
Abstract
Background
The tropical legume Mucuna pruriens (L.) DC. can meet three agricultural needs: low-cost protein, high-value medicines, and green manure or cover crops. But like other underutilized crops, it needs more modern breeding resources. Identifying marker-trait associations (MTAs) can facilitate marker-assisted breeding and crop improvement. Recent studies have demonstrated the feasibility of identifying MTAs using a small number of accessions (< 100). We have characterized a panel of 70 M. pruriens accessions across two consecutive years and performed association analysis for 16 phenotypic traits related to seed (seed length, seed width, seed thickness, seed yield per plant, hundred seed weight); pod (pod length, pod width, number of pods per cluster, number of pods per plant); inflorescence (inflorescence length, flower buds per inflorescence, flower length, pedicel length), and biochemical attributes (L-DOPA, total protein, total carbohydrate), using 66 genic-microsatellite markers following mixed linear model.
Results
The results showed significant phenotypic (P < 0.05) and genetic diversity (Shannon’s information index, I = 0.62) in our germplasm collection. Many tested traits were highly heritable (broad-sense heritability ranging from 42.86 to 99.93%). A total of 15 MTAs was detected at an adjusted significance level of P < 5.55 × 10− 3 for nine traits (seed length, seed thickness, seed width, hundred seed weight, seed yield per plant, inflorescence length, flower buds per inflorescence, flower length, and petiole length), contributed by 10 SSR markers (MPU_19, MPU_42, MPU_54, MPU_57, MPU_58, MPU_83, MPU_89, MPU_108, MPU_111, and MPU_122.) with phenotypic variance explained (PVE) ranging from 14.7 to 31.1%. Out of the ten trait-associated markers, the BLAST analysis revealed putative functions of seven markers, except MPU_57, MPU_58, and MPU_83.
Conclusion
Fifteen MTAs identified for important traits with phenotypic variance explained > 10% from mixed linear model offer a solid resource base for improving this crop. This is the first report on association mapping in M. pruriens and our results are expected to assist with marker-assisted breeding and identifying candidate genes in this promising legume.
Similar content being viewed by others


Background
Tapping the potential of neglected and underutilized species is vital to reduce overreliance on staples and improve global food security. The prospects of Mucuna pruriens (L.) DC. for tropical agriculture is well recognized (Pugalenthi et al. 2005). Its important traits include edible protein and the production of L-DOPA (l-3,4-dihydroxyphenylalanine), used in treating Parkinson’s disease. Identification of marker-trait association can accelerate marker-assisted breeding programs and propel its use for commercial cultivation.
M. pruriens is a member of the phaseloid clade of Leguminosae/Fabaceae. It is a self-pollinated annual species with a diploid genome (2n = 2x = 22) of approximately 1281 to 1361 Mbp (Sastrapradja et al. 1974; Sathyanarayana et al. 2017) and occurs both in the wild (var. pruriens and var. hirsuta) and cultivated (var. utilis) forms (Wilmot-Dear 1987; Sasidharan 2004). Earlier works reported wide-ranging taxonomic confusions within this species (Duke 1981). The taxonomic revision carried out by Wilmot Dear (1987) showed that many of the previously reported species (M. aterrima, M. cochinchinensis, M. hassjoo, M. nivea, and M. utilis) as mere varieties of M. pruriens suggesting their inclusion in one of three botanical varieties [viz., var. pruriens and var. hirsuta (wild forms) and var. utilis (cultivated forms)]. Further examination of these varieties based on morphometric and RAPD markers found close genetic similarities and breeding compatibility between var. hirsuta and var. pruriens, proposing their merger into a single botanical variety (var. pruriens) (Leelambika and Sathyanarayana 2011). However, clear affirmation on this from combined classical and modern taxonomic tool is awaited, and until then, var. hirsuta conceivably needs to be maintained separately to keep uniformity in literature. Regarding agronomic benefits, with a seed-to-seed duration of 90–120 days, M. pruriens flourishes well under acidic soil (pH < 5.8), elevation below 1600 m, annual rainfall > 400 mm, warm (19–27 °C) and moist tropical climatic conditions (Pugalenthi et al. 2005; Kumar and Saha 2013). The plant is native to Eastern India and Southern China (Duke 1981; Wilmot-Dear 1987) but is now distributed across Asia, the Americas, the West Indies, Africa, and the Pacific Islands (Fung et al. 2011). In Southeast Asia, it is mainly found in India, Nepal, Bangladesh, Myanmar, Sri Lanka, and Malaysia (Fung et al. 2011; Kumar and Saha 2013). Within the Indian sub-continent, its distribution ranges from the lower Himalayan range to the entire tropical plains of India (Muralia and Pathak 2003). It is adapted to less fertile and dry soils (Siddhuraju et al. 2000), produces a seed yield of about 1.3–2.4 t/ha (Kumwenda and Gilbert 1998; Gurumoorthi et al. 2003), and possesses disease-resistant (Eilitta et al. 2002), nematicidal (Carsky and Ndikawa 1998) as well as allelopathic properties (Fujii et al. 1991). M. pruriens (var. utilis) contains high seed protein (20–30%) akin to other edible legumes such as soybean (35–40%), chickpea (18.3–25.2%), and pigeon pea (19–21.7%) (Kumar et al. 1991; Hira and Chopra 1995; Mang et al. 2016) and thus offers a cheap source of edible protein.
Genetic diversity is a critical determinant in the success of any crop improvement program. In M. pruriens, earlier works found wide-ranging phenotypic and trait-specific variability in different germplasm collections (Mahesh and Sathyanarayana 2011, 2015; Sathyanarayana et al. 2012). DNA markers such as randomly amplified polymorphic DNA (RAPD) (Padmesh et al. 2006; Leelambika et al. 2010; Patil et al. 2016), inter simple sequence repeat (ISSR) (Patil et al. 2016; Chinapolaiah et al. 2018), amplified fragment length polymorphism (AFLP) (Sathyanarayana et al. 2011; Mahesh and Sathyanarayana 2015; Tripathi et al. 2018) and simple sequence repeat (SSR) (Shetty et al. 2015; Sathyanarayana et al. 2017; Kumar et al. 2019) have reinforced these findings. Regarding sequence information, the first transcriptome analysis reported de novo assembly, functional annotation, and differential gene expression among pod, leaf, and root tissues (Sathyanarayana et al. 2017). In another study that followed, detailed transcriptome analysis was carried out along with biochemical characterization to track the presumptive biosynthetic pathway genes associated with L-DOPA production (Singh et al. 2018). More recently, Yuan et al. (2021) have generated sequencing of the plastid genome of 152,119 bp, while a whole genome sequencing effort by Hao et al. (2022) has created 500.49 Mb sequence data across 11 chromosomes.
In molecular breeding, studies aiming at developing linkage maps and tagging agronomic traits using AFLP markers were initiated (Capo-chichi et al. 2004; Mahesh et al. 2016). However, these efforts didn’t advance beyond preliminary findings, perhaps due to resource constraints often faced by such underutilized crops. It is well known that conventional linkage analysis is both time and resource-intensive due to the requirement of bi-parental populations. It also suffers from efficacy issues due to low-resolution and limited allelic variation (Flint-Garcia et al. 2005; Yu and Buckler 2006; Oraguzie and Wilcox 2007; Abdurakhmonov and Abdurakhmov 2008). Alternatively, association mapping offers a powerful method for identifying marker-trait association as it determines the relationship between phenotypic variations and genetic polymorphisms considering naturally occurring or historical recombination (Ambreen et al. 2018). It has been successfully used for identifying marker-trait associations (MTAs) in different legume species (Liu et al. 2017; Singh et al. 2017; Zhao et al. 2017; Vaijayanthi et al. 2018). However, no study has explored this opportunity in M. pruriens. Among the molecular markers, EST-SSRs have been proven reliable for association analysis as they are directly linked with the genes expressing a trait (Varshney et al. 2005). Several species-specific EST-SSR markers have recently been developed using RNA-seq analysis in M. pruriens (Sathyanarayana et al. 2017). Plant breeders also often choose the SSR markers due to their co-dominant nature, multiallelic expression, high polymorphism, and greater genome abundance (Kalia et al. 2011).
