
Abstract
Primary familial brain calcification (PFBC) is a genetic neurological disorder characterized by symmetric brain calcifications that manifest with variable neurological symptoms. This study aimed to explore the genetic basis of PFBC and elucidate the underlying pathophysiological mechanisms. Six patients from four pedigrees with brain calcification were enrolled. Whole-exome sequencing identified two novel homozygous variants, c.488G > T (p.W163L) and c.2135G > A (p.W712*), within the myogenesis regulating glycosidase (MYORG) gene. Cerebellar ataxia (n = 5) and pyramidal signs (n = 4) were predominant symptoms, with significant clinical heterogeneity noted even within the same family. An autopsy of one patient revealed extensive brainstem calcifications, sparing the cerebral cortex, and marked by calcifications predominantly in capillaries and arterioles. The pathological study suggested morphological alterations characterized by shortened foot processes within astrocytes in regions with pronounced calcification and decreased immunoreactivity of AQP4. The morphology of astrocytes in regions without calcification remains preserved. Neuronal loss and gliosis were observed in the basal ganglia, thalamus, brainstem, cerebellum, and dentate nucleus. Notably, olivary hypertrophy, a previously undescribed feature in MYORG-PFBC, was discovered. Neuroimaging showed reduced blood flow in the cerebellum, highlighting the extent of cerebellar involvement. Among perivascular cells constituting the blood-brain barrier (BBB) and neurovascular unit, MYORG is most highly expressed in astrocytes. Astrocytes are integral components of the BBB, and their dysfunction can precipitate BBB disruption, potentially leading to brain calcification and subsequent neuronal loss. This study presents two novel homozygous variants in the MYORG gene and highlights the pivotal role of astrocytes in the development of brain calcifications, providing insights into the pathophysiological mechanisms underlying PFBC associated with MYORG variants.
Similar content being viewed by others

Introduction
Brain calcification is a common phenomenon that occurs in many individuals, particularly the elderly [30]. This condition has a multifaceted etiology, involving cellular senescence, oxidative stress, calcium and phosphorus metabolism, mitochondrial dysfunction, infectious and autoimmune factors, and blood–brain barrier (BBB) breakdown. Although rare, there are cases of brain calcification caused by genetic abnormalities, leading to heredofamilial presentations [6, 30, 43]. Primary familial brain calcification (PFBC) is a rare disease characterized by bilateral symmetric calcifications in various brain regions, predominantly in the basal ganglia, with six genes identified as causative: SLC20A2, XPR1, PDGFB, PDGFRB, myogenesis regulating glycosidase (MYORG), and JAM2 [6, 43].
Herein, MYORG causes autosomal recessive PFBC (AR-PFBC) [46], accounting for approximately 13% of PFBC cases, making it the most common gene associated with AR-PFBC [4]. MYORG variants cause calcifications not only in the basal ganglia but also in the cerebellum, thalamus, cerebral white matter, and brainstem [4, 7, 13]. In addition, the symptoms of PFBC associated with the MYORG gene are more widespread than those caused by other genes [4, 13]. The pathophysiology of PFBC may vary among genes [43]. Functional experiments in animals and the localization of Myorg RNA, mainly within astrocytes, suggest that the MYORG-PFBC pathology involves astrocyte dysfunction, affecting the neurovascular unit (NVU) and compromising the BBB [31, 46]. However, no human pathological examination of MYORG-PFBC has been conducted to elucidate its characteristic features and the mechanisms leading to calcification.
In this study, we conducted genetic testing on six patients with brain calcification and identified two novel homozygous variants within the MYORG gene. Furthermore, we comprehensively analyzed their clinical and imaging findings. We also performed an autopsy on one case, which enabled us to conduct detailed brain histological examinations using optical microscopy and immunostaining techniques. Our investigation provides the original report on the pathological features of PFBC patients carrying MYORG variants.
Methods
Whole-exome sequencing
Genomic DNA was analyzed using whole-exome sequencing (WES) with the Illumina Hiseq 2000/2500 platform (Illumina Inc., San Diego, CA, USA) or Ion Proton (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The WES data were aligned to the human genome reference data (NCBI37/hg19), and variant calling was performed using Burrows–Wheeler Aligner and SAM tools. The variants were annotated using the CLC Genomics Workbench (Qiagen, Hilden, Germany) and an in-house script. All candidate variants were validated using Sanger sequencing.
Variant interpretation and protein stability
Variants were checked against two public control databases, namely, Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org) [18] and Japanese Multi Omics Reference Panel (jMorp; https://jmorp.megabank.tohoku.ac.jp), as well as our in-house WES database. Furthermore, several in silico tools were used to predict the pathogenicity of detected variants, including SIFT (http://sift.jcvi.org) [26], PROVEAN (http://provean.jcvi.org/index.php) [9], PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) [2], MutationTaster (http://www.mutationtaster.org) [37], FATHMM (http://fathmm.biocompute.org.uk) [38], CADD (https://cadd.gs.washington.edu) [21], and BayesDel (http://bjfenglab.org). To evaluate the evolutionary conservation of the MYORG candidate variants, nine protein sequences were retrieved from COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi). The three-dimensional (3D) structures of both wild-type and p.W163L variants in the MYORG protein were determined and visualized using DynaMut (https://biosig.lab.uq.edu.au/dynamut/) [35]. The expression levels of MYORG RNA in the adult mouse and human brain were assessed using single-cell RNAseq data from (http://betsholtzlab.org/VascularSingleCells/database.html) [16, 41] and (https://twc-stanford.shinyapps.io/human_bbb/) [44]. The detected variants were evaluated according to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP criteria), and the ClinGen Sequence Variant Interpretation (https://clinicalgenome.org/working-groups/sequence-variant-interpretation/) [1, 5, 11, 33, 34]. Details of the variant classifications are provided in Sup Table 1.
Neuroimaging
Computed tomography (CT), magnetic resonance imaging (MRI), and nuclear imaging were conducted at the clinical facilities. MRI was performed using a 1.5 Tesla scanner. CT scans were evaluated using axial slices of the medulla, pons, midbrain, basal ganglia, and parietal lobes. Single-photon emission computed tomography (SPECT) was performed on both F1-II-3 and F3-II-2. For cerebral blood flow evaluation, Technetium-99 m ethyl cysteinate dimer (99mTc-ECD) SPECT was used for F1-II-3, while N-isopropyl-I-123-p-iodoamphetamine (123-IMP) SPECT was used for F3-II-2. Dopamine transporter evaluation was conducted using 123I ioflupane (123I-FP-CIT).
