tests, Dunn test, and ANOVA as mentioned throughout the text were used to verify statistical significance. Spearman correlation coefficient was used to determine the correlation between relative abundance over time for species for each group, and the top 10 positive and negative results were plotted in R. Statistical significance was determined by 999 permutations, and p values were corrected using FDR; p values below 0.05 were considered significant. To assess the power of the study, we carried out a post hoc statistical power analysis. We used the ‘simr’ package (v1.0.7) and ‘lme4’ package (v1.1) in R, which determines the power for linear mixed effects models based on Monte Carlo simulations. Briefly, we first fitted the model using the function ‘lmer’ on the second axis of the PCoA with Bray–Curtis distance (Fig. 1B), with the following parameters: ‘Axis.2 ~ Group*time + (1|id)’, where ‘Axis.2’ is the second axis of the PCoA, ‘Group’ is the grouping variable in our study, and ‘id’ is the identity of the infant (i.e., the random variable). Then, we created a fitted lmer model using the function ‘makeLmer’ with the above formula and used the output of lmer. Finally, we assessed statistical power for delivery mode using the ‘powerSim’ function as follows: ‘powerSim(model, nsim = 1000, test = fcompare(Axis.2 ~ time))’, where ‘model’ is the fitter lmer model. We obtained a statistical power of 96.10% with alpha = 0.05 and 1000 simulations.

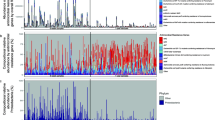
A Alpha-diversity as measured by Shannon’s diversity index between all time points in the three study groups. P values were calculated using the Wilcoxon test, with p.adjusted values < 0.05 used as the significance threshold. B PCoA plot using Bray–Curtis and unweighted UniFrac distance metrics; with ellipses drawn with each grouping variable namely time-point, antibiotic, delivery, and study groups (confidence interval: 90%). P value and R2 values were obtained from PERMANOVA using pairwiseAdonis. C Taxonomic distribution of infant gut microbiota at class level using relative abundance for each group over time from week 1 to year 2
Results
To characterise the microbiome and resistome development in early life, metagenomic data of two hundred and eight stool samples from 45 infants were divided into three groups based on their mode of delivery and antibiotic exposure in the first week of life, i.e., vaginal delivery/no-antibiotic (VDnoab); C-Section/no-antibiotic (CSnoab); C-Section/antibiotic (CSab). This study included samples at five time points from week 1, week 4, week 8, week 24, and year 2 of age. Altogether 10 infants were born by CS and administered antibiotics in the first 4 days of life, with gentamycin and benzylpenicillin being the most commonly administered. Gestational age and birth weight were similar across all groups (Table 1).
Antibiotic use affects microbial composition and decreases diversity
Metagenomics analysis revealed increased richness and alpha-diversity (Shannon index) over time in all groups (Fig. 1A, 1B), with significant differences between all early time points (week 1, week 4, week 8, and week 24) and the final time-point, year 2. The significant difference (p.adj-value = 0.029 Wilcoxon test with BH) in alpha-diversity (Shannon index) observed between week 1 and week 4 in the CSab group, which was not observed in the other groups, shows an increase in microbial diversity in very early life possibly due to the effect of antibiotic administration (Fig. 1A). The CSab group had low initial microbial diversity (Shannon index) when compared with the other groups (Figure S1A), with a significant difference observed at week 1 between CSab and CSnoab (p.adj-value = 0.03, Wilcoxon test with BH) groups and at week 24 between CSab and CSnoab (p.adj-value = 0.039, Wilcoxon test with BH); however, no significant differences were observed otherwise (Figure S1A).
We then calculated beta-diversity using Bray–Curtis dissimilarity and Unifrac distance and visualised it using principal coordinate analysis (PCoA) to look at differences between microbial profiles (Fig. 1B). Using Bray–Curtis distances, the CSab group showed a distinct microbial composition compared to CSnoab (p value = 0.0105, PERMANOVA) and VDnoab (p value = 0.0060, PERMANOVA) groups. Beta-diversity also varied between time points (p value = 0.001, R2 = 0.11; Adonis2), by mode of delivery (p value = 0.003, R2 = 0.013; Adonis2), by duration of breastfeeding (p value = 0.001, R2 = 0.06), and antibiotic use (p value = 0.002, R2 = 0.016; Adonis2) in early life; with antibiotic use explaining slightly higher variation between the groups after the time and feeding variables. Microbial communities were mainly structured by time-point, and significant differences were observed between the CSab and VDnoab groups at week 1, week 4, and week 8 (p values = 0.024, 0.048, 0.018, respectively, Adonis2) and between VDnoab and CSnoab groups at week 1 (p value = 0.075) (Figure S1B). To determine the effect of delivery mode and antibiotic use on the microbial composition, PERMANOVA was performed using pairwiseAdonis in R on microbial composition data at each time-point. At week 1, delivery mode and antibiotic usage caused significant variations in the microbial composition of infants, with delivery mode explaining higher variance (pairwiseAdonis). The effect of the mode of delivery decreased over time, but antibiotic use in the first week of life was a significant variable affecting microbial composition in infants up to week 24 (Figure S2B).
Microbial composition of infants in early life evolves with age
Given the significant effect of delivery mode and early antibiotic exposure on microbial diversity, the colonisation pattern in the three groups was examined. The phyla Firmicutes and Proteobacteria were prevalent in CS-born infants, with the highest relative abundance in the CSab group (Figure S2A). Actinobacteria abundance was relatively high in VD infants, with high relative abundance in the no-antibiotic-treated groups and lowest in the CSab group from week 1 (At week 1: p.adj value: VDnoab to CSab = 0.0005, Dunn test with BH). A detailed look at each time point demonstrated the absence of phylum Bacteroidota in the CS-born infants at week 1 with higher relative abundance in the no-antibiotic groups (p.adj-value: VDnoab to CSnoab = 0.003, VDnoab to CSab = 0.008, Dunn test with BH). At weeks 1 and 4, Firmicutes were relatively higher in the CS-born infants (CSnoab) (Figure S2A) (at week 1: p.adj-value: CSnoab to VDnoab = 0.009, at week 4: p.adj-value: CSab to CSnoab = 0.005 and CSab to VDnoab = 0.002, Dunn test with BH).
We further examined the mean relative abundances of the classes (Fig. 1C) and the top 10 most abundant species for all groups longitudinally (Fig. 2A). Commensals such as Bifidobacterium species including B. bifidum, B. breve, B. longum, B. kashiwanohense, Bacteroides/Phocaeicola (Phocaeicola vulgatus), Collinsella aerofaciens, and E. coli abundances were highest in the VDnoab group at week 1 and week 4, and generally, an increasing trend was seen for all groups until week 24. Interestingly, Bifidobacterium dentium abundance was highest in the CSab group, granting this species an undisclosed benefit in adverse circumstances like the administration of antibiotics. A low prevalence of commensals was observed in CS-born infants, with higher initial relative abundance of opportunistic bacteria in the CSab group belonging to Proteobacteria and Firmicutes such as Klebsiella pneumoniae, Ruminococcus gnavus, Veillonella parvula, Raoultella ornithinolytica, and Enterobacter cloacae complex, which decreased over time (Fig. 2A, C). The prevalence of the top 10 species occurring over time in the three groups is provided in Table 2.