For association mapping, core collections have often been effective, but core collections of adequate size are generally available only for selected/mainstream crop species (Liu et al. 2017; Zhao et al. 2017; Ambreem et al. 2018). For crops that lack a large and established core collection, a diverse panel of accessions from the germplasm collection can be used, provided they demonstrate the presence of high genetic variance, low kinship association among its individuals, and lack of strong population structure (Hu et al. 2014; Mahajan et al. 2017). Association analysis is usually carried out on a large number of accessions (> 300). But in cases where the numbers are limited, it is still possible to explore MTAs (Soumya et al. 2021). Recently, several genome-wide association studies (GWAS) have been successfully carried out in population sizes between 60 and 150 (Lehnert et al. 2018; Ma et al. 2018; Rohilla et al. 2020). Taking cues from these studies, in the present work, we evaluated a panel of seventy diverse M. pruriens accessions for suitability as an association mapping panel and performed association analysis on sixteen agronomically important traits using microsatellite markers.
Materials and methods
Plant material and phenotypic evaluation
As an association mapping panel, we used a subset of 70 M. pruriens accessions from our germplasm collection. This comprised 11 accessions of M. pruriens var. utilis (cultivated variety), 28 of var. pruriens (wild variety), and 31 of var. hirsuta (wild variety). They represented three large geographic regions in India - Eastern, Southern, and West-Central India (Table S1). Sixteen traits related to seed (seed length, seed width, seed thickness, seed yield per plant, hundred seed weight); pod (pod length, pod width, number of pods per cluster, number of pods per plant); inflorescence (inflorescence length, flower buds per inflorescence, flower length, pedicel length), and biochemical attributes (L-DOPA, total protein, total carbohydrate) were evaluated for two consecutive years (2014 and 2015). We used manual measurements using a vernier caliper, weighing balance, and counting as relevant for the trait for data scoring. The L-DOPA content was estimated as per the method of Daxenbichler et al. (1972). The total protein and carbohydrate contents were estimated using Lowry’s (Lowry et al. 1951) and Anthrone’s (Hedge and Hofreiter 1962) methods, respectively. All statistical analyses, such as one-way analysis of variance (ANOVA), correlation, and principal component analysis (PCA), were performed using the R program (R Core Team 2014).
Estimation of variance components and broad-sense heritability
To estimate the variance, genotypic and phenotypic coefficients of variation were evaluated using Syukur et al. (2012) as follows:
σ2G = (MSG – MSE) / r.
σ2P = σ2G + σ2E/r.
Where, σ2G = genotypic variance, σ2P = phenotypic variance, σ2E = environmental variance (error mean square from the analysis of variance); MSG = mean square of genotypes; MSE = error mean square; r = number of replications.
The broad-sense heritability for each trait was estimated according to Allard (1960) as follows:
H2 = (σ2G/ σ2 P) × 100.
Where, H2 = heritability in broad-sense; σ2G = genotypic variance; σ2P = phenotypic variance.
DNA isolation and SSR genotyping
Leaf tissues from 15-day-old seedlings were used for the DNA isolation as described previously (Doyle and Doyle 1990). Genotyping was done using a sub-set of 90 species-specific genic SSR primer pairs (Table S2) chosen from Sathyanarayana et al. (2017). Polymerase chain reaction (PCR) mixture included template DNA (50ng/µl), primers (1µM each), dNTPs (2.5mM), Taq polymerase (1U), PCR buffer (1X), and MgCl2 (1.5mM), maintaining the final volume of 25 µl. DNA amplification condition comprised of initial denaturation at 94 °C for 3 min followed by 35 cycles of 94°C for 30s, appropriate annealing temperature (Tm) for 30 s, and an extension at 72 °C for 20 s with a final extension of 7 min at 72 °C. Subsequently, one (1) µl each of PCR amplicons generated with different dye-labeled primers were mixed with 2.95 µl distilled water, 7 µl of formamide and 0.05 µl of the GeneScan™ 500 LIZ® Size Standard. Denatured DNA was size fractioned with capillary electrophoresis. As each SSR primer-pair produced multiple amplicons, primer-pairs are referred to as SSR markers and, amplicons as bands or alleles from here onwards. We applied stringent filtering criteria (minor allele frequency > 0.05 and missing percentage less than 20%) for the marker bands produced. Those SSRs fulfilling these criteria were retained and used for further analysis.
Marker attributes and genetic diversity
Marker attributes such as polymorphism information content (PIC) and major allele frequency (MAF) were calculated by PowerMarker v 3.25 (Liu and Muse 2005). The total number of alleles (Na), effective number of alleles (Ne), observed heterozygosity (Ho), expected heterozygosity (He), percentage of polymorphic loci (%P), gene flow (Nm), and Hardy-Weinberg equilibrium (HWE) were assessed by GenAlEx v 6.5 (Peakall and Smouse 2012). HP-Rare v 1.0 (Kalinowski 2005) was used to determine private/rare allelic richness per locus (Rp) based on rarefaction approach. The genetic diversity indices such as - Shannon’s information index (I), Nei’s gene diversity (h), and total genetic diversity (HT) were determined by using POPGENE v 1.32 (Yeh et al. 1999).
Population structure, relative kinship, and genetic relationship
Population structure was analyzed using the Bayesian clustering method implemented in STRUCTURE v 2.3.4 (Pritchard et al. 2000). For each accession, the proportion of ancestral contribution was estimated using the admixture model and correlated allele frequencies. K-values ranging from 1 to 10 were tested with ten independent replications, 100,000 lengths of the burn-in period, and 200,000 Markov Chain Monte Carlo (MCMC) repetitions for each K. The optimal K-value was obtained using STRUCTURE analysis results with STRUCTURE HARVESTER (Earl 2012). Accessions were assigned to a subgroup depending on the Q-value (membership proportion), and if the Q-value was < 80%, they were termed admixtures. Molecular variance (AMOVA) and pairwise FST of geographic population and subpopulations were analyzed using GenAlEx v 6.5 (Peakall and Smouse 2012) with 1,000 permutations. The kinship coefficients (Fij; individual level) were calculated following Loiselle et al. (1995) to estimate the relatedness between the individuals. TASSEL v 5.0 (Bradbury et al. 2007) was used to generate the kinship coefficient matrix among all pairs of accessions. The kinship heat map was obtained using GAPIT, R package (Wang et al. 2014). Genetic relationships based on the distance were arrived at through the construction of an unrooted neighbor-joining (NJ) dendrogram and principal coordinate analysis (PCoA) using Darwin v 6.0 (Perrier and Jacquemoud-Collet 2006). The reliability of the NJ dendrogram was tested with the bootstrap value of 1000 replicates.
Association mapping and annotation of the MTAs
TASSEL v 5.0 (Bradbury et al. 2007) was used for the association analysis. A mixed linear model (MLM) was employed to determine the MTAs. The MLM is considered superior over the general linear model (GLM), as GLM incorporates only population structure (Q matrix), which often results in false-positives; instead, MLM incorporates both Q and K matrix, which overcomes this limitation (Yu et al. 2006). Generally, in such studies, Bonferroni multiple test correction (P = 0.05/n; n is the number of markers used in the study) is applied to obtain the threshold P-value as it provides a stringent cut-off to avoid false positives. However, it assumes that the loci are independent, which is not always true, given that certain loci may be in linkage disequilibrium. Therefore, to avoid loss of beneficial MTAs, we also used a less stringent criterion, P = 1/n as an alternative cut-off in addition to Bonferroni correction. This approach has been beneficially employed in some earlier studies (Li et al. 2012; Yang et al. 2014; Xu et al. 2018). The Manhattan plots and quantile-quantile (Q-Q) plots for MTAs were generated using the CMplot R package (Yin et al. 2021). The putative function of each trait-associated marker was determined using the Arabidopsis thaliana (L.) Heynh. genome database (https://www.arabidopsis.org). For this, the transcripts of SSR markers were used as a query sequence and matched against the reference genome sequences of A. thaliana with the BLAST tool. Then, the top hit was selected as the putative gene/function of the respective marker. An e-value ≤ 1e-05 was used as a threshold.