Histological and immunochemical analysis
An autopsy was performed on patient II-1 from Family 1. The brain extracted during the procedure was fixed in 20% buffered formalin. Coronal cuts were made through the cerebral hemispheres, transverse cuts through the brainstem, and sagittal cuts through the cerebellum. Subsequently, the regions of interest were trimmed, including the primary motor cortex; isocortex of the frontal, temporal, parietal, and occipital lobes; subcortical white matter of the frontal and temporal lobes; anterior cingulate gyrus; insular cortex; hippocampus; amygdala; basal ganglia; thalamus; midbrain; pons; medulla oblongata; and cerebellum. These tissue samples were then paraffin-embedded for further analysis.
Sections with 9-µm thickness were prepared for staining with hematoxylin–eosin (HE), Klüver–Barrera, Gallyas–Braak, von Kossa, Dahr, and Berlin blue. Furthermore, sections with 4.5-µm thickness were specifically prepared for immunohistochemistry analysis. The primary antibodies used for immunohistochemistry were anti-α-synuclein (polyclonal rabbit, 1:1000; Sigma Aldrich, St. Louis, MO), anti-β-amyloid (clone 6 F/3D; monoclonal mouse, 1:100; Dako, Glostrup, Denmark), anti-phosphorylated tau (AT8; monoclonal mouse, 1:5000; Fujirebio Europe, Ghent, Belgium), anti-phosphorylated transactive response DNA-binding protein 43 kDa (pTDP-43, polyclonal rabbit, 1:5000; Cosmo Bio, Tokyo, Japan), anti-glial fibrillary acid protein (monoclonal, 1:400; Dako, Glostrup, Denmark), anti-polyglutamin-expansion disease marker (5TF1-1C2, monoclonal mouse, 1:3000, Millipore, Darmstadt, Germany), anti-AQP4 (CL0178, monoclonal mouse, 1:800, Sigma-Aldrich, Germany), and anti-Iba1 (polyclonal rabbit, 1:8000, WAKO Chemical), and anti-CD31 (JC70A, monoclonal mouse, 1:120, Dako, Glostrup, Denmark).
Results
Clinical features
Family 1
The proband (referred to as F1-II-1 in Fig. 1) in this family was a Japanese man, the eldest among three siblings. He was diagnosed with diabetes mellitus at 30 years old, which was accompanied by diabetic retinopathy and nephropathy. At the age of 34, he began experiencing gait instability and difficulties with fine motor skills. At the age of 39, consultation with a neurologist revealed dysarthria with scanning speech, cerebellar ataxia, trunk ataxia, and pyramidal tract signs predominantly affecting the right side of limbs. Cognitive function assessment using the Revised Hasegawa’s Dementia Scale (HDS-R) yielded a score of 28/30 points (normal range: 21–30). At the age of 46, progressive limb deterioration and trunk cerebellar ataxia necessitated the use of a wheelchair. At the age of 49, chorea and dystonia mainly affecting the upper limbs became evident. Simultaneously, the proband underwent endoscopic gastrostomy to address dysphagia. Unfortunately, he passed away at the age of 53 due to aspiration pneumonia complications.

Genetic analysis of the MYORG gene in four families. A: Family pedigrees of ascertained individuals and detected MYORG variants. Family 1: F1-I-1 and F1-I-2 are heterozygous carriers of c.488G > T whereas F1-II-1, F1-II-2, and F1-II-3 are homozygous carriers of c.488G > T in MYORG. Families 2, 3, and 4: homozygous c.2135G > A variant in the MYORG gene was detected in all probands (F2-II-1, F3-II-2, and F4-II-1) and in an asymptomatic individual, F2-II-4. B: Sanger sequencing reveals c.488G > T (p.W163L) in Family 1 and c.2135G > A (p.W712*) in Families 2, 3, and 4. Black arrows indicate homozygous variants. C: Conservation analysis of p.W163L and p.W712* variants in MYORG. Both residues are highly conserved across multiple species. Refseq, reference sequence
F1-II-2 (a 55-year-old man) and F1-II-3 (a 52-year-old man) are younger siblings of F1-II-1. F1-II-2 has a history of cerebral hemorrhage in the left frontal lobe and demonstrated reduced tendon reflexes; however, no other neurological abnormalities were observed. F1-II-3 developed hand tremors at the age of 36 and was diagnosed with speech articulation disorders at the age of 41. Consultation with a neurologist revealed mild speech articulation disorders, cerebellar limb ataxia, trunk ataxia, and limb rigidity. F1-II-3 has shown an extremely slow progression and, after 16 years since onset, continues with daily activities.
Their parents (F1-I-1 and F1-I-2) also underwent neurological examinations, but no evident neurological abnormalities were observed.
Family 2
The proband (F2-II-1) was a 49-year-old Japanese woman, the eldest of four siblings. Although her parents were not consanguineous, they both originated from the same small island. Neither her parents nor siblings exhibited any neurological abnormalities. The proband experienced articulation difficulties and struggled with clear pronunciation at 45 years old. At the age of 46, a head CT scan revealed brain calcifications. At the age of 47, she had difficulty in walking. Neurological evaluation at 48 years old revealed saccadic ocular movement, ataxic dysarthria, increased tendon reflexes in the limbs, cerebellar ataxia, and gait disturbances. Notably, dysarthria, parkinsonism, muscle weakness, sensory impairment, and autonomic dysfunction were not observed. Her HDS-R and Mini-Mental State Examination (MMSE) scores were 27 and 25/30, respectively, both falling within the normal range. Blood tests revealed normal levels of calcium, phosphate, and parathyroid hormone.
Family 3
The proband (F3-II-2) was a 51-year-old Japanese man. At the age of 24, he underwent a head radiological examination, which revealed brain calcification. At the age of 28, he presented with dysarthria after a cerebral hemorrhage, which gradually improved over time and eventually resolved. At the age of 43, depressive symptoms emerged; thus, antidepressant medication was initiated. At the age of 51, neurological examination revealed gait disturbances, fatigue, mild cerebellar ataxia, increased tendon reflexes in the limbs, autonomic nervous system failure, and bipolar disorder. The Wechsler Adult Intelligence Scale indicated a full intelligence quotient score of 93, with a decrease in the performance intelligence quotient score to 57. In the cognitive function tests, his HDS-R and MMSE scores were 29 and 28, respectively, both falling within the normal range.