A Relative abundance (%) showing the top 10 species per group over time from week 1 up to year 2, where time is in weeks. B Plots show differential abundance analysis using Songbird with VDnoab group as reference run using the formula C(Group, Treatment('VDnoab')). The plot on the left side corresponds to the CSnoab group as treatment while that on the right depicts the CSab group as treatment against the reference group. In both cases, negative values (blue bars) represent the association to the reference group (here VDnoab) while positive values (red bars) represent the association to the treatment group; here CSnoab (on left plot) and CSab (right side plot). C Heatmap representing the association of bacterial abundance with time. Results from Spearman correlation analysis using the relative abundance of species for each group to time-point. P values were adjusted using BH, those < 0.05 were considered significant, and corresponding species and their coefficients were plotted. In the plot, blue bands denote decreasing with time (negative coefficient), red bands denote positive coefficient which means increasing over time, while grey means NA denoting not detected significantly as top 10 in that group
Stronger association and colonisation of CS-born babies with pathobionts
To further determine the association of each group and time with bacterial taxa, microbial relative abundance (species) data were associated with longitudinal time-point using Spearman correlation for each group (Fig. 2C). The top 10 positive and negative coefficients derived after filtering for significant p.adjusted values were then plotted (Wilcoxon test and BH method). In general, facultative anaerobes (such as Staphylococcus, Streptococcus, Enterococcus) known to colonise the gut in the first week of life showed a decreasing trend over time, while obligate anaerobes (Blautia, Bifidobacterium, Anaerostipes, Bacteroides, Eggerthella, Ruminococcus, Eubacterium) showed a growing trend with time [53]. The taxa B. bifidum, Faecalibacterium prausnitzii, Blautia (Blautia wexlerae), Bacteroides (Bacteroides fragilis), Eubacterium (Eubacterium rectale, Eubacterium hallii), Anaerostipes (Anaerostipes hadrus), and Ruminococcus (Ruminococcus bromii) were found to have positive associations and increased in all three groups over time (Fig. 2C).
Species that were significantly different between any two groups overall were identified using the Songbird differential abundance analysis tool. Briefly, Songbird results showed a stronger association of Bifidobacterium species to the VDnoab group, but when CSnoab to CSab were compared, Bifidobacterium species were more associated with the CSnoab group (Table S2). When the VDnoab group was used as a reference (Fig. 2B), several commensals with high relative abundance and prevalence (Fig. 2A and Table 2) were associated with it, some of which are involved in folate, lactate, and butyrate production such as Bifidobacterium adolescentis, Ruthenibacterium lactatiformans, Erysipelatoclostridium ramosum, B. longum, Lactobacillus paragasseri, B. kashiwanohense, C. aerofaciens, B. fragilis, Parabacteroides distasonis, and Phocaeicola dorei. Conversely, a mix of opportunistic bacteria and commensals including Clostridium perfringens, Veillonella (sp seminalis and parvula), Bifidobacterium pseudocatenulatum, Streptococcus salivarius, E. coli, Enterococcus faecalis, and R. bromii was associated with the CSnoab group. Similarly, the CSab group was more associated with B. dentium, Ruminococcus gnavus, E. faecalis, V. parvula, Faecalibacterium prausnitzii, and K. pneumoniae.
When comparing associations between CSnoab vs CSab groups (Figure S2C), we observed similar results as above where B. dentium, V. parvula, K. pneumoniae, F. prausnitzii, B. breve, E. faecalis, and R. gnavus showed a stronger association to the CSab group (Figure S2C).
Antibiotic administration in early life is strongly associated with the resistome profile
To further analyse the data from a genome perspective, we reconstructed a total of 1448 medium-quality prokaryotic MAGs corresponding to eight phyla, 1441 known species (GTDB-k), and 49 unknown species. Using universal markers, we reconstructed a phylogenetic tree using PhyloPhlan (Fig. 3).

Phylogenetic tree created using Phylophlan from 1448 medium-quality MAGs generated in this study. Each branch represents a MAG with (from innermost to outermost ring): phylum level distribution of the MAGs, antibiotic use in early life and delivery mode, groups variable in study, succeeded by time-point and log-transformed genome counts of ARG abundance per MAG as examined using ABRicate with CARD
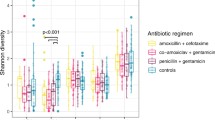
We annotated the MAGs using the CARD database with ABRicate. The presence of 213 ARGs was detected in 1024 MAGs. Resistance was observed against 26 different classes of antibiotics in the infant cohort, which included antimicrobials belonging to ‘critically important’, ‘highly important’, and ‘important’ categories (as categorised by WHO: https://apps.who.int/iris/bitstream/handle/10665/312266/9789241515528-eng.pdf). The highest ARG distribution was in the phylum Proteobacteria followed by Actinobacteria and Firmicutes (Fig. 3), with Proteobacteria possessing the highest abundance of unique ARGs (182). The majority of genera contributing to this ARG abundance included several taxa belonging to Gammaproteobacteria namely Escherichia, Klebsiella, Enterobacter, Raoultella, Citrobacter, and others including Enterococcus, Staphylococcus, Streptococcus, and Bifidobacterium. Interestingly, species belonging to Escherichia contributed to the largest number of ARG abundance accounting for 56% of the total number of ARGs detected (146 different ARGs present out of 213 detected) (Figure S3 A). Results from the read-based approach using RGI with CARD further confirmed the resistome profile distribution in infants with a high abundance of resistance genes observed for aminoglycoside, MDR, macrolide, beta-lactam, tetracycline, and fluoroquinolone antibiotics (Fig. 4A). Functional characterisation of ARGs showed that antibiotic target alteration was the most common type, followed by antibiotic inactivation and antibiotic efflux as mechanisms for antibiotic resistance. The distribution and abundance pattern of ARGs in the study’s three groups was investigated, and a decrease in alpha-diversity from week 1 to year 2 (p.adjusted values using Wilcoxon test with BH: for CSab = 0.01, for CSnoab = 0.012, and for VDnoab = 0.0005), week 4 to year 2 for CSab group only (p.adjusted value = 0.01, Wilcoxon test with BH), week 8 to year 2 (p.adjusted values using Wilcoxon test with BH: for CSnoab = 0.01 and for VDnoab = 0.00031) and week 24 to year 2 (p.adjusted values: for CSab = 0.03, for CSnoab = 0.00026, and for VDnoab = 0.00052, Wilcoxon test with BH) was observed; however, no other differences were seen (Fig. 4B). PERMANOVA demonstrated discrete clustering between the CSab and VDnoab groups (p value = 0.03, PERMANOVA) using a non-metric multidimensional scaling (NMDS) plot with Bray–Curtis distance matrix (Figure S3 B). Antibiotic use in early life and time both had a significant impact on the ARG diversity (antibiotic exposure: p value = 0.007, time: p value = 0.001, PERMANOVA) but no significant effect of mode of delivery was seen.