Results
Phenotypic variability
The one-way ANOVA revealed significant variability (P < 0.05) for most phenotypic traits evaluated in our association mapping panel, except for pedicel length, flower length, and L-DOPA content. Inflorescence length and flower length showed the highest (CV = 84.97%) and the lowest variations (CV = 6.90%) (Table 1). The values of correlation coefficients indicated a significantly high positive correlation (R > 0.70) between some important seed, pod, and inflorescence traits (Fig. 1). For instance, hundred seed weight was strongly correlated (R > 0.70) with seed thickness, seed width, seed length, and pod length, and moderately correlated (0.30 < R < 0.70) with pod width and flower length. A similar trend was observed for seed yield as well.

Correlation matrix of sixteen phenotypic traits in M. pruriens association mapping panel. Note: The intensity of color ranges from blue (positive) to brown (negative) and the size of the circles show strength of significant correlation (P < 0.05) IL: Inflorescence length; FBpI: Flower buds per inflorescence; FL: Flower length; PedL: Pedicel length; PodL: Pod length; PodW: Pod width; NPpC: Number of pods per cluster; NPpPt: Number of pods per plant; SL: Seed length; SW: Seed width; ST: Seed thickness; SYpPt: Seed yield per plant; HSW: Hundred seed weight; LDp: Total L-DOPA; TPr: Total protein; TCr: Total carbohydrate; ×: Statistically non-significant correlation values
In principal component analysis (PCA), the first five principal components (PCs) explained 81.39% of the total phenotypic variance, of which PC1 accounted for 34.15%, and PC2 accounted for 24.68% (Fig. 2) (Table S3). The resultant scatter plot distinctly separated the cultivated and wild varieties. However, we couldn’t find clear-cut separation for the two wild varieties - var. pruriens and var. hirsuta (Fig. 2). We observed moderate to high estimates (42.86–99.93%) of broad-sense heritability (H2) for the 16 phenotypic traits (Table 1). Traits such as the number of pods per cluster, number of pods per plant, hundred seed weight, seed yield per plant, etc., showed high heritability values ranging from 93.34 to 99.93%. Among the biochemical traits, total protein (98.58%) and total carbohydrate (95.71%) recorded high heritability, and L-DOPA content revealed a moderately high heritability (62.50%).

Scatter plot of M. pruriens accessions based on sixteen phenotypic traits from the principal component analysis (PCA).Note: Red circles, green triangles and blue squares represent M. pruriens var. hirsuta, var. pruriens, and var. utilis, respectively
Genetic diversity
Initially, 90 markers tested on 70 accessions of M. pruriens (Table S4) produced 298 alleles with an average of 3.39 alleles per locus. At this stage, we applied a stringent filtering criterion to retain only 180 SSR alleles contributed by 66 markers based on minor allele frequency > 0.05, along with 61 accessions, which showed maximum missing site < 20% for further analysis. The major allele frequency (MAF, revealed by filtered SSR data) varied from 0.50 to 0.94, with an average of 0.73 indicating high genetic diversity and polymorphism at the observed loci. The heterozygosity (Ho) values ranged from 0.00 to 0.59 with a mean value of 0.12, and the gene diversity (He) varied from 0.03 (MPU_122) to 0.73 (MPU_42) with a mean value of 0.36. The summary of the marker attributes at different loci and individual markers retained after stringent filtration are given in Table 2 and Table S5, respectively.
The polymorphism information content (PIC) value, which provides relative informativeness of each marker, ranged between 0.09 (for SSR marker MPU_22) and 0.38 (MPU_52) with an average of 0.29 (Table S5). The significant proportion of SSR markers in our study was moderately polymorphic (43 SSR markers with PIC-values ranging from 0.26 to 0.38, Table S5). The mean and private/rare allelic richness per marker were 2.82 and 0.06, respectively. The majority of the SSR markers deviated from the Hardy-Weinberg equilibrium (HWE) at a significance level of P < 0.001, and very few markers deviated at P < 0.01 (MPU_46 and MPU_115) and P < 0.05 (MPU_92) (Table S5). The SSR markers MPU_122 and MPU_130 showed no significant deviation.
Shannon’s information index (I = 0.62) and Nei’s gene diversity (h = 0.32) suggest high genetic diversity within our association mapping panel. The total gene diversity value (HT = 0.34) (Table S5) substantiated this finding. For more perceptive analyses, we subdivided the association panel accessions into two groups: the first one based on three regional gene pools (Eastern, Central-West, and Southern India) and the second one on three botanical varieties (var. utilis, var. pruriens, and var. hirsuta). We independently estimated genetic diversity statistics for each gene pool (Table 3). In the case of regional gene pools, I varied from 0.56 to 0.67, and h ranged from 0.31 to 0.34 (Table 3). Accessions from West-Central India (I = 0.67, h = 0.34) and Southern India (I = 0.64, h = 0.34) were more diverse as compared to ones from Eastern India (I = 0.56, h = 0.31). The percentage of polymorphic SSR loci was 96.97% for the accessions of South India, followed by 89.39% and 87.88% for the accessions of West-Central and Eastern India, respectively. Among the three varietal gene pools, I varied from 0.44 to 0.72, and h ranged from 0.28 to 0.36 (Table 3). Wild varieties, var. pruriens (I = 0.72, h = 0.36) and var. hirsuta (I = 0.62, h = 0.34), were genetically more diverse than the cultivated var. utilis (I = 0.44, h = 0.28). The percentage of polymorphic SSR loci was 98.48%, 96.97%, and 72.73% in var. pruriens, var. hirsuta, and var. utilis.
Population structure
STRUCTURE analysis (∆K vs. K plot) revealed a sharp peak at K = 2, indicating the presence of two genetic groups (MpSTR-I and MpSTR-II) in our association mapping panel (Fig. 3a and b). Based on the geographical affiliation of accessions, subgroup-1 (designated as MpSTR-I) was composed of 17 accessions, of which 14 were derived from Eastern India, two from West-Central India, and one from Southern India. Similarly, subgroup-2 (designated as MpSTR-II) was composed of 44 accessions, of which 34 were derived from Southern India, nine from West-Central India, and one from Eastern India. This indicated that the majority of accessions belonging to Eastern India were present in MpSTR-I, and those belonging to Southern India were present in MpSTR-II. However, accessions from West-Central India did not form an independent subgroup but merged with one of the two major clusters. Based on the varietal affiliation, MpSTR-I was composed of five accessions of var. utilis and 12 accessions of var. pruriens. Likewise, MpSTR-II was composed of 25 accessions of var. hairsuta, 16 accessions of var. pruriens, and three accessions of var. utilis. Thus, independent grouping based on variety was also absent. Out of the total accession present in both of the subgroups, 48 accessions showed > 80% of shared ancestry, and 13 were admixtures with < 80% of shared ancestry (Fig. 3b).