Family 4
The proband (F4-II-1) was a 66-year-old Japanese woman from non-consanguineous parents. Her parents originated from a small island (distinct from Family 2). She had seven younger siblings, none of whom exhibited neurological abnormalities. At the age of 50, despite being asymptomatic, brain MRI revealed calcifications in the basal ganglia and dentate nucleus. At around 64 years old, she began experiencing dysarthria and difficulties in fine motor skills. At the age of 66, she started stumbling and experiencing frequent falls while walking. Due to the progressive worsening of her dysarthria, she underwent a neurological evaluation, which revealed dysarthria, dysphagia, and saccade ocular movements. The finger-to-nose test indicated bilateral cerebellar ataxia. Her tendon reflexes were normal, and her Babinski reflex was negative. Furthermore, limb muscle strength and cognitive function (MMSE: 26 points) were normal. No signs of parkinsonism were evident. Blood tests indicated normal levels of calcium, phosphate, and parathyroid hormone.
The clinical symptoms of all the affected cases within Families 1–4 are shown in Table 1.
Neuroimaging
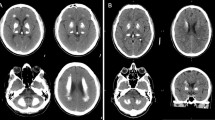
Brain CT images were available for six cases (F1-II-1, F1-II-2, F1-II-3, F2-II-1, F3-II-2, and F4-II-1) with homozygous variants in the MYORG gene (Fig. 2). In all six cases, calcifications were found in the basal ganglia, cerebellar dentate nucleus, and cerebral white matter. In addition, calcifications in the thalamus were observed in all cases except for F4-II-1. F1-II-1, F1-II-3, and F3-II-2 also showed calcifications in the brainstem, which were symmetric in all cases.

Brain CT imaging of 6 patients with MYORG variants. Brain CT imaging depicts symmetrical calcifications in various brain regions across six cases. All cases exhibit calcifications in the basal ganglia, cerebellar dentate nucleus, and cerebral white matter. Midbrain, pontine, and medullary calcifications are observed in F1-II-1 and F1-II-3. Periventricular calcifications are observed in F3-II-2, F1-II-3, F2-II-1, F3-II-2, and F4-II-1. The most severe calcifications are observed in F1-II-1
Although F1-II-2 was asymptomatic, brain CT revealed the presence of calcifications. However, the CT of F1-II-2 did not reveal calcifications in the brainstem, and the severity of the calcifications was milder than that in siblings F1-II-1 and F1-II-3. Variations in the degree and location of calcifications within the same family were noted. F1-II-1, who presented with the most severe clinical symptoms, also exhibited the highest degree of calcifications in the brain CT. The bilateral inferior olivary of F1-II-1 revealed no abnormal signal in the brain MRI (Fig. S2).
SPECT was performed on both F1-II-3 and F3-II-2. Both cases exhibited reduced blood flow in the bilateral cerebellum and brainstem. In F1-II-3, a reduction in the blood flow to the frontal lobe was also observed. Furthermore, in F3-II-2, a 123I-FP-CIT (DaTSCAN) was conducted. The specific binding ratios were 7.33 and 7.43 on the right and left, respectively, indicating preserved dopamine transporter levels (Fig. 3).

Brain perfusion and dopamine transporter visualization. A: Single-photon emission computed tomography imaging of F1-II-3 (99mTc-ECD) and F3-II-2 (123I-IMP). Blood flow reduction in the bilateral cerebellum and brainstem is observed in both cases. B: Dopamine transporter scintigraphy of F3-II-2 using 123I-FP-CIT (DaTSCAN). Uptake in the bilateral putamen is preserved, with a specific binding ratio of 7.33 on the right side and 7.43 on the left side
Genetic findings
MYORG variants and interpretations
Two novel homozygous variants were identified within four families from the MYORG gene (NM_020702.5) (Fig. 1A), namely, c.488G > T (p.W163L) from Family 1 and c.2135G > A (p.W712*) from Families 2, 3, and 4.
Pedigree analysis was conducted for Families 1 and 2 (Fig. 1B). In Family 1, the co-segregation of the MYORG variant and clinical phenotypes was confirmed, where asymptomatic individuals F1-I-1 and F1-I-2 were found to be heterozygous carriers of p.W163L. In Family 2, the parents (F2-I-1 and F2-I-2) were heterozygous carriers of p.W712*, whereas the younger sisters (F2-II-2 and F2-II-3) carried wild-type alleles. However, the homozygous p.W712* was found to be shared by the younger brother of the proband (F2-II-4), who presented with no subjective symptoms. Unfortunately, a CT scan for F2-II-4, which was crucial for assessing the status of this individual, was unavailable. Therefore, the possibility of presymptomatic brain calcification cannot be ruled out.
These two MYORG variants were not registered as homozygous in either public control databases or our in-house database and were predicted to be pathogenic in multiple in silico analyses (Table 2). The residues of these variants are highly conserved across multiple species (Fig. 1C). According to the ACMG/AMP criteria, c.488G > T and c.2135G > A were classified as likely pathogenic variants (Table 2).
Stability analysis and expression of MYORG
The ΔΔG for the MYORG c.488G > T variant was predicted to be − 2.699 kcal/mol by mCSM, − 2.480 kcal/mol by SDM, and − 2.719 kcal/mol by DUET, all indicating protein destabilization (Table 2, Fig. S1A). The scRNA-Seq data of vascular and vessel-associated cells in the adult mouse brain revealed that the MYORG mRNA is predominantly expressed in astrocytes (Fig. S1B). A transcriptomic dataset of human brain vascular and perivascular cell types showed that MYORG is most abundantly expressed in the astrocytes of the cortex, but is also distributed in ependymal, venous, capillary, and arterial cells (Fig. S1C).