A Box plot representing the total resistome profile of the infants included in this study, with antibiotic class names of penams and aminoglycosides retained in MDR classes to facilitate understanding, as these are the classes of antibiotics administered to infants in the CSab group. Antibiotic classes were detected using RGI with CARD. The abundance of antibiotic classes was normalised to cpm and log-transformed. B Alpha-diversity plot using Shannon diversity index for ARG diversity for each group between all the time points. P value of < 0.05 was considered significant (Wilcoxon test). C MaAsLin2 differential abundance analysis coefficients obtained by running MaAsLin2 overall and at week 1 and week 4 using Group as a fixed effect with each group (VDnoab, CSnoab, and CSab) as reference per run along with the parameters min_prevalence = 0.1 and min_abundance = 0
When looking at antibiotic resistance for all the classes with VDnoab as reference (Figure S4), the CSab group showed resistance to several antibiotic classes overall and at week 1 and week 4 including aminoglycoside, beta-lactams and penams, MDR including aminoglycosides, which were drugs administered to the infants during the first week of life. Interestingly, at week 4, CSab showed significantly high resistance to several antibiotic classes with reference to the VDnoab group while the CSnoab group showed none. These differences could be due to the transient nature of the many taxa that colonised during early life in the CSnoab group, which might have reduced in abundance over time. However, in the CSab group, antibiotic exposure in the first 4 days of life resulted in selective pressure that might result in the presence of these ARG-containing bacteria, which were detected at week 4, and then possibly decreased over time (Figure S4).
Additionally, 20 classes of differentially abundant antibiotic resistance classes were detected using MaAsLin2. MaAsLin2 was run using fixed effect as a group variable with each group (VDnoab, CSnoab, CSab) as a reference each time and subject number as a random effect for the overall dataset. Overall, at week 1, and at week 4, the CSab group was strongly (positively) associated with genes conferring resistance to aminoglycoside, beta-lactams, penams, MDR, elfamycin, fluoroquinolone, tetracycline, and macrolide when compared with the other groups. Some of these antibiotic classes such as penams, and aminoglycosides are the same class as those administered to infants in this group during the first 4 days of life (Fig. 4 C). Interestingly, infants in the other two groups, CSnoab and VDnoab also showed positive associations to certain antibiotic classes even at week 1 and week 4 when no antibiotics were administered during the first week of life.
Strain persistence is increased in vaginal delivery and antibiotic naïve groups
To understand the prevalence patterns of the resistome in the infants gut, the antibiotic resistance observed due to early life antibiotic exposure was investigated to check if it persists in bacterial strains up to 2 years of age. inStrain was used with MAGs to identify identical strains and examine their persistence in infants in early life. Briefly, bins were dereplicated at 98% whole-genome average nucleotide identity (gANI) to obtain unique strains, which were mapped onto shotgun reads. If the genomic region demonstrated more than 99.99% population-level ANI (popANI) identity in two samples, the strains were considered identical in the two samples. Based on the classification of persistent, non-persistent, and early colonisers (adapted from [43]), a higher number of early colonisers were present in the no-antibiotic groups, with the highest detected in the VDnoab group (Fig. 5B). Further, 98% of the detected strains were early colonisers, of which 45% were non-persistent colonisers. Of the early colonisers, 38% in the CSnoab group, 50% in the VDnoab group, and 48% in the CSab group were non-persistent colonisers (Fig. 5B). The highest number of persistent colonisers that remained until two years of age was in the VDnoab group, which could be due to the higher number of strains inherited maternally by vertical transmission such as those belonging to the Bacteroidota phylum in this group. Overall, due to their abundant appearance in early life in healthy infants and critical role in carbohydrate metabolism in early life, bifidobacteria constituted the genera that contributed to the maximum percentage of persistent colonisers. Bacteroides were generally persistent colonisers, with very few non-persistent colonisers, while majorly, Proteobacteria along with some Actinobacteria and Firmicutes were among the top non-persistent colonisers (Figure S5, S6, and S7). Based on the stringent criteria to define strains, we consider that ARG abundance detected within persistent colonisers are the same genes that appeared in early life and persisted until year 2. Investigating the top 10 strains carrying ARGs revealed that all strains in the no-antibiotic groups belonged to the genera Escherichia, while those in the CSab group belonged to a mixture of taxa from Gammaproteobacteria, including Escherichia, Klebsiella, and Raoultella; with none persisting up to 2 years of age. A closer look at the top 10 strains per group and antibiotic class showed a wide variety of early colonisers belonging to genera Escherichia, Enterococcus, Bifidobacterium, Streptococcus, Collinsella, Phocaeicola, and Bacteroides among others; the majority of these strains were non-persistent colonisers, with most present until week 8 and some until week 24 (Fig. 5A).

A Persistence of strains carrying the top 10 highest abundance of AMR per group and class in infants up to 2 years of age. The right side of the plot depicts abundance as detected by ABRicate in the form of a heatmap. The strains are colour coded based on the phylum to which they belong, as detected by GTDB. B Bar plot showing the percent distribution of the genera Bifidobacterium, Bacteroides/Parabacteroides/Phocaeicola, and others as early colonisers (further divided into persistent colonisers and non-persistent colonisers) and late colonisers for each of the three groups in the study
Functional profiles of the microbiota in infants are influenced by antibiotic exposure and age
Differences in microbial composition, relative abundance, and resistome might infer variation in the functional capabilities of the microbiota. Thus, we investigated differences in the functional ability of gut microbiota in the three study groups VDnoab, CSnoab, and CSab from both the MAGs and reads perspectives. Carbohydrate metabolism including HMO and galactooligosaccharides (GOS) degradation are important functions for infant gut microbiota in early life especially when fed breast milk. Overall CAZyme glycoside hydrolase (GH) abundance inferred from the MAGs was significantly higher in the VDnoab group as compared to CSnoab and CSab groups (p.adj = < 0.0001 and 0.015, respectively; Wilcoxon test using BH method) and CSnoab group had higher GH abundance compared to the CSab group (p value = 0.0033) at week 1. At week 4, a significant difference was observed between VDnoab and CSnoab and VDnoab and CSab (p value = 3.3e − 06, 9.7e − 06, respectively). CAZyme analysis was also performed for selective GH related to HMO, GOS, and fructooligosaccharides (FOS) degradation enzyme families due to their importance in early life. We observed that the VDnoab group had significantly different overall CAZyme composition (p.adj = 0.012, PERMANOVA) while no other differences were observed otherwise between groups for overall or selective GH composition. Further, CAZyme composition differed significantly based on time with all early time points clustering separately from year 2 for both overall and selective CAZymes (PERMANOVA). Furthermore, delivery mode had a significant impact on the overall and selective GH composition (p.adj = 0.019 and 0.039, respectively, PERMANOVA), while antibiotic use only affected the overall GH composition (p.adj = 0.019). The phyla Actinobacteria and Bacteroidota had the highest percent contribution to GH abundance and it was highest in the VDnoab group. At the same time, Firmicutes and Proteobacteria contributed to a higher percentage in the CS-born infants (Fig. 6A). GH abundance related to HMOs and GOS decreased over time in all three groups, and the VDnoab group had significantly higher abundance than the CSnoab group (p value = 0.0035, Wilcoxon test).