Population structure of M. pruriens association mapping panel inferred using STRUCTURE software (a) Hypothetical sub-population estimation using ∆K-values (K = 2) indicating two subpopulations (b) Population structure at K = 2 based on inferred ancestry (Q-matrix) in which two sub-populations are indicated as MpSTR-I (red color) and MpSTR-II (green color)
To ascertain the partitioning of variation, we performed AMOVA on three population groups assumed based on (a) geographic distribution, (b) varietal affiliation, and (c) subgroups identified in STRUCTURE analysis (Table 4). All three revealed higher within gene pool variance than among gene pools. The ratios for within- to between-gene pool variance were 93:7 in the case of geographical distribution, 97:3 for varietal affiliations, and 89:11 for structure-derived sub-populations. We estimated population differentiation (FST) within each group separately. The results revealed low to moderate differentiation in groups (pairwise FST) based on the varietal affiliation (FST = 0.032) and geographic origin (FST= 0.070) and high genetic differentiation (FST= 0.105) in groups based on STRUCTURE analysis (Table 4).
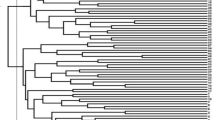
We constructed the NJ dendrogram using a simple matching coefficient to determine the genetic relationship and performed PCoA. The NJ algorithm revealed two main clusters, MpNJ-I and MpNJ-II (Fig. 4). Similar to STRUCTRE results, most of the 28 accessions in MpNJ-I were derived from Southern India (25 accessions). However, considerable mixing was observed among the 33 accessions in MpNJ-II, as 15 belonged to Eastern India, nine were from West-Central India, and nine were from Southern India. The accessions in the NJ tree and PCoA plots (PC1 vs. PC2) are color-coded from the information of two subgroups identified using the STRUCTURE analysis (MpSTR-I and MpSTR-II). We didn’t find any indication of grouping based on geographic origin or varietal affiliation, as accessions representing these groups were generally mixed.

Neighbor joining (NJ) dendrogram based on the genetic distance. Note: Red and green color indicates subgroup-1 (MpSTR-I) and subgroup-2 (MpSTR-II) from STRUCTURE analysis
In principal coordinate analysis (PCoA), the principal axes 1 and 2 explained 28 and 26% of the total variance (Fig. 5). The dispersion was relatively homogeneous in all four quadrants, signifying the diverse nature of the accessions. The clustering pattern in PCoA largely corresponded with that of the NJ-dendrogram. We also estimated the relative kinship between these accessions to measure their relatedness. About 55.29% of kinship coefficient (Fij) values between any two accessions were within 0 to 0.05, and there was a subsequent reduction in frequency with an increase in kinship value (Fig. 6). The heat map (Fig. 7) revealed substantial differences among the accessions.

Scatter plot depicting dispersion of M. pruriens accessions based on principal coordinate analysis. Note: Red and green color indicates subgroup-1 (MpSTR-I) and subgroup-2 (MpSTR-II) from STRUCTURE analysis

Distribution of global pairwise kinship coefficients (Fij) of M. pruriens association panel

Heat map of kinship matrix generated for M. pruriens association mapping panel based on 180 filtered SSR markers data. Dendrogram are shown on the top and left side of the figure
Association mapping
For association analysis, we employed the mixed linear model (MLM), which integrates both population structure (Q matrix) and kinship (K matrix) to avoid false-positive MTAs. Only two MTAs involving two SSR markers (MPU_83 and MPU_122) passed the stringent Bonferroni adjusted threshold P < 2.77 × 10− 4 (P = 0.05/180, where 180 is the number of markers used in the analysis). These two associations were for the seed yield per plant (phenotypic variance explained, PVE = 31.12%) and the pedicel length (PVE = 25.07%) (Fig. 8 a-d).

Manhattan plot (P-value) for MLM depicting significant marker-trait associations at adjusted Bonferroni threshold P < 2.77 × 10− 4(a) Seed yield per plant (b) Pedicel length; Quantile-Quantile (Q-Q) plot of MLM (c) Seed yield per plant (d) Pedicel length
We also used a less stringent criterion of P < 5.55 × 10− 3 (1/180) as the cut-off for the reasons explained in the method section. By applying, a total of 15 additional MTAs related to inflorescence length, flower buds per inflorescence, flower length, pedicel length, seed length, seed width, seed thickness, hundred seed weight, and seed yield per plant were identified (Table 5). The PVE for these MTAs ranged from 14.72 to 31.12%. These MTAs were contributed mainly by 10 SSR markers: MPU_19, MPU_42, MPU_54, MPU_57, MPU_58, MPU_83, MPU_89, MPU_108, MPU_111, and MPU_122. We found four MTAs contributing to a hundred seed weight, two each for seed length, seed width, pedicel length, and at least one each for inflorescence length, flower buds per inflorescence, flower length, seed thickness, and seed yield per plant. The highest PVE (31.12%) was observed in MTA between SSR marker MPU_83 and an important agronomic trait, seed yield per plant. This crucial MTA was also spotted at Bonferroni adjusted threshold P < 2.77 × 10− 4 (Fig. 8a). The Manhattan and Q-Q plots of MTAs at P < 5.55 × 10− 3 are given in Fig. S1 and S2, and the MTAs significant at threshold P < 0.05 and PVE > 10% are provided in Table S6.
The association analysis also revealed four SSR markers associated with multiple agronomic traits (P < 5.55 × 10− 3; PVE > 10%). Marker MPU_83 was not only associated with the seed length and seed width but also with the seed yield per plant. SSR loci MPU_54 was associated with seed thickness and hundred seed weight. MPU_42 was associated with seed length and seed width, and MPU_19 was associated with flower length and flower buds per inflorescence. These MTAs were justified by high significant correlations (P < 0.05) values between these traits of agronomic importance (Fig. 1).
Annotation of marker-trait associations
To test the reliability of our MTAs, we determined the putative gene/function for each trait-associated marker (P < 5.55 × 10− 3; PVE > 10%) by annotating the related SSR primers using the Arabidopsis thaliana (L.) Heynh. genome database. Out of the ten trait-associated markers, the BLAST analysis annotated seven markers viz., MPU_19, MPU_42, MPU_54, MPU_89, MPU_108, MPU_111, and MPU_122 (Table 6). Notably, the SSR marker MPU_19, associated with flower length and flower buds per inflorescence, was annotated as 4-coumarate-CoA ligase 2, and MPU_42, associated with seed length and seed width, was annotated for Zinc transporter 7-precursors. Likewise, the SSR marker MPU_54 associated with traits such as seed thickness and hundred seed weight was annotated for sucrose non-fermenting 1- related protein kinase 2 (SnRK2) gene, and MPU_89 associated with hundred seed weight was annotated as 3-dehydroquinate synthase. The SSR MPU_108, associated with hundred seed weight, matched with NAC domain-containing protein, a plant-specific transcription factor (TF) associated with multiple aspects of stress and development. Further, MPU_111 (hundred seed weight) and MPU_122 (pedicel length) were annotated with Ubiquitin-specific protease and LEUNIG homolog (LUG and LUH), respectively.
Discussion
Phenotypic diversity and heritability
M. pruriens is an underutilized legume species; hence, its germplasm is maintained by a few international and national research institutes/organizations such as the International Institute of Tropical Agriculture (IITA), Nigeria; United States Department of Agriculture (USDA), the USA; and the National Bureau of Plant Genetic Resources (NBPGR), India (Jorge et al. 2007). We used the most diverse and informative available germplasm collection, containing 70 accessions, as an association mapping panel. We found significant variability for most of the flower, pod, seed, and biochemical characteristics in our association mapping panel. This is consistent with earlier reports in different M. pruriens germplasm (Gurumoorthi et al. 2003; Kalidass and Mohan 2011; Sathyanarayana et al. 2012). The majority of the seed and pod-based traits revealed a significant positive correlation, possibly due to the critical role played by the pod in seed development in terms of protection and nutrient source (Bennett et al. 2011). Several accessions such as IC-369,144 (27.89%), 500,113-MH (28.05%), 500,126-KA (28.55%), 500,135-KA (28.76%), 500,178-MH (29.11%), 500,184-MH (29.19%), and 500,190-MH (29.12%) showed a seed protein content akin to soybean (35–40%; Michelfelder 2009). These stocks will be helpful in breeding for protein content. But, unlike previous studies (Tripathi et al. 2018; Kumar et al. 2019), we found less variability for the L-DOPA content in our germplasm. The estimation method used (Daxenbichler et al. 1972) in this study is old and laborious; thus, it is plausible that poor recovery and associated shortcomings might have skewed this experiment. Moderately high heritability (H2 > 60%) was observed for different traits in our study. Two earlier studies found analogous observations for traits such as pod length, pod width, pod weight, hundred seed weight, seed yield per plant, and inflorescence length (Hadapad et al. 2018; Chinapolaiah et al. 2019). Our results reinforce them. High heritability means less influence of the environmental factors rendering phenotypic selection reliable, besides contributing to high additive effect in the breeding programs (Tiwari et al. 2011; Rosmania et al. 2016).