Pathological findings of brain tissue from F1-II-1
The brain weighed 895 g before fixation. Macroscopic examination revealed diffuse atrophy of the cerebrum (Fig. 4A), brainstem (Fig. S3A-C), and cerebellum (Fig. S3D). Coronal sections exhibited brownish discoloration with gritty consistency in the basal ganglia and thalamus (Fig. 4A). A similar gritty consistency was observed in the cut surfaces of the brainstem and cerebellum. Mineralization was detected in multiple lesions, including the basal ganglia (Fig. 4), thalamus, brainstem, cerebellum, and cerebral white matter, as shown by HE, von Kossa, and Berlin blue staining (Fig. S3E-K). Notably, mineralization was predominantly noted in the capillaries (Fig. 4D, E) and in the tunica media of medium- and small-caliber arteries and arterioles (Fig. 4G-K). In the basal ganglia, the most pronounced mineralization of vessel walls was observed in the region of the medial and intermediate lenticulostriate arteries (Fig. 4B) of the perforating branches of the middle cerebral artery. Mild mineralization was observed in the distal portion of the lateral lenticulostriate artery, whereas severe mineralization was noted in the posterior limb of the internal capsule, thalamus, and hippocampus. The cerebral white matter showed mild mineralization of vessel walls (Fig. 4G). The cerebral cortices were relatively preserved, and mineralization was not detected, except for the hippocampus. The brainstem demonstrated marked mineralization, neuronal loss, and gliosis in various regions, including the substantia nigra, red nucleus (Fig. S3E), superior colliculus, periaqueductal gray matter, pontine and medullary tegmentum, pontine and medullary reticular formation (Fig. S3F, G), and middle cerebellar peduncles. Pseudohypertrophy of the bilateral inferior olivary nuclei was observed (Fig. S3C, H). In the cerebellum, the molecular and granular cell layers exhibited marked mineralization; moreover, the dentate nucleus showed severe mineralization with neuronal loss and gliosis (Fig. S3I, J, K). Neuronal cell mineralization was observed in the basal ganglia (Fig. 4F). Atherosclerotic lesions in brain blood vessels and histological brain ischemic changes were scarce. Notably, astrocytes exhibited marked cellular hypertrophy associated with shortened and enlarged foot processes in regions with severe mineralization (Fig. 5C). Meanwhile, similar astrocytic changes were not found in regions without mineralization (Fig. 5D). AQP4 immunoreactivity is decreased (Fig. 5E) in the area with severe mineralization (Fig. 5A), compared to that (Fig. 5F) in the region without mineralization (Fig. 5B). Iba1 immunoreactive hypertrophic microglia surrounds the mineralized capillary (Fig. 5G, arrow) in the area with severe mineralization. Iba1 immunoreactive microglia attaches the capillary wall (Fig. 5H, arrow) in the region without mineralization. CD31 immunoreactivity is observed inside the globular mineralization (Fig. S4). Astrocytic cellular hypertrophy associated with shortened foot processes was also observed in the subcortical white matter with mineralization (Fig. S5). However, these morphological changes were not found in the cortices without mineralization (Fig. S5). Anti-1C2 immunohistochemistry revealed no significant findings. The tau deposition was minimal, Braak AT8 stage 1, and no amyloid plaques or α-synuclein deposits were identified.

Macro- and microscopic findings of patient F1-II-1. Coronal sections show diffuse brain atrophy in the cerebrum (A). Mineralization and rarefaction of the caudate head (asterisk) and pallidum (B). Mineralization in the area (asterisk in B) (C) and capillary (D). Prominent mineralization around the capillaries (E). Mineralization (arrows) in neuron in the pallidum (F). Mineralization within the walls of the intracerebral arteries in periventricular white matter (G). In the intracerebral arteries, mineralization clusters outer side of the smooth muscle layer (H, I, J) with iron deposition (K). Staining: B-D, von Kossa; E, G-J, hematoxylin–eosin; F, Klüver–Barrera; K, Berlin blue. Scale bar: A, 2 cm; B, 4 mm; C, 100 μm; D, F, H-J, 10 μm; E, 50 μm; G, 500 μm; K, 20 μm

Immunohistochemical findings of caudate head of patient F1-II-1. Pronounced mineralization region (A) and area without mineralization (B) in the caudate head. Astrocytic cell bodies (arrows) appear enlarged, accompanied by shortening, and distortion processes (C) in the region (A). The morphology of astrocytes in the region (B) remains preserved (D). AQP4 immunoreactivity is decreased (E) in the region (A), compared to that (F) in the region (B). Iba1 immunoreactive microglia surrounds the mineralized capillary (arrow, G) in the area (A). Iba1 immunoreactive microglia attaches the capillary wall (arrow, H) in the region (B). Staining: A, B, von Kossa; C, D, anti-glial fibrillary acid protein antibody; E, F, anti-AQP4 antibody; G, H, anti-Iba1 antibody. Scale bar: A-F, 20 μm; G–H, 10 μm
The summary of the neuropathological findings is shown in Table 3.
Discussion
In this study, two novel variants in the MYORG gene, namely, c.488G > T (p.W163L) and c.2135G > A (p.W712*), were identified in six patients from four families with brain calcifications (Fig. 1A, B). Both variants were classified as likely pathogenic according to the ACMG-AMP guidelines. The homozygous nonsense variant p.W712*, located at the end of MYORG, would result in the production of a truncated protein. This variant represents the closest proximity to the 3’UTR of MYORG identified thus far.
Patients with PFBC commonly experience extrapyramidal and psychiatric symptoms as well as cognitive dysfunction [4]. However, in MYORG-PFBC, there is an additional higher incidence of involuntary movements, cerebellar ataxia, pyramidal signs, and dysarthria compared with other PFBC-related genes [3, 13]. Among these symptoms, dysarthria has been reported to be a particular characteristic symptom of MYORG-PFBC [4, 13]. In this study, cerebellar ataxia was noted in five cases, whereas pyramidal signs and dysarthria were observed in four, which is consistent with previous observations. Moreover, all four cases with dysarthria had cerebellar ataxia, with two exhibiting scanning speech or ataxic dysarthria. Nonetheless, brainstem calcification was found in only two of them. It is hypothesized that the etiology of dysarthria is attributed not to extrapyramidal or brainstem pathologies but to the involvement of cerebellar cortex and dentate nucleus associated with calcification. Clinical phenotype and severity greatly varied among three siblings with homozygous variants in Family 1, with F1-II-1 showing the most severe symptoms. The diversity suggests the influence of additional genetic or environmental factors on the severity of the phenotype and calcification.