A Bar plot depicting percent contribution of each of the detected phyla to cpm normalized abundance of CAZymes important for HMO, GOS, and FOS degradation in infants. The plot presents the percent distribution for each phyla for cpm normalized abundance at each time point for all three groups VDnoab, CSnoab, and CSab. B Pathway abundance data for unstratified pathways as detected from HUMAnN3 was categorised into higher MetaCyc pathways. cpm abundance was log-transformed, and a significant difference in abundance between the groups was calculated using the Wilcoxon test with BH using the VDnoab group as a reference. P.adj-values (as denoted by *) < 0.05 are considered significant. C PCoA plot using Bray–Curtis distance for stratified pathway abundance data from HUMAnN3, showing distinct clustering of CSab from the other two groups
Pathway relative abundance from HUMAnN3 was analysed by clustering pathways to higher order MetaCyc pathways. Based on MetaCyc pathways (unstratified) from HUMAnN3 results (Fig. 6B), only aldehyde degradation was observed to be significantly different (lower) in the CSab group compared to the VDnoab group. The antibiotic-treated infants showed a stronger association with pyrimidine nucleotide biosynthesis, ubiquinol (electron carrier biosynthesis), and aromatic compound degradation (toluene, catechol, protocatechuate) pathways, with essential pathways such as glycolysis, phospholipid biosynthesis, nucleic acid processing, carbohydrate degradation, and amino acid biosynthesis increased over time (Table 3). Similarly, NMDS analysis with Bray–Curtis showed a significant difference between week 1 and all later time points, and year 2 and all earlier time points (p values < 0.05, Adonis2).
A closer look at the stratified counterpart of pathway abundance data from HUMAnN3 showed study variables including groups, delivery mode, antibiotic exposure in early life, and time-point having an effect on pathway abundance with significant clustering observed between VDnoab and CSab and between CSnoab and CSab groups (Fig. 6C) (pairwise Adonis, PERMANOVA: p values = 0.003 and 0.045, respectively, for groups, p value = 0.01 by mode of delivery, p value = 0.001 by early antibiotic exposure, and p value = 0.001 by time point, PERMANOVA). Based on R2 values from Adonis, time variable and early antibiotic exposure (R2: 0.15, 0.015, respectively) had the highest influence on separating samples based on pathway abundance; this along with distinct clustering of the CSab group from the no-antibiotic groups point towards the impact of antibiotics in early life on the functionality of the microbiota. Many pathways including those related to sugar metabolism, purine and pyrimidine biosynthesis, and amino acid biosynthesis were associated with the antibiotic-treated group. These differentially abundant pathways were mostly driven by B. dentium species, Ruminococcus, and Faecalibacterium species (Table S3).
Discussion
We analysed the longitudinal resistome and taxonomic profiles of infants in early-life grouped by early antibiotic use and delivery mode. Results from this study show that the resistome diversity decreases over age from week 1 to year 2, while the microbial diversity increases over time. This is in congruence with previous studies; however, most studies report these profiles only up to year 1 and not many study the resistome profiles as early as week 1. These data also showed the presence of selective antibiotic resistance classes which were administered to the infants in the antibiotic-exposed group at week 1 and week 4. Although the antibiotic in question was only administered in the first 4 days of life, the effect was seen further as age progressed, highlighting the importance of further development of strong stewardship programmes in early life. Additionally, our data also suggest that antibiotic use in early life results in amplification of the resistome profile by selecting MDR bacteria. In this study, antibiotic exposure in early life and mode of delivery were seen to influence microbial diversity and richness between samples longitudinally, with antibiotic exposure having the greatest effect.
A stronger association and higher prevalence of Bifidobacterium and Bacteroides were detected in vaginally delivered infants, while CS-born infants had higher relative abundance of potential pathogens including Enterococcus spp. and species of Enterobacteriaceae which was similar to previous reports [7, 19, 21, 22, 54]. CS-born infants are more prone to negative health outcomes such as allergy, asthma, obesity, and diabetes development in later life [55, 56]. Additionally, colonisation patterns of CS infants with bacteria belonging to the category of ESKAPE (E. faecium, S. aureus, K. pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) pathogens are of concern as most bacteria in this group are MDR and are known to act as potential pathogens or are pathobionts [57, 58], thus making any futuristic treatments more difficult to curtail in later life. Many Bifidobacterium and Bacteroidota spp. showed lower or delayed colonisation in CS-born infants, with a pronounced effect in the CSab group, which is analogous to previous reports [19, 59, 60], except for B. dentium which was seen to have high relative abundance in the CSab group. Recent studies have shown B. dentium in high abundance in infants belonging to the intrapartum antibiotic prophylaxis (IAP) group [61, 62] or those fed with donor breast milk [63]. Our result showing high B. dentium in the CSab group is in agreement with these reports and further studies are needed to understand the advantageous growth of this Bifidobacterium spp. under sub-optimal circumstances. The beneficial and protective roles of Bifidobacterium and Bacteroidota spp. in the infant gut, such as fermenting HMOs, maintaining barrier function, enhancing immune responses, colonisation resistance, and competing for nutrients against pathogens are well established [60,61,62, 64,65,66,67]. Decreased colonisation of these beneficial taxa is associated with adverse outcomes such as necrotising enterocolitis and late-onset sepsis in the preterm population [68, 69], thus demanding further attention for studies in infants born by CS and exposed to antibiotics.
A diverse range of ARGs and MDR bacteria was detected in all three groups, with decreasing ARG diversity seen over time. The most common antibiotic classes observed were MDR, which includes resistance to penams, fluoroquinolones, peptides, aminocoumarin, aminoglycosides, and tetracyclines. This is in line with many reports suggesting the infant gut microbiota is a reservoir for ARGs [11, 12, 70]. The top abundance of ARGs was detected in Escherichia, Klebsiella, Enterobacter, Bifidobacterium, Raoultella, and other Proteobacteria and Firmicutes species, with the highest association of antibiotic classes to the CSab group. Our findings are consistent with previous studies that report a high abundance of ARGs in several taxa belonging to Gammaproteobacteria [12, 17, 71,72,73]. The CSab group also showed the presence of ARGs and stronger associations with classes of antibiotics administered to the infants in this group, clustering separately from the no-antibiotic groups, further pointing towards the fact that antibiotic exposure contributed to ARG abundance more than the delivery mode.
This study found a higher frequency of ARGs in the CSab group, followed by the CSnoab and VDnoab groups, primarily due to this group’s antibiotic exposure in early life. This can be attributed to the fact that several commensals and pathobionts harbour a set of ARGs, which could be inherited or transferred by horizontal gene transfer in the highly populated environment in the gut. Additionally, the use of antibiotics in early life alters the microbial composition and results in the selection of bacteria whose ARG reservoir includes genes capable of conferring resistance to the class of antibiotic in question. The selection of such bacteria resistant to the antibiotic in question may possess several other ARGs leading to the presence and detection of MDR bacteria. These pathobionts can switch to their opportunistic pathogenic side and can lead to infections that are difficult to treat due to the plethora of ARGs they contain. Disturbed microbial colonisation due to delivery mode and early antibiotic exposure can lead to colonisation by more hospital-acquired strains. These nosocomial strains are enriched with a diverse resistome profile [74,75,76,77]. Furthermore, the high frequency of ARGs found in this study was associated with their high abundance in Proteobacteria. Increased abundance of specific taxa in this group (such as some Klebsiella, Enterobacter, Raoultella species) is reported to be associated with several functional variations and difficult-to-treat infections [78,79,80,81]. The establishment of such strains from infancy in the gut can lead to complications in treatment in later life. Highly regulated antibiotic stewardship programmes are thus necessary along with probable therapeutic administration of probiotics where possible. Due to the stringent parameters used to define a strain, we were able to examine the persistence of resistance in the strains and observed that the top 10 strains per class and group showed very little to no persistence up to two years of age. Strains that carried the highest ARG abundance mostly belonged to the genera Enterococcus, Escherichia, Veillonella, Streptococcus, Bifidobacterium, and Bacteroidetes. Moreover, Bifidobacterium strains and Bacteroidota strains were observed to be major persistent colonisers, which sustain up to two years of age with more persistence observed in the non-antibiotic groups with the highest in the vaginally delivered infants.