Genetic diversity, population structure, and kinship analysis
Initially, we used 90 SSR markers on 70 M. pruriens accessions, but after stringent filtration, only 66 SSR markers and 61 accessions were retained for further analysis. These markers showed a moderate PIC value (0.25 < PIC < 0.50). This corroborates with the earlier report in M. pruriens (Kumar et al. 2019), albeit with a slightly higher allelic range and allele per locus. The result endorses the choice of SSR markers for our study. The Shannon’s information index suggests high genetic diversity (I = 0.62). This is higher than the values reported using AFLP (0.34) and SSR (0.47) markers in earlier studies (Tripathi et al. 2018; Kumar et al. 2019). In a predominantly self-pollinating species like M. pruriens, we expect lower levels of genetic diversity. However, higher values recorded here and in some earlier studies (summarized in Sathyanarayana et al. 2016) categorically point to the presence of out-crossing in this species, as Padmesh et al. (2006) reported. This is partly supported by a high average gene flow among the population groups (Nm = 3.03). Thus, further studies on the pollination mechanism in M. pruriens can throw more light on the drivers of genetic diversity in this species. Among the three geographical regions, accessions from West-Central and Southern India revealed higher diversity than that of Eastern India, consistent with Kumar et al. (2019). Of the three botanical varieties, wild varieties (var. pruriens and var. hirsuta) were more diverse than the cultivated variety (var. utilis) – the finding often documented in many earlier studies (Leelambika and Sathyanarayana 2011; Sathyanarayana et al. 2012; Tripathi et al. 2018; Kumar et al. 2019). Nonetheless, wide phenotypic and genetic diversity in our germplasm collection signifies its utility for the association analysis.
The low to moderate FST values (0.032, 0.070) in M. pruriens varietal and geographical populations indicated less divergence. The population structure analyses from the distance-based NJ-tree, PCoA, and Bayesian-based STRUCTURE largely conformed to each other in suggesting the minimal influence of the geographical origin and/or the varietal affiliation on the grouping of the accessions. High gene flow estimates (Nm = 3.03), the presence of genetic admixtures, and higher within-population variance in AMOVA further concur with this. These observations are also chronicled in different germplasm collections in earlier studies (summarized in Sathyanarayana et al. 2016). Together, these studies and our results suggest one or more explanations: extensive pollen flow across long geographic distances, lack of inter-varietal barriers for hybridization, long seed dispersal, and high seed germination rates. The species thus appears to be highly adaptable, as evident from its broad and diverse distribution range. Alternatively, this might suggest incomplete lineage sorting during its diversification, as observed in safflower (Carthamus tinctorius L.) (Ambreen et al. 2018). However, we caution that our experiments are based on the limited sample size. Future studies must examine more samples from a broad geographical range to confirm these hypotheses. Further, we noticed a low pairwise kinship estimates, suggesting a weak relationship between most M. pruriens accessions in our association mapping panel. Low population divergence and kinship estimates further support the suitability of our panel for association analysis.
Association mapping and significant marker-trait associations
Among the recent approaches used for fine-scale mapping of the desirable traits, association mapping has produced fast and reliable results in several legume taxa. This includes markers for seed-related traits in peanuts (Arachis hypogaea L.) (Zhao et al. 2017); iron and zinc concentration in lentils (Lens culinaris Medikus) (Singh et al. 2017); frost tolerance in peas (Pisum sativum L.) (Liu et al. 2017); flowering, pod yield per plant and fresh pod per pant in lablab [Lablab purpureus (L.) Sweet] (Vaijayanthi et al. 2018); crude protein and mineral concentration in alfalfa (Medicago sativa L.) (Jia et al. 2017); etc. However, in M. pruriens, association mapping has never been used (Lepcha et al. 2019), perhaps due to a lack of resources. Thus, we attempted association mapping of key agronomic traits in M. pruriens in the present study. Usually, a large number of accessions (> 300) are used in association studies. In comparison, our number is small (61). However, several recent studies have demonstrated the feasibility of finding the right marker-trait associations even using a small number of accessions (Lehnert et al. 2018; Ma et al. 2018; Rohilla et al. 2020; Soumya et al. 2021).
We identified 15 significant MTAs (P < 5.55 × 10− 3) using MLM, with PVE ranging from 14.7%to 31.1%. These associations also benefitted by the moderate to high heritability observed for all the trait-associated markers (H2 ranging from 42.86 to 97.27%). Four SSR markers were associated with multiple traits. They are mainly comprised of seed or inflorescence traits that are highly correlated. Such an account of a single marker associated with various traits can be attributed to closely linked QTLs affecting different traits (Rakshit et al. 2010). Alternatively, it may also be due to the pleiotropic effect of the linked QTLs on other traits (Miller and Rawlings 1967).
Annotation of SSR markers associated with the traits
We investigated the putative functions of the trait-associated markers by annotating against corresponding genes/loci in the Arabidopsis thaliana (L.) Heynh. database (https://www.arabidopsis.org). The results produced some useful insights. For instance, the SSR marker MPU_111, associated with a hundred seed weight, corresponded with the ubiquitin-specific protease 23 gene in A. thaliana. Another gene belonging to the same family in rice (Oryza sativa L.), ubiquitin-specific protease 23, is known to exert a positive regulatory influence on grain width and size (Shi et al. 2019). SSR marker MPU_54 associated with traits such as seed thickness and hundred seed weight was annotated for sucrose non-fermenting 1-related protein kinase 2 (SnRK2) gene. This gene is involved in the abscisic acid (ABA) signaling during seed germination, dormancy, and seedling growth and has a central role in plant stress signal transduction (Feng et al. 2018). The SSR MPU_108, associated with a hundred seed weight, was traced to NAC domain-containing protein, a plant-specific transcription factor (TF) associated with the multiple aspects of stress and development. It is reported that three NAC TF encoding genes (ONAC020, ONAC026, and ONAC023) are expressed at very high levels during the seed development in rice and exhibit a strong association with the seed size or weight with the sequence variations located in the upstream regulatory region (Mathew et al. 2016). SSR marker-MPU_122 associated with pedicel length was annotated for LEUNIG homolog (LUG and LUH). Likely, these two proteins act cooperatively to coordinate inflorescence architecture through their influence on auxin biosynthesis, transport, and perception (Douglas et al. 2017). The marker MPU_89 associated with hundred seed weight was annotated to the 3-dehydroquinate synthase gene, which catalyzes the transformation of the seven-carbon sugar 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAH7P) into the 3-dehydroquinate (DHQ) (Negron et al. 2011). This pathway controls the production of precursors of aromatic amino acids in several prokaryotes, fungi, and plants (Ganem 1978). Scrutinizing its relationship with L-DOPA biosynthesis in M. pruriens might open fascinating insights into the regulation of aromatic amino acids, particularly tyrosine production during L-DOPA production. Thus, MTAs identified in the present study are candidates for experimental studies and upon validation will be useful for improving the seed yield and related economic traits in M. pruriens. To our understanding, this is the first report on marker-trait association based on association mapping in any Mucuna species from anywhere in the world.