In PFBC, calcifications predominantly occur around the BBB and NVU, surrounding blood vessels rather than neurons, as reported by previous autopsy studies [17, 20, 23, 25, 29, 42]. Our autopsy case demonstrated calcification in nearby capillaries, which is a similar pattern to previous autopsy data. Brain calcification is presumed to involve multiple mechanisms, with one primary mechanism involving mineral deposition around blood vessels because of BBB disruption [29]. The complex structure of the BBB includes the endothelium cells, pericytes, basal membrane, vascular smooth muscle cells, and astrocytes [48]. Among these perivascular cells constituting the BBB, MYORG is most highly expressed in astrocytes (Fig. S1B, C) [46], whereas the other PFBC causative genes (SLC20A2, XPR1, PDGFB, PDGFRB, and JAM2) were mainly expressed in perivascular cells, respectively [43]. Astrocytes, through their endfeet, serve as a crucial link between the concentrated neurons of the brain parenchyma and blood circulation at the NVU [14]. Previous studies have demonstrated reactive astrocytes in autopsied patients with PFBC [20, 29] and reactive astrocyte surrounding calcification in the mouse model of PFBC [19, 31, 47]. These astrocytic impairments, suggesting oxidative stress and inflammation, are likely to be a significant factor in the calcification observed in PFBC. The present study demonstrates that astrocyte alterations, such as swelling or shortening of foot processes, were found in areas with pronounced calcifications associated with decreased immunoreactivity of AQP4. Moreover, these changes could not be found in areas without calcifications. Although foot process extension is typically observed in reactive astrocytes, these morphological changes in astrocytes in our patient differ from those of reactive astrocytes. Taken together, astrocytes play a pivotal role in brain calcification in MYORG-PFBC, and a loss-of-function mechanism caused by MYORG variants contributes to the co-occurrence of morphological alterations of astrocytes and calcification. These morphological changes of astrocytes may disrupt the BBB and NVU, potentially resulting in brain calcification and subsequent neuronal loss.
The MYORG gene is associated with a broader distribution of brain calcifications compared to other genes that cause PFBC [4]. In PFBC, calcifications frequently appear in the basal ganglia, thalamus, and cerebellar dentate nucleus but rarely in other regions. However, MYORG-PFBC often causes widespread brain calcifications, particularly in the brainstem [7]. In this study, brainstem calcification was noted in three of six cases. Our autopsy findings indicated pronounced calcification and neuronal loss within the brainstem. Interestingly, a diversity in the severity of calcifications within the brainstem was observed, ranging from severe calcification in the brainstem tegmentum and the medullary reticular formation to mild calcification in the oculomotor complex, pontine nucleus, and dorsal motor nucleus of the vagus, with the pyramids remaining unaffected. Furthermore, both the cerebellum and basal ganglia exhibited extensive calcifications and neuronal loss, whereas the pathological change in cerebral cortices was relatively mild. In previous studies, cerebral cortex calcifications were observed, albeit mild, in the CT scans of MYORG-PFBC cases. Meanwhile, cerebral cortex calcifications have been frequently observed in patients with SLC20A2-PFBC [20] and genetically undiagnosed PFBC [23, 25, 29, 42]. Thus, this pattern of calcification, characterized by extensive involvement of the brainstem and sparing of the cerebral cortex, could potentially be a distinctive sign of MYORG-PFBC. The underlying mechanisms leading to the distribution of calcification and high prevalence of brainstem calcifications in MYORG-PFBC remain to be elucidated. The expression of MYORG RNA has been found distributed throughout the brain in zebrafish, mouse, and human (https://www.proteinatlas.org) [40, 46, 49]. Despite the abundant distribution of MYORG RNA and the widespread presence of astrocytes throughout the brain, these factors alone do not explain the observed calcification pattern of MYORG-PFBC. It is hypothesized that the heterogeneous composition of the brainstem, featuring a greater prevalence of gray matter with a higher number of capillaries [32] and finer capillary diameters compared with white matter [15], contributes to the predisposition for calcifications [13]. Nonetheless, this hypothesis does not account for the absence of calcifications in the cerebral cortex, which indicates that the current understanding is insufficient to fully explain the distribution of calcifications, highlighting the need for further investigation into the pathogenicity of brain calcification.
Recent studies have demonstrated a connection between homozygous MYORG variants and the onset of stroke [13, 27, 45]. In our study, two cases (F1-II-2 and F3-II-2) had a documented history of cerebral hemorrhage, consistent with previous findings. However, another patient F1-II-1, devoid of any stroke history and subjected to autopsy, exhibited no hemorrhagic or ischemic changes, and vascular arteriosclerosis was notably mild. Brain calcification has been associated with the presence of diffuse neurofibrillary tangles, which can lead to presenile dementia [24]. In addition, Lewy bodies have been reported in patients with PFBC [28, 36], and disruptions in the BBB and NVU may contribute to the pathogenesis of degenerative diseases such as Alzheimer’s and Parkinson’s diseases [39, 50]. In this study, pathological analysis of F1-II-1 revealed no deposits of β-amyloid, α-synuclein, polyglutamine, and negligible tau. To sum up, aside from PFBC, the brain autopsy study on F1-II-1 did not find any protein accumulations suggestive of other neurodegenerative diseases.
Despite the absence of abnormal signals or enlargement in the inferior olivary nucleus on MRI (Fig. S2), brain autopsy of F1-II-1 revealed bilateral olivary hypertrophy (Fig. 4H and P). Considering the potential of high signal fading over time in the inferior olivary nucleus on MRI [12] coupled with calcification-induced brainstem atrophy, there is a likelihood of olivary hypertrophy underestimation in MYORG-PFBC when solely relying on MRI. Olivary hypertrophy has been typically associated with disruptions in the Guillain–Mollaret triangle, potentially leading to progressive ataxia and palatal tremor [10]. To date, the co-occurrence of brain calcification and olivary hypertrophy remains rare, except in patients with autosomal dominant spinocerebellar ataxia type 20, which could develop both dentate nucleus calcification and olivary hypertrophy [22]. Nevertheless, olivary hypertrophy has not yet been described in patients with MYORG-PFBC. The neuronal loss in the dentate nucleus, red nucleus, and pontine tegmentum, aligning with the Guillain–Mollaret pathway, may be induced by profound vascular calcification in these areas and lead to olivary hypertrophy.
Meanwhile, the SPECT analysis in our two cases revealed the predominance of reduced cerebellar and brainstem blood flow, with no significant reduction in basal ganglia and cerebral lobe perfusion (Fig. 3A). Furthermore, 123I-FP-CIT analysis in one case revealed normal dopamine transporter levels. Studies on MYORG-PFBC cases using radioisotopes are scarce, with SPECT showing reduced blood flow in the frontal lobe, basal ganglia, and thalamus in one case [8] as well as normal and reduced dopamine transporter levels (123I-FP-CIT) in two and one case, respectively [8, 13]. To the best of our knowledge, this is the first study to illustrate a reduction in cerebellar blood flow through radioisotopes.
This study has several limitations that warrant consideration. First, the pathological findings discussed were derived from a single case, potentially limiting their generalizability to the broader population of individuals with MYORG-PFBC. Second, the utilization of SPECT and 123I-FP-CIT examinations was limited to only a few cases, highlighting the need for additional research involving a larger cohort to validate the findings. Third, the observations regarding astrocyte changes solely rely on pathological assessments, indicating the need for more comprehensive molecular biological investigations.