Though overall GH abundance was significantly different and higher in the VDnoab group and lowest in the CSab group at week 1 and week 4, this difference was not prominent when selective GH related to HMO, FOS, and GOS enzyme families were considered. Further, the percent contribution of various phyla to select GH functions was observed to vary between groups, prominently seen in early time points. This could be because functions like HMO degradation are essential and redundant in early life and colonisation is not solely dependent on feeding habits, mode of delivery, or antibiotic exposure. While Bifidobacterium and Bacteroides are recognised as proficient HMO degraders [82, 83], their scarcity or non-existence in infants does not negate the critical role of HMO degradation, which can still be performed by other bacterial species present in these infants. Similarly, unstratified metaCyC pathways did not show significant differences when the CS-born infant groups were compared to the VDnoab group while based on stratified pathway abundance data CSab was significantly different from the other two groups. This could be because the specific microbial composition of each group contributes to similar functional attributes. Furthermore, strong associations of perinatal factors such as antibiotic treatment with aromatic compound degradation pathways, amino acid biosynthesis, cofactor and coenzyme biosynthesis, nucleotide biosynthesis (both purine and pyrimidine), and carbohydrate degradation pathways were observed. Similar associations such as increased nucleotide biosynthesis pathways and carbohydrate degradation were reported earlier in antibiotic-treated groups [84, 85]. Such changes in metabolic potential can affect children’s health in later life, and detailed studies will help understand these changes and their influence.
Some strengths of this study include examining several factors such as delivery mode, antibiotic use in early life, and age on the dynamic nature of microbial colonisation pattern with read- and assembly-based approaches using shotgun sequencing. We also inspected and observed the taxa in infants that act as persistent colonisers and colonise the gut up to 2 years of age. This study was limited due to a small cohort of 45 infants and limited sample numbers between time points. Further, clinical metadata of antibiotic exposure relating to all time points of sample collection or similar data from the mothers were missing, which narrows down the hypotheses that could be validated in the study. Thus, future studies with detailed clinical metadata and more time points with smaller intervals will help unfold the complex relationship between study variables, microbial and resistome profile and functionality of the microbiota.
Conclusion
This study highlights the effects of antibiotic exposure in early life and mode of delivery on infant gut microbiota in a longitudinal setting. Treatment with a combination of benzylpenicillin and gentamicin in early life has a stronger influence over the type and order of microbial colonisation in infants in early life than delivery mode. Such disturbed colonisation, along with a diverse resistance reservoir impact the functional capability of the infant gut microbiota, including but not limited to the impairment that can be caused to other crucial functional capacities such as colonisation resistance, metabolism, maintaining gut permeability, and a healthy immune response. Thus, more studies with larger cohorts are needed to better understand ARGs’ persistence and impact on microbial functional capability with more certainty. Moreover, new alternatives/strategies need to be developed and used where needed to restore the microbial ecosystem, maintain a healthy microenvironment, restore their functional powers, and reduce the use of prophylactic antibiotics during the crucial infancy stage.
Availability of data and materials
The metagenomics data generated as a part of this study is available on NCBI with the SRA accession number PRJNA971895, available here: https://www.ncbi.nlm.nih.gov/sra/PRJNA971895.
References
Arrieta M-C, Stiemsma LT, Amenyogbe N, Brown EM, Finlay B. The intestinal microbiome in early life: health and disease. Front Immunol. 2014;5(September):427. https://doi.org/10.3389/fimmu.2014.00427.
Pickard JM, Zeng MY, Caruso R, Núñez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279(1):70–89. https://doi.org/10.1111/imr.12567.
Dinan TG, Cryan JF. Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol. 2017;595(2):489–503. https://doi.org/10.1113/JP273106.
Walker WA. Initial intestinal colonization in the human infant and immune homeostasis. Ann Nutr Metab. 2013;63(Suppl. 2):8–15. https://doi.org/10.1159/000354907.
Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30(6):492–506. https://doi.org/10.1038/s41422-020-0332-7.
Cong X, Xu W, Janton S, Henderson WA, Matson A, McGrath JM, Maas K, Graf J. Gut microbiome developmental patterns in early life of preterm infants: Impacts of feeding and gender. PLoS ONE. 2016;11(4):1–19. https://doi.org/10.1371/journal.pone.0152751.
Dominguez-bello MG, Costello EK, Contreras M, Magris M, Hidalgo G. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–5. https://doi.org/10.1073/pnas.1002601107.
Pärnänen KMM, Hultman J, Markkanen M, Satokari R, Rautava S, Lamendella R, Wright J, McLimans CJ, Kelleher SL, Virta MP. Early-life formula feeding is associated with infant gut microbiota alterations and an increased antibiotic resistance load. Am J Clin Nutr. 2022;115(2):407–21. https://doi.org/10.1093/ajcn/nqab353.
Arboleya S, Saturio S, Gueimonde M. Impact of intrapartum antibiotics on the developing microbiota: a review. Microbiome Res Rep. 2022;1(3):22. https://doi.org/10.20517/mrr.2022.04
Patangia DV, Ryan CA, Dempsey E, Stanton C, Ross RP. Vertical transfer of antibiotics and antibiotic resistant strains across the mother/baby axis. Trends Microbiol. 2022;30(1):47–56. https://doi.org/10.1016/j.tim.2021.05.006.
Tapiainen T, Koivusaari P, Brinkac L, Lorenzi HA, Salo J, Renko M, Pruikkonen H, Pokka T, Li W, Nelson K, Pirttilä AM, Tejesvi MV. Impact of intrapartum and postnatal antibiotics on the gut microbiome and emergence of antimicrobial resistance in infants. Sci Rep. 2019;9(1):10635. https://doi.org/10.1038/s41598-019-46964-5.
Pärnänen K, Karkman A, Hultman J, Lyra C, Bengtsson-Palme J, Larsson DGJ, Rautava S, Isolauri E, Salminen S, Kumar H, Satokari R, Virta M. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat Commun. 2018;9(1):1–11. https://doi.org/10.1038/s41467-018-06393-w.
Klassert TE, Zubiria-Barrera C, Kankel S, Stock M, Neubert R, Lorenzo-Diaz F, Doehring N, Driesch D, Fischer D, Slevogt H. Early bacterial colonization and antibiotic resistance gene acquisition in newborns. Front Cell Infect Microbiol. 2020;10(July):1–9. https://doi.org/10.3389/fcimb.2020.00332.
Li X, Stokholm J, Brejnrod A, Vestergaard GA, Russel J, Trivedi U, Thorsen J, Gupta S, Hjelmsø MH, Shah SA, Rasmussen MA, Bisgaard H, Sørensen SJ. The infant gut resistome associates with E. coli, environmental exposures, gut microbiome maturity, and asthma-associated bacterial composition. Cell Host Microbe. 2021;29(6):975-987.e4. https://doi.org/10.1016/j.chom.2021.03.017.
Gibson MK, Wang B, Ahmadi S, Burnham CD, Tarr PI, Warner BB, Dantas G. Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol. 2016;301903:1–25. https://doi.org/10.1038/nmicrobiol.2016.24.Developmental.