However, our study has the following limitations: (a) Firstly, we have used a smaller number of accessions and SSR markers as the species has limited germplasm and genomic resources, and (b) Secondly, we have used single environment data for our analysis. So, there is a need to analyze the stability and utility of these marker-trait associations with more accessions and markers under additional environments through multi-location trials. Future studies must also focus on developing MTAs for other vital traits relevant to this crop, particularly indeterminate growth habit - a significant detriment in commercial cultivation of this crop and deduce the location of these MTAs on different linkage groups using bi-parental populations.
Conclusion
In summary, a high phenotypic/genotypic diversity, low population divergence, moderate to high heritability of the evaluated traits, and low kinship estimates in our association mapping panel confirm its utility for association mapping. Fifteen MTAs identified for important traits with PVE > 10% from MLM offer a solid resource base for improving this crop. Their reliability tested through annotation against the Arabidopsis genome database lends further credence to this. Thus, the results from this study offer significant groundwork for future marker-assisted breeding and mining candidate genes for important agronomic traits in M. pruriens, a promising underutilized legume for the tropics.
Data availability
The datasets generated during and/or analyzed during the current study are available in the supplementary information section.
References
Abdurakhmonov IY, Abdukarimov A (2008) Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int J Plant Genom 2008:574927. https://doi.org/10.1155/2008/574927
Allard RW (1960) Principles of plant breeding. John Wiley and Sonc Inc., New York
Ambreen H, Kumar S, Kumar A, Agarwal M, Jagannath A, Goel S (2018) Association mapping for important agronomic traits in safflower (Carthamus tinctorius L.) core collection using microsatellite markers. Front Plant Sci 9:402. https://doi.org/10.3389/fpls.2018.00402
Bennett EJ, Roberts JA, Wagstaff C (2011) The role of the pod in seed development: strategies for manipulating yield. New Phytol 190:838–853. https://doi.org/10.1111/j.1469-8137.2011.03714.x
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635. https://doi.org/10.1093/bioinformatics/btm308
Capo-chichi LJA, Morton CM, Weaver DB (2004) An intraspecific genetic map of velvet bean (Mucuna sp.) based on AFLP markers. Theor Appl Genet 108:814–821. https://doi.org/10.1007/s00122-003-1493-8
Carsky RJ, Ndikawa R (1998) Identification of cover crops for the semi-arid savannah zone of West Africa. In: Buckles D, Eteka A, Osiname M, Galiba M, Galiano G (eds) Cover crops in West Africa - contributing to sustainable agriculture. International Development Research Centre (IDRC), Ottawa, pp 179–187
Chinapolaiah A, Bindu HH, Khadke GN, Manjesh GN, Rao NH, Kumar SS, Suthar MK (2018) Genetic diversity analysis in underutilized medicinal climber Mucuna pruriens (L.) DC. Germplasm revealed by inter simple sequence repeats markers. Legume Res 43:32–37. https://doi.org/10.18805/LR-3954
Chinapolaiah A, Bindu KH, Manjesh GN, Thondaiman V, Shivakumara KT (2019) Genetic variability, correlation and path analysis for yield and biochemical traits in velvet bean [Mucuna pruriens (L)]. J Pharmacogn Phytochem 8:2698–2704
R Core Team (2014) R: A language and environment for statistical computing. R foundation for statistical computing, Vienna, Austria. http://www.R-project.org/
Daxenbichler ME, Van Etten CH, Earle FR, Tallent WH (1972) L-DOPA recovery from Mucuna seed. J Agric Food Chem 20:1046–1048
Douglas SJ, Li B, Kliebenstein DJ, Nambara E, Riggs CD (2017) A novel filamentous flower mutant suppresses brevipedicellus developmental defects and modulates glucosinolate and auxin levels. PLoS ONE 12:e0177045. https://doi.org/10.1371/journal.pone.0177045
Doyle JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Duke (1981) Handbook of legumes of world economic importance. Plenum, New York, pp 170–184
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conser Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eilitta M, Bressani R, Carew LB, Carsky RJ, Flores M, Gilbert R, Huyck L, St. Laurent L, Szabo NJ (2002) Mucuna as a food and feed crop: an overview. In: Flores BM, Eilitta M, Myhrman R, Carew LB, Carsky RJ (eds) Food and feed from Mucuna: current uses and the Way Forward, Workshop. CIDICCO, CIEPCA and World Hunger Research Center, Tegucigalpa, pp 18–47
Feng J, Wang L, Wu Y, Luo Q, Zhang Y, Qiu D, Han J, Su P, Xiong Z, Chang J, Yang G, He G (2018) TaSnRK2.9, sucrose non-fermenting 1-relatedprotein kinase gene, positively regulates plant response to drought and salt stress in transgenic tobacco. Front Plant Sci 9:2003. https://doi.org/10.3389/fpls.2018.02003
Flint-Garcia SA, Thuillet AC, Yu J, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064. https://doi.org/10.1111/j.1365-313X.2005.02591.x
Fujii Y, Shibuya T, Usami Y (1991) Allelopathic effect of M. pruriens on the appearance of weeds. J Res Weed Sci 36:43–49
Fung SY, Tan NH, Sim SM, Marinello E, Guerranti R, Aguiyi JC (2011) M. Pruriens Linn. Seed extract pretreatment protects against cardiorespiratory and neuromuscular depressant effects of Naja sputatrix (Javan spitting cobra) venom in rats. Indian J Exp Biol 49:254–259
Ganem B (1978) From glucose to aromatics: recent developments in natural products of the shikimic acid pathway. Tetrahedron 34:3353–3383
Grotz N, Fox T, Connolly E, Park W, Guerinot ML, Eide D (1998) Identification of a family of zinc transporter genes from Arabidopsis that respond to zinc deficiency. Proc Natl Acad Sci 95:7220–7224. https://doi.org/10.1073/pnas.95.12.7220
Gurumoorthi P, Kumar SS, Vadivel V, Janardhanan K (2003) Studies on agrobotanical characters of different accessions of velvet bean collected from western ghats, South India. Trop Subtrop Agroecosyst 2:105–115
Hadapad B, Ravi CS, Shivaprasad M, Bindu H, Nadukeri S, Devaraju (2018) Genetic variability and correlation studies for quantitative and qualitative traits in velvet bean (Mucuna pruriensL.) Genotypes in rubber plantation under hill zone of Karnataka. J Pharmacogn Phytochem SP3:83–90
Hamberger B, Hahlbrock K (2004) The 4-Coumarate: CoA ligase gene family in Arabidopsis thaliana comprises one rare, sinapate-activating and three commonly occurring isoenzymes. Proc Natl Acad Sci 101:2209–2214. https://doi.org/10.1073/pnas.0307307101
Hedge JE, Hofreiter BT (1962) Determination of reducing sugars and carbohydrates. In: Whistler RL, BeMiller JN (eds) Methods in carbohydrate chemistry. Academic, New York, pp 380–394
Hira CK, Chopra N (1995) Effects of roasting on protein quality of chickpea (Cicer arietinum) and peanut (Arachis hypogaea). J Food Sci Technol 32:501–503
Hu Z, Zhang D, Zhang G, Kan G, Hong D, Yu D (2014) Association mapping of yield-traits and SSR markers in wild soybean (Glycine soja Sieb. And Zucc). Breed Sci 63:441–449. https://doi.org/10.1270/jsbbs.63.441
Jia C, Wu X, Chen M, Wang Y, Liu X, Gong P, Xu O, Wang X, Gao H, Wang Z (2017) Identification of genetic loci associated with crude protein and mineral concentrations in alfalfa (Medicago sativa) using association mapping. BMC Plant Biol 17:97. https://doi.org/10.1186/s12870-017-1047-x
Jorge M, Eilitta M, Proud F (2007) Mucuna species: recent advances in application of biotechnology. Fruit Veg Cereal Sci Biotech 1:80–94
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK (2011) Microsatellite markers: an overview of the recent progress in plants. https://doi.org/10.1007/s10681-010-0286-9. Euphytica
Kalidass C, Mohan VR (2011) Nutritional and anti-nutritional composition of itching bean [Mucuna pruriens (L.) DC. Var. Pruriens]: an underutilized tribal pulse in western ghats, Tamil Nadu. Trop Subtrop Agroecosyst 14:279–293.