In summary, this study introduces two novel and likely pathogenic MYORG variants and provides clinical and neuroimaging profiles of PFBC from four Japanese families. Our findings show remarkable phenotypic diversity among patients carrying identical variants, even within the same family. This underscores the complex nature of the disease. We also observe reduced blood flow in the cerebellum and brainstem in the SPECT study and olivary hypertrophy in an autopsy case. Notably, our autopsy study represents the original report demonstrating the validation of morphological alterations and decreased immunoreactivity of AQP4 within astrocytes in regions with pronounced calcification, improving our understanding of the MYORG-PFBC pathogenesis. Among perivascular cells constituting the BBB and NVU, MYORG is most highly expressed in astrocytes. These findings highlight the pivotal role of astrocytes in the disruption of BBB in brain calcification, providing significant insights into the pathophysiology and phenotypic manifestation of PFBC.
Data availability
Datasets are not readily available due to ethical and privacy restrictions. Requests should be directed to the corresponding author.
References
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM (2018) Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 39:1517–1524. https://doi.org/10.1002/humu.23626
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Arkadir D, Lossos A, Rahat D, Abu Snineh M, Schueler-Furman O, Nitschke S, Minassian BA, Sadaka Y, Lerer I, Tabach Y, Meiner V (2019) MYORG is associated with recessive primary familial brain calcification. Ann Clin Transl Neurol 6:106–113. https://doi.org/10.1002/acn3.684
Balck A, Schaake S, Kuhnke NS, Domingo A, Madoev H, Margolesky J, Dobricic V, Alvarez-Fischer D, Laabs BH, Kasten M, Luo W, Nicolas G, Marras C, Lohmann K, Klein C, Westenberger A (2021) Genotype–phenotype relations in primary familial brain calcification: systematic MDSGene review. Mov Disord 36:2468–2480. https://doi.org/10.1002/mds.28753
Biesecker LG, Byrne AB, Harrison SM, Pesaran T, Schäffer AA, Shirts BH, Tavtigian SV, Rehm HL (2024) ClinGen guidance for use of the PP1/BS4 co-segregation and PP4 phenotype specificity criteria for sequence variant pathogenicity classification. Am J Hum Genet 111:24–38. https://doi.org/10.1016/j.ajhg.2023.11.009
Carecchio M, Mainardi M, Bonato G (2023) The clinical and genetic spectrum of primary familial brain calcification. J Neurol 270:3270–3277. https://doi.org/10.1007/s00415-023-11650-0
Chelban V, Carecchio M, Rea G, Bowirrat A, Kirmani S, Magistrelli L, Efthymiou S, Schottlaender L, Vandrovcova J, Salpietro V, Salsano E, Pareyson D, Chiapparini L, Jan F, Ibrahim S, Khan F, Qarnain Z, Groppa S, Bajaj N, Balint B, Bhatia KP, Lees A, Morrison PJ, Wood NW, Garavaglia B, Houlden H (2020) MYORG-related disease is associated with central pontine calcifications and atypical parkinsonism. Neurol Genet 6. https://doi.org/10.1212/NXG.0000000000000399
Chen SY, Lin WC, Chang YY, Lin TK, Lan MY (2020) Brain hypoperfusion and nigrostriatal dopaminergic dysfunction in primary familial brain calcification caused by novel MYORG variants: Case report. BMC Neurol 20. https://doi.org/10.1186/s12883-020-01910-1
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7. https://doi.org/10.1371/journal.pone.0046688
Gautier JC, Blackwood W (1961) Enlargement of the inferior olivary nucleus in association with lesions of the central tegmental tract or dentate nucleus. Brain 84:341–361. https://doi.org/10.1093/brain/84.3.341
Gelb BD, Cavé H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, Rauen KA, Williams B, Zenker M, Vincent LM (2018) ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Sci 20:1334–1345. https://doi.org/10.1038/gim.2018.3
Goyal M, Versnick E, Tuite P, Saint Cyr J, Kucharczyk W, Montanera W, Willinsky R, Mikulis D (2000) Hypertrophic Olivary Degeneration: Metaanalysis of the temporal evolution of MR Findings. Am J Neuroradiol 21:1073–1077
Grangeon L, Wallon D, Charbonnier C, Quenez O, Richard AC, Rousseau S, Budowski C, Lebouvier T, Corbille AG, Vidailhet M, Méneret A, Roze E, Anheim M, Tranchant C, Favrole P, Antoine JC, Defebvre L, Ayrignac X, Labauge P, Pariente J, Clanet M, Maltête D, Rovelet-Lecrux A, Boland A, Deleuze JF, Favrole P, Verny C, Krystkowiak P, Chamard L, Moutton S, Goizet C, Ferec C, Timsit S, Schaeffer S, Derache N, Defer G, Durif F, Sellal F, Rouaud O, Thauvin-Robinet C, Cubizolle S, Sauvée M, Leblanc A, Demas A, Poisson A, Tournier-Lasserve E, Hervé D, Chabriat H, Grolez G, Carriere N, Defebvre L, Lebouvier T, Witjas T, Azulay JP, Fluchere F, Didic M, Nguyen K, Charif M, Ayrignac X, Lionnet C, Marelli-Tosi C, Gaud S, Rouaud T, Laurens B, Folgoas E, Isidor B, Chiesa J, Pallix-Guyot M, Gaillard N, Olivier N, Jurici S, Marey I, Charles P, Ewenczyck C, Durr A, Hubsch C, Meneret A, Vidailhet M, Nadjar Y, Le Ber I, Grabli D, Roze E, Navarro V, Mecharles-Darrigol S, Lagarde J, Sarazin M, Vérin M, Lefaucheur R, Maltête D, Hannequin D, Martinaud O, Guyant-Maréchal L, Nicolas G, Frebourg T, Richard AC, Campion D, Guillin O, Yger M, Anheim M, Renaud M, Tranchant C, Rudolf G, Crétin B, Mallaret M, Pariente J, Ory-Magne F, Frebourg T, Campion D, Nicolas G (2019) Biallelic MYORG mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 142:1573–1586. https://doi.org/10.1093/brain/awz095
Harder DR, Zhang C, Gebremedhiin D (2002) Astrocytes function in matching Blood Flow to metabolic activity. Physiology 17:27–31. https://doi.org/10.1152/physiologyonline.2002.17.1.27
Hase Y, Ding R, Harrison G, Hawthorne E, King A, Gettings S, Platten C, Stevenson W, Craggs LJL, Kalaria RN (2019) White matter capillaries in vascular and neurodegenerative dementias. Acta Neuropathol Commun 7:16. https://doi.org/10.1186/s40478-019-0666-x