Gosalbes MJ, Vallès Y, Jiménez-Hernández N, Balle C, Riva P, Miravet-Verde S, De Vries LE, Llop S, Agerso Y, Sørensen SJ, Ballester F, Francino MP. High frequencies of antibiotic resistance genes in infants’ meconium and early fecal samples. J Dev Orig Health Dis. 2016;7(1):35–44. https://doi.org/10.1017/S2040174415001506.
Lebeaux RM, Coker MO, Dade EF, Palys TJ, Morrison HG, Ross BD, Baker ER, Karagas MR, Madan JC, Hoen AG. The infant gut resistome is associated with E. coli and early-life exposures. BMC Microbiol. 2021;21(1):201. https://doi.org/10.1186/s12866-021-02129-x.
Oliveira J, Reygaert WC. Gram Negative Bacteria. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538213/
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, Khan MT, Zhang J, Li J, Xiao L, Al-Aama J, Zhang D, Lee YS, Kotowska D, Colding C, Jun W. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host and Microbe. 2015;17(5):690–703. https://doi.org/10.1016/j.chom.2015.04.004.
Moore AM, Patel S, Forsberg KJ, Wang B, Bentley G, Razia Y, Qin X, Tarr PI, Dantas G. Pediatric fecal microbiota harbor diverse and novel antibiotic resistance genes. PLoS ONE. 2013;8(11):e78822. https://doi.org/10.1371/journal.pone.0078822.
Reyman M, van Houten MA, van Baarle D, Bosch AATM, Man WH, Chu MLJN, Arp K, Watson RL, Sanders EAM, Fuentes S, Bogaert D. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun. 2019;10(1):1–12. https://doi.org/10.1038/s41467-019-13014-7.
Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, Kumar N, Stares MD, Rodger A, Brocklehurst P, Field N, Lawley TD. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. 2019;574(7776):117–21. https://doi.org/10.1038/s41586-019-1560-1.
World Health Organization. Antimicrobial resistance: global report on surveillance. World Health Organization. 2014. https://apps.who.int/iris/handle/10665/112642
Laxminarayan R, Matsoso P, Pant S, Brower C, Røttingen JA, Klugman K, Davies S. Access to effective antimicrobials: a worldwide challenge. The Lancet. 2016;387(10014):168–75. https://doi.org/10.1016/S0140-6736(15)00474-2.
Jaime Ramirez, Francisco Guarner, Luis Bustos Fernandez, Aldo Maruy, Lucia Sdepanian Vera, Henry Cohen. Antibiotics as major disruptors of gut microbiota. Front Cell Infect Microbiol. 2020;10:572912. https://doi.org/10.3389/fcimb.2020.572912.
Fjalstad JW, Esaiassen E, Juvet LK, van den Anker JN, Klingenberg C. Antibiotic therapy in neonates and impact on gut microbiota and antibiotic resistance development: a systematic review. J Antimicrob Chemother. 2018;73(3):569–80. https://doi.org/10.1093/jac/dkx426.
Aversa Z, Atkinson EJ, Schafer MJ, Theiler RN, Rocca WA, Blaser MJ, LeBrasseur NK. Association of infant antibiotic exposure with childhood health outcomes. Mayo Clin Proc. 2021;96(1):66–77. https://doi.org/10.1016/j.mayocp.2020.07.019.
Betrán AP, Ye J, Moller A-B, Zhang J, Gülmezoglu AM, Torloni MR. The increasing trend in caesarean section rates: global, regional and national estimates: 1990–2014. PLoS ONE. 2016;11(2): e0148343. https://doi.org/10.1371/journal.pone.0148343.
Liu R, Lin L, Wang D. Antimicrobial prophylaxis in caesarean section delivery. Exp Ther Med. 2016;12(2):961–4. https://doi.org/10.3892/etm.2016.3350.
Patangia DV, Anthony Ryan C, Dempsey E, Paul Ross R, Stanton C. Impact of antibiotics on the human microbiome and consequences for host health. Microbiol open. 2022;11(1):e1260. https://doi.org/10.1002/mbo3.1260. (PMID: 35212478; PMCID: PMC8756738).
Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8:343ra81. https://doi.org/10.1126/scitranslmed.aad0917.
Hill CJ, Lynch DB, Murphy K, et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome. 2017;5:4. https://doi.org/10.1186/s40168-016-0213-y.
Yu Z, Morrison M. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 2004;36(5):808–12. https://doi.org/10.2144/04365ST04.
Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, Valles-Colomer M, Weingart G, Zhang Y, Zolfo M, Huttenhower C, Franzosa EA, Segata N. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 2021;10:1–42. https://doi.org/10.7554/eLife.65088.
Olbrich M, Künstner A, Busch H. MBECS: microbiome batch effects correction suite. BMC Bioinformatics. 2023;24:182. https://doi.org/10.1186/s12859-023-05252-w.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27(5):824–34. https://doi.org/10.1101/gr.213959.116.
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6(1):158. https://doi.org/10.1186/s40168-018-0541-1.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55. https://doi.org/10.1101/gr.186072.114.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2019;36(6):1925–7. https://doi.org/10.1093/bioinformatics/btz848.
Asnicar F, Thomas AM, Beghini F, Mengoni C, Manara S, Manghi P, Zhu Q, Bolzan M, Cumbo F, May U, Sanders JG, Zolfo M, Kopylova E, Pasolli E, Knight R, Mirarab S, Huttenhower C, Segata N. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 2020;11(1):2500. https://doi.org/10.1038/s41467-020-16366-7.
Huang L, Zhang H, Wu P, Entwistle S, Li X, Yohe T, Yi H, Yang Z, Yin Y. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018;46(D1):D516–21. https://doi.org/10.1093/nar/gkx894.
Olm MR, Crits-Christoph A, Bouma-Gregson K, Firek BA, Morowitz MJ, Banfield JF. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat Biotechnol. 2021;39(6):727–36. https://doi.org/10.1038/s41587-020-00797-0.
Lou YC, Olm MR, Diamond S, Crits-Christoph A, Firek BA, Baker R, Morowitz MJ, Banfield JF. Infant gut strain persistence is associated with maternal origin, phylogeny, and traits including surface adhesion and iron acquisition. Cell Reports Medicine. 2021;2(9):100393. https://doi.org/10.1016/j.xcrm.2021.100393.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, Doshi S, Courtot M, Lo R, Williams LE, Frye JG, Elsayegh T, Sardar D, Westman EL, Pawlowski AC, McArthur AG. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Research. 2017;45(D1):D566–73. https://doi.org/10.1093/nar/gkw1004.
Chen L, Zheng D, Liu B, Yang J, Jin Q. VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016;44(D1):D694–7. https://doi.org/10.1093/nar/gkv1239.
Carattoli A, Zankari E, García-Fernández A, Voldby Larsen M, Lund O, Villa L, Møller Aarestrup F, Hasman H. In silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58(7):3895–903. https://doi.org/10.1128/AAC.02412-14.