Kalinowski ST (2005) HP-Rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kumar P, Saha S (2013) An updated review on taxonomy, phytochemistry, pharmacology and toxicology of Macuna pruriens. J Pharmacogn Phytochem 2:306–314
Kumar S, Singh GK, Kumar R, Bhatia NK, Awasthi CP (1991) Variation in quality traits of pigeon pea (Cajanus cajan L. Millsp.) Varieties. J Food Sci Technol 28:173–174
Kumar PR, Sundeep S, Sathyanarayana N (2019) Microsatellite analysis reveals low inter-population differentiation in velvet bean (Mucuna pruriens var. utilis) of India. Nucleus 63:35–45. https://doi.org/10.1007/s13237-019-00276-1
Kumwenda JDT, Gilbert RA (1998) Biomass production by legume green manures on exhausted soils in Malawi: A soil fertility network trial. In: Waddington SR, Murwira HK, Kumwenda JDT, Hikwa D and Tagwira F (ed) Soil fertility research for maize-based farming systems in Malawi and Zimbabwe. Proceedings of the soil fertility network results and planning workshop, Mutare, pp 85–86
Leelambika M, Sathyanarayana N (2011) Genetic characterization of Indian Mucuna (Leguminosae) species using morphometric and random amplification of polymorphic DNA (RAPD) approaches. Plant Biosyst 145:786–797. https://doi.org/10.1080/11263504.2011.610924
Leelambika M, Mahesh S, Jaheer M, Sathyanarayana N (2010) Comparative evaluation of genetic diversity among Indian Mucuna species using morphometric, biochemical and molecular approaches. World J Agric Sci 6:568–578
Lehnert H, Serfling A, Friedt W, Ordon F (2018) Genome-wide association studies reveal genomic region associated with response of wheat (wheat (Triticum aestivum L.) to mycorrhizae under drought stress conditions. Front Plant Sci 9:1728. https://doi.org/10.3389/fpls.2018.01728
Lepcha P, Kumar PR, Sathyanarayana N (2019) Exploring genomics research in the context of some underutilized legumes-a review. In: Banerjee R, Kumar GV, Kumar SPJ (eds) OMICS-based approaches in plant biotechnology. Scrivener Publishing, Beverly, pp 1–8. https://doi.org/10.1002/9781119509967.ch1
Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Hao X, Zhang P, Zhao J, Liu Y, Wang G, Li J, Yan J (2012) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45:43–50. https://doi.org/10.1038/ng.2484
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129. https://doi.org/10.1093/bioinformatics/bti282
Liu R, Fang L, Yang T, Zhang X, Hu J, Zhang H, Han W, Hua Z, Hao J, Zong X (2017) Marker-trait association analysis of frost tolerance of 672 worldwide pea (Pisum sativum L.) collections. https://doi.org/10.1038/s41598-017-06222-y. Sci Rep7:5919
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 1:1420–1425. https://doi.org/10.2307/2445869
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with folin phenol reagent. J Biol Chem 193:265–275
Ma L, Liu M, Yan Y, Qing C, Zhang X, Zhang Y, Long Y, Wang L, Pan L, Zou C, Li Z, Wang Y, Peng H, Pan G, Jiang Z, Shen Y (2018) Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front Plant Sci 9:561. https://doi.org/10.3389/fpls.2018.00561
Mahajan R, Zargar SM, Salgotra RK, Singh R, Wani AA, Nazir M, Sofi PA (2017) Linkage disequilibrium based association mapping of micronutrients in common bean (Phaseolus vulgaris L.): a collection of Jammu & Kashmir, India. 3 Biotech 7:295. https://doi.org/10.1007/s13205-017-0928-x
Mahesh S, Sathyanarayana N (2011) The genotype X environment interaction and stability analysis for L-DOPA trait in M. pruriens seeds. Indian J Genet Pl Br 71:279–282
Mahesh S, Sathyanarayana N (2015) Intra-specific variability for salinity tolerance in Indian Mucuna pruriens (L.) DC. Germplasm. J Crop Sci Biotechnol 18:181–194. https://doi.org/10.1007/s12892-015-0019-7
Mahesh S, Leelambika M, Jaheer M, Anithakumari AM, Sathyanarayana N (2016) Genetic mapping and QTL analysis of agronomic traits in Indian Mucuna pruriens using an intraspecific F2 population. J Genet 95:35–44. https://doi.org/10.1007/s12041-015-0602-1
Mang YD, Njintang YN, Abdou BA, Scher J, Bernard C, Mbofung MC (2016) Dehulling reduces toxicity and improves in vivo biological value of proteins in vegetal milk derived from two mucuna (Mucuna pruriens L.) seeds varieties. J Food Sci Technol 53:2548–2557
Mathew IE, Das S, Mahto A, Agarwal P (2016) Three rice NAC transcription factors heteromerize and are associated with seed size. Front Plant Sci 7:1638. https://doi.org/10.3389/fpls.2016.01638
Michelfelder AJ (2009) Soy: a complete source of protein. Am Fam Physician 79:43–47
Miller PA, Rawlings JO (1967) Breakup of initial linkage blocks through intermating in a cotton breeding population. Crop Sci 7:199–204
Milner MJ, Seamon J, Craft E, Kochian LVJ (2013) Transport properties of members of the ZIP family in plants and their role in zn and mn homeostasis. Exp Bot 64:369–381. https://doi.org/10.1093/jxb/ers315
Muralia S, Pathak AK (2003) Database of medicinal plant used in Ayurveda: Medicinal and aromatic plants cultivation and uses. pp 185–187
Negron L, Patchett ML, Parker EJ (2011) Expression, purification, and characterization of dehydroquinatesynthase from Pyrococcus furiosus. Enzyme Res 2011:134893. https://doi.org/10.4061/2011/134893
Oraguzie NC, Wilcox PL (2007) An overview of association mapping. In: Oraguzie NC, Rikkerink EHA, Gardiner SE (eds) Association Mapping in plants. Springer, New York, pp 1–9. https://doi.org/10.1007/978-0-387-36011-9_1
Padmesh P, Reji JV, Dhar JM, Seeni D (2006) Estimation of genetic diversity in varieties of Mucuna pruriens using RAPD. Biol Plant 50:367–372. https://doi.org/10.1007/s10535-006-0051-z
Patil RR, Pawar KD, Rane MR, Yadav SR, Bapat VA, Jadhav JP (2016) Assessment of genetic diversity in Mucuna species of India using randomly amplified polymorphic DNA and inter simple sequence repeat markers. Physiol Mol Biol Plants 22:207–217. https://doi.org/10.1007/s12298-016-0361-3
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Perrier X, Jacquemoud-collet JP (2006) DARwin software, version 5.0.158. Department Systems Biologiques (BIOS), CIRAD, Montpellier
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Pugalenthi M, Vadivel V, Siddhuraju P (2005) Alternative food/feed perspectives of and underutilized legume M. pruriensvar. utilis- a review. Plant Foods Hum Nutr 60:201–218. https://doi.org/10.1007/s11130-005-8620-4
Rakshit A, Rakshit S, Singh J, Chopra SK, Balyan HS, Gupta PK, Bhat S (2010) Association of AFLP and SSR markers with agronomic and fiber quality traits in Gossypium hirsutum L. J Genet 89:155–162. https://doi.org/10.1007/s12041-010-0055-5
Rohilla M, Singh N, Mazumder A, Sen P, Roy P, Chowdhury D, Singh NK, Mondal TK (2020) Genome-wide association studies using 50 K rice genic SNP chip unveil genetic architecture for anaerobic germination of deep-water rice population of Assam, India. Mol Genet Genomics 295:1211–1226. https://doi.org/10.1007/s00438-020-01690-w