He L, Vanlandewijck M, Mae AM, Andrae J, Ando K, Gaudio DF, Nahar K, Lebouvier T, Lavina B, Gouveia L, Sun Y, Raschperger E, Segerstolpe A, Liu J, Gustafsson S, rasanen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C (2018) Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci Data 5:1–11. https://doi.org/10.1038/sdata.2018.160
Iwase T, Yoshida M, Hashizume Y, Yazawa I, Takahashi S, Ando T, Ikeda T, Nokura K (2019) Intracranial vascular calcification with extensive white matter changes in an autopsy case of pseudopseudohypoparathyroidism. Neuropathology 39:39–46. https://doi.org/10.1111/neup.12518
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Aguilar Salinas CA, Ahmad T, Albert CM, Ardissino D, Atzmon G, Barnard J, Beaugerie L, Benjamin EJ, Boehnke M, Bonnycastle LL, Bottinger EP, Bowden DW, Bown MJ, Chambers JC, Chan JC, Chasman D, Cho J, Chung MK, Cohen B, Correa A, Dabelea D, Daly MJ, Darbar D, Duggirala R, Dupuis J, Ellinor PT, Elosua R, Erdmann J, Esko T, Färkkilä M, Florez J, Franke A, Getz G, Glaser B, Glatt SJ, Goldstein D, Gonzalez C, Groop L, Haiman C, Hanis C, Harms M, Hiltunen M, Holi MM, Hultman CM, Kallela M, Kaprio J, Kathiresan S, Kim BJ, Kim YJ, Kirov G, Kooner J, Koskinen S, Krumholz HM, Kugathasan S, Kwak SH, Laakso M, Lehtimäki T, Loos RJF, Lubitz SA, Ma RCW, MacArthur DG, Marrugat J, Mattila KM, McCarroll S, McCarthy MI, McGovern D, McPherson R, Meigs JB, Melander O, Metspalu A, Neale BM, Nilsson PM, O’Donovan MC, Ongur D, Orozco L, Owen MJ, Palmer CNA, Palotie A, Park KS, Pato C, Pulver AE, Rahman N, Remes AM, Rioux JD, Ripatti S, Roden DM, Saleheen D, Salomaa V, Samani NJ, Scharf J, Schunkert H, Shoemaker MB, Sklar P, Soininen H, Sokol H, Spector T, Sullivan PF, Suvisaari J, Tai ES, Teo YY, Tiinamaija T, Tsuang M, Turner D, Tusie-Luna T, Vartiainen E, Watkins H, Weersma RK, Wessman M, Wilson JG, Xavier RJ (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581:434–443. https://doi.org/10.1038/s41586-020-2308-7
Keller A, Westenberger A, Sobrido MJ, García-Murias M, Domingo A, Sears RL, Lemos RR, Ordoñez-Ugalde A, Nicolas G, Da Cunha JEG, Rushing EJ, Hugelshofer M, Wurnig MC, Kaech A, Reimann R, Lohmann K, Dobričić V, Carracedo A, Petrović I, Miyasaki JM, Abakumova I, Mäe MA, Raschperger E, Zatz M, Zschiedrich K, Klepper J, Spiteri E, Prieto JM, Navas I, Preuss M, Dering C, Janković M, Paucar M, Svenningsson P, Saliminejad K, Khorshid HRK, Novaković I, Aguzzi A, Boss A, Le Ber I, Defer G, Hannequin D, Kostić VS, Campion D, Geschwind DH, Coppola G, Betsholtz C, Klein C, Oliveira JRM (2013) Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 45:1077–1082. https://doi.org/10.1038/ng.2723
Kimura T, Miura T, Aoki K, Saito S, Hondo H, Konno T, Uchiyama A, Ikeuchi T, Takahashi H, Kakita A (2016) Familial idiopathic basal ganglia calcification: histopathologic features of an autopsied patient with an SLC20A2 mutation. Neuropathology 36:365–371. https://doi.org/10.1111/neup.12280
Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. https://doi.org/10.1038/ng.2892
Knight MA, Gardner RJMK, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E (2004) Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain 127:1172–1181. https://doi.org/10.1093/brain/awh139
Kobayashi S, Yamadori I, Miki H, Ohmori M (1987) Idiopathic nonarteriosclerotic cerebral calcification (Fahr’s disease): an electron microscopic study. Acta Neuropathol 73:62–66. https://doi.org/10.1007/BF00695503
Kosaka K (1994) Diffuse neurofibrillary tangles with calcification: a new presenile dementia. J Neurol Neurosurg Psychiatry 57:594–596. https://doi.org/10.1136/jnnp.57.5.594
Kozik M, Kulczycki J (1978) Laser-spectrographic analysis of the Cation Content in Fahr’s syndrome. Arch Psychiat Nervenkr 225:135–142. https://doi.org/10.1007/BF00343397
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1082. https://doi.org/10.1038/nprot.2009.86
Li Y, Fang W, Long W, Zhao G (2022) Ischemic stroke in a patient with Fahr’s disease carrying biallelic mutations in the MYORG gene. Neurosciences 27:270–274. https://doi.org/10.17712/nsj.2022.4.20220047
Manyam BV, Walters AS, Keller IA, Ghobrial M (2001) Parkinsonism associated with autosomal dominant bilateral striopallidodentate calcinosis. Parkinsonism Relat Disord 7:289–295. https://doi.org/10.1016/S1353-8020(00)00036-5
Miklossy J, Mackenzie IR, Dorovini-Zis K, Calne DB, Wszolek ZK, Klegeris A, McGeer PL (2005) Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol 109:643–653. https://doi.org/10.1007/s00401-005-1007-7
Monfrini E, Arienti F, Rinchetti P, Lotti F, Riboldi GM (2023) Brain calcifications: genetic, molecular, and clinical aspects. Int J Mol Sci 24. https://doi.org/10.3390/ijms24108995
Nahar K, Lebouvier T, Andaloussi Mäe M, Konzer A, Bergquist J, Zarb Y, Johansson B, Betsholtz C, Vanlandewijck M (2020) Astrocyte–microglial association and matrix composition are common events in the natural history of primary familial brain calcification. Brain Pathol 30:446–464. https://doi.org/10.1111/bpa.12787
Norenberg MD (2010) Central Pontine myelinolysis: historical and mechanistic considerations. Metab Brain Dis 25:97–106. https://doi.org/10.1007/s11011-010-9175-0
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell-Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE, Tayoun AA, Berg JS, Cutting GR, Ellard S, Kang P, Karbassi I, Mester J, Pesaran T, Plon SE, Rehm HL, Strande NT, Topper S (2022) Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet 109:2163–2177. https://doi.org/10.1016/j.ajhg.2022.10.013
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Sci 17:405–424. https://doi.org/10.1038/gim.2015.30
Rodrigues CHM, Pires DEV, Ascher DB (2018) DynaMut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46:W350–W355. https://doi.org/10.1093/nar/gky300