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., Huynh, W., Nguyen, A.-L. V, Cheng, A. A., Liu, S., Min, S. Y., Miroshnichenko, A., Tran, H.-K., Werfalli, R. E., Nasir, J. A., Oloni, M., Speicher, D. J., Florescu, A., Singh, B., … McArthur, A. G. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019;48(D1), D517–D525. https://doi.org/10.1093/nar/gkz935
Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, Chatterjee S, Thompson KN, Wilkinson JE, Subramanian A, Lu Y, Waldron L, Paulson JN, Franzosa EA, Bravo HC, Huttenhower C. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol. 2021;17(11):e1009442. https://doi.org/10.1371/journal.pcbi.1009442.
Morton JT, Marotz C, Washburne A, Silverman J, Zaramela LS, Edlund A, Zengler K, Knight R. Establishing microbial composition measurement standards with reference frames. Nat Commun. 2019;10(1):2719. https://doi.org/10.1038/s41467-019-10656-5.
Oksanen J, Guillaume Blanchet F, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, M. Stevens HH, Szoecs E, Wagner H. vegan: Community Ecology Package. R package version 2.5–7. 2020. https://CRAN.R-project.org/package=vegan
Mcmurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8(4):e61217. https://doi.org/10.1371/journal.pone.0061217.
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Robertson RC, Manges AR, Finlay BB, Prendergast AJ. The human microbiome and child growth - first 1000 days and beyond. Trends Microbiol. 2019;27(2):131–47. https://doi.org/10.1016/j.tim.2018.09.008. (Epub 2018 Oct 24. PMID: 30529020).
Adlerberth I, Lindberg E, Åberg N, Hesselmar B, Saalman R, Strannegård I-L, Wold AE. Reduced enterobacterial and increased Staphylococcal colonization of the infantile bowel: an effect of hygienic lifestyle? Pediatr Res. 2006;59(1):96–101. https://doi.org/10.1203/01.pdr.0000191137.12774.b2.
Cardwell CR, Stene LC, Joner G, Cinek O, Svensson J, Goldacre MJ, Parslow RC, Pozzilli P, Brigis G, Stoyanov D, Urbonaite B, Sipetić S, Schober E, Ionescu-Tirgoviste C, Devoti G, de Beaufort CE, Buschard K, Patterson CC. Caesarean section is associated with an increased risk of childhood-onset type 1 diabetes mellitus: a meta-analysis of observational studies. Diabetologia. 2008;51(5):726–35. https://doi.org/10.1007/s00125-008-0941-z. (Epub 2008 Feb 22. PMID: 18292986).
Słabuszewska-Jóźwiak A, Szymański JK, Ciebiera M, Sarecka-Hujar B, Jakiel G. Pediatrics consequences of caesarean section-a systematic review and meta-analysis. Int J Environ Res Public Health. 2020;17(21):8031. https://doi.org/10.3390/ijerph17218031. (PMID: 33142727).
Ma Y, Wang C, Li Y, Li J, Wan Q, Chen J, Tay FR, Niu L. Considerations and caveats in combating ESKAPE pathogens against nosocomial infections. Advanced Science. 2020;7(1):1901872. https://doi.org/10.1002/advs.201901872.
Santajit S, Indrawattana N. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed Res Int. 2016;2016(1155):1–8. https://doi.org/10.1155/2016/2475067.
Dogra S, Sakwinska O, Soh S-E, Ngom-Bru C, Brück WM, Berger B, Brüssow H, Lee YS, Yap F, Chong Y-S, Godfrey KM, Holbrook JD. Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. MBio. 2015;6(1):1–9. https://doi.org/10.1128/mBio.02419-14.
Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, Björkstén B, Engstrand L, Andersson AF. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by Caesarean section. Gut. 2014;63(4):559–66. https://doi.org/10.1136/gutjnl-2012-303249.
Saturio S, Nogacka AM, Alvarado-Jasso GM, Salazar N, de los Reyes-Gavilán CG, Gueimonde M, Arboleya S. Role of bifidobacteria on infant health. Microorganisms. 2021;9(12):2415. https://doi.org/10.3390/microorganisms9122415.
Saturio S, Suárez M, Mancabelli L, Fernández N, Mantecón L, de los Reyes-Gavilán CG, Ventura M, Gueimonde M, Arboleya S, Solís G. Effect of intrapartum antibiotics prophylaxis on the bifidobacterial establishment within the neonatal gut. Microorganisms. 2021;9(9):1867. https://doi.org/10.3390/microorganisms9091867.
Arboleya S, Saturio S, Suárez M, Fernández N, Mancabelli L, de los Reyes-Gavilán CG, Ventura M, Solís G, Gueimonde M. Donated human milk as a determinant factor for the gut bifidobacterial ecology in premature babies. Microorganisms. 2020;8(5):760. https://doi.org/10.3390/microorganisms8050760.
O’Callaghan, A., & van Sinderen, D. Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol. 2016;7(JUN). https://doi.org/10.3389/fmicb.2016.00925
Stewart CJ, Embleton ND, Marrs ECL, Smith DP, Fofanova T, Nelson A, Skeath T, Perry JD, Petrosino JF, Berrington JE, Cummings SP. Longitudinal development of the gut microbiome and metabolome in preterm neonates with late onset sepsis and healthy controls. Microbiome. 2017;5(1):75. https://doi.org/10.1186/s40168-017-0295-1.
Tamana SK, Tun HM, Konya T, Chari RS, Field CJ, Guttman DS, Becker AB, Moraes TJ, Turvey SE, Subbarao P, Sears MR, Pei J, Scott JA, Mandhane PJ, Kozyrskyj AL. Bacteroides-dominant gut microbiome of late infancy is associated with enhanced neurodevelopment. Gut Microbes. 2021;13(1):1–17. https://doi.org/10.1080/19490976.2021.1930875.
Zafar H, Saier MH. Gut Bacteroides species in health and disease. Gut Microbes. 2021;13(1):1–20. https://doi.org/10.1080/19490976.2020.1848158.
Dobbler PT, Procianoy RS, Mai V, Silveira RC, Corso AL, Rojas BS, Roesch LFW. Low microbial diversity and abnormal microbial succession is associated with necrotizing enterocolitis in preterm infants. Front Microbiol. 2017;8:1–12. https://doi.org/10.3389/fmicb.2017.02243.
Patel RM, Denning PW. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr Res. 2015;78(3):232–8. https://doi.org/10.1038/pr.2015.97.
Moore AM, Ahmadi S, Patel S, et al. Gut resistome development in healthy twin pairs in the first year of life. Microbiome. 2015;3:27. https://doi.org/10.1186/s40168-015-0090-9.
Casaburi G, Duar RM, Vance DP, Mitchell R, Contreras L, Frese SA, Smilowitz JT, Underwood MA. Early-life gut microbiome modulation reduces the abundance of antibiotic-resistant bacteria. Antimicrob Resist Infect Control. 2019;8(1):131. https://doi.org/10.1186/s13756-019-0583-6.
Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, Tarr PI, Warner BB, Dantas G. Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat Microbiol. 2019;4(12):2285–97. https://doi.org/10.1038/s41564-019-0550-2.
Bailey JK, Pinyon JL, Anantham S, Hall RM. Commensal Escherichia coli of healthy humans: a reservoir for antibiotic-resistance determinants. J Med Microbiol. 2010;59(11):1331–9. https://doi.org/10.1099/jmm.0.022475-0.
Raplee I, Walker L, Xu L, Surathu A, Chockalingam A, Stewart S, Han X, Rouse R, Li Z. Emergence of nosocomial associated opportunistic pathogens in the gut microbiome after antibiotic treatment. Antimicrob Resist Infect Control. 2021;10(1):36. https://doi.org/10.1186/s13756-021-00903-0.