Sasidharan N (2004) Biodiversity documentation for Kerala Part 6: flowering plants. Kerala Forest Research Institute (KFRI), Peechi, India
Sastrapradja S, Sastrapradja D, Aminah SH, Lubis I, Idris S (1974) Morphological and cytological investigation on some species of Mucuna (Papilionaceae). Ann Bogor 5:173–178
Sathyanarayana N, Leelambika M, Mahesh S, Jaheer M (2011) AFLP assessment of genetic diversity among Indian Mucuna accessions. Physiol Mol Biol Plants 17:171–180. https://doi.org/10.1007/s12298-011-0058-6
Sathyanarayana N, Mahesh S, Jaheer M, Leelambika M (2012) Genetic diversity of wild and cultivated Mucuna pruriens (L.) DC. Accessions analyzed using thirty morpho-agronomical characters. Trop Subtrop Agroecosyst 15:249–259
Sathyanarayana N, Mahesh S, Leelambika M, Jaheer M, Chopra R, Rashmi KV (2016) Role of genetic resources and molecular markers in M. pruriens (L.) DC. Improvement. Plant Genet Resour 14:270–282. https://doi.org/10.1017/S1479262116000071
Sathyanarayana N, Pittala RK, Tripathi PK, Chopra R, Singh HR, Belamkar V, Bhardwaj PK, Doyle JJ, Egan AN (2017) Transcriptomic resources for the medicinal legume Mucuna pruriens: de novo transcriptome assembly, annotation, identification and validation of EST-SSR markers. BMC Genom 18:409. https://doi.org/10.1186/s12864-017-3780-9
Shetty P, Sharma S, Sathyanarayana N (2015) Exploiting legume EST data bases for the development of gene-derived SSR-markers in medicinal legume Mucuna pruriensL. (DC). Electron J Plant Breed 6:1041–1051
Shi C, Ren Y, Liu L, Wang F, Zhang H, Tian P, Pan T, Wang Y, Jing R, Liu T, Wu F, Lin Q, Lei C, Zhang X, Zhu S, Guo X, Wang J, Zhao Z, Wang J, Zhai H, Cheng Z, Wan J (2019) Ubiquitin specific protease 15 has an important role in regulating grain width and size in rice. Plant Physiol 180:381–391. https://doi.org/10.1104/pp.19.00065
Siddhuraju P, Becker K, Makkar HP (2000) Studies on the nutritional composition and antinutritional factors of three different germplasm seed materials of an under-utilized tropical legume, Mucuna pruriens var. utilis. J Agri Food Chem 48:6048–6060
Singh A, Sharma V, Dikshit K, Aski M, Kumar H, Thirunavukkarasu N, Patil BS, Kumar S, Sarkar A (2017) Association mapping unveils favorable alleles for grain iron and zinc concentrations in lentil (Lens culinaris subsp. Culinaris). PLoS ONE 12:e0188296. https://doi.org/10.1371/journal.pone.0188296
Soumya PR, Burridge AJ, Singh N, Batra R, Pandey R, Kalia S, Rai V, Edwards KJ (2021) Population structure and genome-wide association studies in bread wheat for phosphorus efficiency traits using 35 K wheat breeder’s affymetrix array. Sci Rep 11:7601. https://doi.org/10.1038/s41598-021-87182-2
Syukur M, Sujiprihati S, Yunianti R (2012) Teknik PemuliaanTanaman. PenebarSwadaya, Jakarta
Tiwari DK, Pandey P, Tripathi S, Giri SP, Dwivedi JL (2011) Studies on genetic variability for yield components in rice (Oryza sativa L). AAB Bioflux 3:76–81
Tripathi PK, Jena SN, Rana TS, Sathyanarayana N (2018) High levels of gene flow constraints population structure in Mucuna pruriens L. (DC.) Of Northeast India. Plant Gene 15:6–14. https://doi.org/10.1016/j.plgene.2018.05.005
Vaijayanthi PV, Ramesh S, Gowda MB, Rao AM, Keerthi CM (2018) Genome-wide marker-trait association analysis in a core set of dolichos bean germplasm. Plant Genet Resour 3:1–11. https://doi.org/10.1017/S1479262118000163
Varshney RK, Graner A, Sorrells ME (2005) Genomics-assisted breeding for crop improvement. Trends Plant Sci 10:621–630. https://doi.org/10.1016/j.tplants.2005.10.004
Wang Q, Tian F, Pan Y, Buckler ES, Zhang ZA (2014) A SUPER powerful method for genome wide association study. PLoS ONE 9:e107684. https://doi.org/10.1371/journal.pone.0107684
Wilmot-Dear CM (1987) A revision of Mucuna (Leguminosae Phaseoleae) in the Indian sub-continent and Burma. Kew Bull 42:23–46
Xu Y, Yang T, Zhou Y, Yin S, Li P, Liu J, Xu S, Yang Z, Xu C (2018) Genome-wide association mapping of starch pasting properties in maize using single-locus and multi-locus models. Front Plant Sci 9:1311. https://doi.org/10.3389/fpls.2018.01311
Yang N, Lu Y, Yang X, Huang J, Zhou Y, Ali F, Wen W, Liu J, Li J, Yan J (2014) Genome-wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet 10:e1004573. https://doi.org/10.1371/journal.pgen.1004573
Yeh FC, Yang RC, Boyle T, Ye ZH, Mao JX (1999) POPGENE, version 1.32: the user-friendly software for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton, AB
Yin L, Zhang H, Tang Z, Xu J, Yin D, Zhang Z, Yuan Z, Zhu M, Zhao S, Li X, Liu X (2021) rMVP: a memory-efficient, visualization enhanced, and parallel-accelerated tool for genome-wide association study. Genom Proteom Bioinf. https://doi.org/10.1016/j.gpb.2020.10.007
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160. https://doi.org/10.1016/j.copbio.2006.02.003
Yu J, Pressoir G, Briggs WH, Bi IV, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208. https://doi.org/10.1038/ng1702
Zhao J, Huang L, Ren X, Pandey MK, Wu B, Chen Y, Zhou X, Chen W, Xia Y, Li Z, Luo H, Lei Y, VarshneyRK, Liao B, Jiang H (2017) Genetic variation and association mapping of seed-related traits in cultivated peanut (Arachis hypogaea L.) using single-locus simple sequence repeat markers. Front Plant Sci 8:2105. https://doi.org/10.3389/fpls.2017.02105
Acknowledgements
The authors acknowledge financial supports from (a) Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (b) Department of Biotechnology (DBT), Government of India (c) Ministry of Tribal Affairs (MoTA), Government of India and (d) Vice-Chancellor, Sikkim University.
Funding
This work was supported by the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (Grant Number: No. SR/SO/PS/0028/2011); Department of Biotechnology (DBT), Government of India (Grant Number: No.102/IFD/SAN/5149/2012–2013) to NS and Ministry of Tribal Affairs (MoTA), Government of India fellowship to PL (Grant No. NFST-2015-17-ST-SIK-1633 dated 01/04/2016).
Author information
Authors and Affiliations
Contributions
NS conceptualized the study, design experiment, acquired funding, and supervised the study; PL performed data analysis and wrote the original manuscript; MS, LM, and MJ performed field investigation and compiled data; RC investigated the gene annotation; VB reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest in the publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lepcha, P., Shekhar, M., Murugesan, L. et al. Association mapping of important agronomic traits in Mucuna pruriens (L.) DC.. Bot Stud 65, 26 (2024). https://doi.org/10.1186/s40529-024-00421-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40529-024-00421-3