Samden D, Lhatoo B, Perunovic S, Love H, Houlden, Malcolm J, Campbell (2003) Familial idiopathic brain calcification – a New and familial α-Synucleinopathy? Eur Neurol 49:223–226. https://doi.org/10.1159/000070189
Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11:361–362. https://doi.org/10.1038/nmeth.2892
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR (2013) Predicting the Functional, Molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34:57–65. https://doi.org/10.1002/humu.22225
Sweeney MD, Sagare AP, Zlokovic BV (2018) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14:133–150. https://doi.org/10.1038/nrneurol.2017.188
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson IM, Edlund K, Lundberg E, Navani S, Szigyarto CAK, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, Von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, Von Heijne G, Nielsen J, Pontén F (2015) Tissue-based map of the human proteome. Science (1979) 347. https://doi.org/10.1126/science.1260419
Vanlandewijck M, He L, andaloussi Mäe M, andrae J, ando K, Del Gaudio francesca, Nahar K, Lebouvier T, Laviña bàrbara, Gouveia L Sun Ying, raschperger elisabeth, räsänen M, Zarb Yvette, Mochizuki N, Keller Annika, Lendahl urban, betsholtz christer (2018) a molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480. https://doi.org/10.1038/nature25739
Wider C, Dickson DW, Schweitzer KJ, Broderick DF, Wszolek ZK (2009) Familial idiopathic basal ganglia calcification: a challenging clinical-pathological correlation. J Neurol 256:839–842. https://doi.org/10.1007/s00415-009-5025-6
Xu X, Sun H, Luo J, Cheng X, Lv W, Luo W, Chen WJ, Xiong ZQ, Liu JY (2023) The pathology of primary familial brain calcification: implications for treatment. Neurosci Bull 39:659–674. https://doi.org/10.1007/s12264-022-00980-0
Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, Losada PM, Chen MB, Schaum N, Khoury N, Toland A, Calcuttawala K, Shin H, Pálovics R, Shin A, Wang EY, Luo J, Gate D, Schulz-Schaeffer WJ, Chu P, Siegenthaler JA, McNerney MW, Keller A, Wyss-Coray T (2022) A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603:885–892. https://doi.org/10.1038/s41586-021-04369-3
Yang Q, Li J, Jiao B, Weng L (2022) Primary familial brain calcification in a patient with a novel compound heterozygous mutation in MYORG presenting with an acute ischemic stroke: a case report. Ann Transl Med 10:423–423. https://doi.org/10.21037/atm-21-4883
Yao XP, Cheng X, Wang C, Zhao M, Guo XX, Su HZ, Lai LL, Zou XH, Chen XJ, Zhao Y, Dong EL, Lu YQ, Wu S, Li X, Fan G, Yu H, Xu J, Wang N, Xiong ZQ, Chen WJ (2018) Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 98:1116–1123e5. https://doi.org/10.1016/j.neuron.2018.05.037
Zarb Y, Weber-Stadlbauer U, Kirschenbaum D, Kindler DR, Richetto J, Keller D, Rademakers R, Dickson DW, Pasch A, Byzova T, Nahar K, Voigt FF, Helmchen F, Boss A, Aguzzi A, Klohs J, Keller A (2019) Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 142:885–902. https://doi.org/10.1093/brain/awz032
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV (2015) Establishment and dysfunction of the blood-brain barrier. Cell 163:1064–1078. https://doi.org/10.1016/j.cell.2015.10.067
Zhao M, Lin XH, Zeng YH, Su HZ, Wang C, Yang K, Chen YK, Lin BW, Yao XP, Chen WJ (2022) Knockdown of myorg leads to brain calcification in zebrafish. Mol Brain 15. https://doi.org/10.1186/s13041-022-00953-4
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738. https://doi.org/10.1038/nrn3114
Acknowledgements
The authors sincerely thank all the patients and their relatives for receiving genetic tests and pathological examinations in this study. The authors appreciate T Ohnishi at Kagoshima University for their vital technical assistance, and Professor T Tsuzuki at department of surgical pathology Aichi Medical University, School of Medicine for immunohistochemical study. The authors extend their appreciation to the Division of Gene Research, Research Support Centre, Kagoshima University, for the use of their facilities. Finally, the authors thank ENAGO (www.enago.jp) for English language editing.
Funding
This study was partly supported by a Grant-in-Aid from the Research Committee of Ataxia, Health and Labour Sciences Research Grants, Ministry of Health, Labour and Health, Welfare and Labour, Japan (2016100002B). This research was supported by the Japan Agency for Medical Research and Development (Grant Numbers 201442014A, 201442071A, JP14ek0109065, JP20ek0109491 and JP21wm0425019). This research was also supported by JSPS KAKENHI (Grant Numbers JP18H02742, JP20K16604, JP21K15702, JP21H02842, JP22K15713, JP22K07495, JP22K07519, JP23K06931, and JP23K06966).
Author information
Authors and Affiliations
Contributions
TH, YH, and HT conceived the project and designed the study. TH and MS collected the clinical data. TH, JY, AY, and FK analyzed and verified the clinical and genetic data. MY contributed to the evaluation and analysis of pathological examination. MA, EM, YO, JM, and ST aided in interpreting the results and worked on the manuscript. JY contributed to English proofreading. TH drafted the original manuscript, and all co-authors approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study received approval from the institutional review board of Kagoshima University (Application ID: 490). All participants provided their informed consent for their involvement in this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hobara, T., Higuchi, Y., Yoshida, M. et al. Genetic and pathophysiological insights from autopsied patient with primary familial brain calcification: novel MYORG variants and astrocytic implications. acta neuropathol commun 12, 136 (2024). https://doi.org/10.1186/s40478-024-01847-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-024-01847-3