Sukhum KV, Newcomer EP, Cass C, Wallace MA, Johnson C, Fine J, Sax S, Barlet MH, Burnham C-AD, Dantas G, Kwon JH. Antibiotic-resistant organisms establish reservoirs in new hospital built environments and are related to patient blood infection isolates. Comm Med. 2022;2(1):62. https://doi.org/10.1038/s43856-022-00124-5.
Wu D, Jin L, Xie J, Liu H, Zhao J, Ye D, Li X. Inhalable antibiotic resistomes emitted from hospitals: metagenomic insights into bacterial hosts, clinical relevance, and environmental risks. Microbiome. 2022;10(1):19. https://doi.org/10.1186/s40168-021-01197-5.
Lerminiaux NA, Cameron ADS. Horizontal transfer of antibiotic resistance genes in clinical environments. Can J Microbiol. 2019;65(1):34–44. https://doi.org/10.1139/cjm-2018-0275.
Bradley PH, Pollard KS. Proteobacteria explain significant functional variability in the human gut microbiome. Microbiome. 2017;5(1):1–23. https://doi.org/10.1186/s40168-017-0244-z.
Litvak Y, Byndloss MX, Tsolis RM, Bäumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017;39:1–6. https://doi.org/10.1016/j.mib.2017.07.003.
Pammi M, Cope J, Tarr PI, Warner BB, Morrow AL, Mai V, Gregory KE, Kroll JS, McMurtry V, Ferris MJ, Engstrand L, Lilja HE, Hollister EB, Versalovic J, Neu J. Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis: a systematic review and meta-analysis. Microbiome. 2017;5(1):31. https://doi.org/10.1186/s40168-017-0248-8.
Shin N-R, Whon TW, Bae J-W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33(9):496–503. https://doi.org/10.1016/j.tibtech.2015.06.011.
Kunz C, Rudloff S, Baier W, Klein N, Strobel S. Oligosaccharides in human milk: structural, functional, and metabolic aspects. Annu Rev Nutr. 2000;20(1):699–722. https://doi.org/10.1146/annurev.nutr.20.1.699.
Marcobal A, Barboza M, Sonnenburg ED, Pudlo N, Martens EC, Desai P, Lebrilla CB, Weimer BC, Mills DA, German JB, Sonnenburg JL. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe. 2011;10(5):507–14. https://doi.org/10.1016/j.chom.2011.10.007.
Vänni P, Tejesvi MV, Ainonen S, Renko M, Korpela K, Salo J, Paalanne N, Tapiainen T. Delivery mode and perinatal antibiotics influence the predicted metabolic pathways of the gut microbiome. Sci Rep. 2021;11(1):17483. https://doi.org/10.1038/s41598-021-97007-x.
Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, Aagaard KM. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nature Medicine, January. 2017. https://doi.org/10.1038/nm.4272.
Acknowledgements
We would like to thank all the participants in the IMFANTMET and MYNEWGUT cohorts and hospital staff for their help throughout the study. Assistance by Dr. Conall Strain and Dr. Fiona Fouhy with shotgun sequencing is gratefully acknowledged.
Funding
We would also like to acknowledge funding from the European Union—FP7 for MYNEWGUT and ERC, BACtheWINNER, Project No. 101054719, and DAFM for INFANTMET. The authors were funded by the Science Foundation Ireland/APC Microbiome Ireland.
Author information
Authors and Affiliations
Contributions
DP performed DNA extractions and shotgun sequencing library preparation. DP was a major contributor to the writing and editing of the manuscript. DP along with GG performed downstream bioinformatics analysis. GG was involved in manuscript editing. CA-S was involved in the sample collection. ED and TR contributed to supervising, manuscript reviewing, and editing. CS and PR are lead supervisors and are involved in the conceptualization, writing, reviewing, and editing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The infants in this study were recruited as part of INFANTMET (funded by the Department of Agriculture Food and the Marine (DAFM)) and MYNEWGUT (funded by FP7) studies. The ethics were approved by the Cork University Hospital Research Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
A. Microbial diversity of the infant gut as measured using Shannon diversity index between all three groups at each time-point in this study. Significant difference in diversity was observed between CSab and Csnoab at week 1 and 24. B. Beta-diversity using unweighted Unifrac distance as depicted using PCoA at each time point. Figure S2. A. Relative abundance plot showing phylum level distribution in all the study groups at each time-point. B. Percent variance for each time point plotted using R2 values as generated from PERMANOVA for Antibiotic and Mode of delivery variables in the study using unweighted Unifrac distance. C. Plots shows differential abundance analysis using Songbird with CSnoab group as reference run using the formula C(Group, Treatment('CSnoab')). Plot on the left side corresponds to VDnoab group as treatment while that on right depicts CSab group as treatment against the reference group. In both cases negative value (Blue bars) represent association to the reference group (here CSnoab) while positive values (red bars) represent association to the treatment group; here VDnoab (on left plot) and CSab (right side plot). Figure S3. A. Plot showing top 10 species possessing highest abundance (cpm normalised) of ARGs as detected using ABRicate. B. Beta-diversity computed for ARG distribution from RGI using Bray–Curtis distance metrics and plotted using PCoA shows distinct clustering between 1. Groups, 2. Samples based on antibiotic exposure in first four days of life and 3. study time-points. Figure S4. All antibiotic classes as detected using RGI, depicting correlation of each group to AMR class abundance at A. Overall, B. week 1 and C. at week 4. Fluoroquinolone is abbreviated to FQ in the above plot. P-values < 0.05 were considered significant and were generated using Wilcoxon test with VDnoab group as reference. Figure S5. Persistence of all unique strains as observed from inStrains for VDnoab group. Right side of the plot depicts abundance of ARGs as detected by ABRicate in the form of heatmap. Left side of the plot depicts persistence of strains up to two years of age. The strains are colour coded based on the phylum which they belong to, as detected by GTDB. Figure S6. Persistence of all unique strains as observed from inStrains for CSnoab group. Right side of the plot depicts abundance of ARGs as detected by ABRicate in the form of heatmap. Left side of the plot depicts persistence of strains up to two years of age. The strains are colour coded based on the phylum which they belong to, as detected by GTDB. Figure S7. Persistence of all unique strains as observed from inStrains for CSab group. Right side of the plot depicts abundance of ARGs as detected by ABRicate in the form of heatmap. Left side of the plot depicts persistence of strains up to two years of age. The strains are colour coded based on the phylum which they belong to, as detected by GTDB. Table S1. Table shows possible contaminants as flagged from inStrains results. Table S2. Table shows coefficients obtained by running differential abundance analysis using Songbird with CSnoab group as reference run using the formula C(Group, Treatment('CSnoab')). Table S3. Table presenting significant p-values obtained from MaAsLin2 differential abundance analysis for stratified data from HUMAnN3. MaAsLin2 was run using Antibiotic as Fixed effect and Sample as random effect with default parameters of min_abundance = 0.00 and min_prevalance = 0.01.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Patangia, D.V., Grimaud, G., O’Shea, CA. et al. Early life exposure of infants to benzylpenicillin and gentamicin is associated with a persistent amplification of the gut resistome. Microbiome 12, 19 (2024). https://doi.org/10.1186/s40168-023-01732-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40168-023-01732-6