
Abstract
Background
The tumor microenvironment (TME) provides a region for intricate interactions within or between immune and non-immune cells. We aimed to reveal the tissue architecture and comprehensive landscape of cells within the TME of colorectal cancer (CRC).
Methods
Fresh frozen invasive adenocarcinoma of the large intestine tissue from 10× Genomics Datasets was obtained from BioIVT Asterand. The integration of microarray-based spatial transcriptomics (ST) and RNA sequencing (RNA-seq) was applied to characterize gene expression and cell landscape within the TME of CRC tissue architecture. Multiple R packages and deconvolution algorithms including MCPcounter, XCELL, EPIC, and ESTIMATE methods were performed for further immune distribution analysis.
Results
The subpopulations of immune and non-immune cells within the TME of the CRC tissue architecture were appropriately annotated. According to ST and RNA-seq analyses, a heterogeneous spatial atlas of gene distribution and cell landscape was comprehensively characterized. We distinguished between the cancer and stromal regions of CRC tissues. As expected, epithelial cells were located in the cancerous region, whereas fibroblasts were mainly located in the stroma. In addition, the fibroblasts were further subdivided into two subgroups (F1 and F2) according to the differentially expressed genes (DEGs), which were mainly enriched in pathways including hallmark-oxidative-phosphorylation, hallmark-e2f-targets and hallmark-unfolded-protein-response. Furthermore, the top 5 DEGs, SPP1, CXCL10, APOE, APOC1, and LYZ, were found to be closely related to immunoregulation of the TME, methylation, and survival of CRC patients.
Conclusions
This study characterized the heterogeneous spatial landscape of various cell subtypes within the TME of the tissue architecture. The TME-related roles of fibroblast subsets addressed the potential crosstalk among diverse cells.
Similar content being viewed by others


Background
Colorectal cancer (CRC) is a common malignancy and is the second leading cause of cancer-related mortality worldwide. According to an estimation, more than 1.9 million new CRC cases and 935,000 deaths occurred in 2020 [1]. Among CRC cases, early onset disease (at < 50 years of age) accounts for 10%, and its incidence is increasing, particularly in high-income countries. Screening for CRC is now recommended beginning at 45 years of age [2]. CRC can be characterized by symptoms including changes in bowel habits, abdominal pain, and sometimes blood in the stool [3]. The prognosis of CRC has improved due to advancements in treatment, including surgery, chemotherapy, and radiation therapy. However, tumors often develop resistance to treatment because of intratumoral heterogeneity and clonal evolution. CRC has served as a genetic and biological paradigm for the evolution of solid tumors. Genotyping of tumor tissues according to supportive information provided by somatic genetic alterations has become a routine examination in clinical practice [4, 5].
Tumor microenvironment (TME) denotes the non-cancerous cells and components surrounding the tumor cells, including malignant cells, fibroblasts, tumor vasculature, lymphocytes, dendritic cells, and molecules produced and released by them [6, 7]. TME has contributed to revealing and comprehending the roles of non-genetic and non-cellular intrinsic factors in cancer development [8]. Constant cross-talk between tumor cells and the TME plays decisive roles in tumor proliferation, progression, metastasis, and response to therapies [9]. Accordingly, the TME is now considered a therapeutic target in cancer, attracting increasing research and clinical investigation.
Owing to the technological development of next-generation sequencing- and imaging-based approaches, spatial transcriptomics (ST) can now be used to comprehensively elucidate the expression levels of all or selected genes throughout the tissue space. Biological insights into a range of disease contexts can be achieved by integrating ST and single-cell RNA sequencing (scRNA-seq) [10, 11]. Emerging studies have investigated the spatial heterogeneity in pancreatic ductal adenocarcinoma [12], bone marrow niche organization [13], liver [14], squamous cell carcinoma [15], mammalian spermatogenesis [16], breast cancer [17], lung cancer [18], prostate cancer [19], dorsal root ganglia [20], cervical squamous cell carcinoma [21], esophageal squamous cell carcinoma [22], and colorectal cancer [23]. Although previous studies have focused on the spatial heterogeneity of the TME in colorectal cancer, cancer-associated fibroblasts (CAF) have received much attention because comprehensive insights into diverse subpopulations are insufficient and more supportive data are needed.
Given these considerations, we conducted an analysis to characterize the heterogeneous spatial landscape of various subpopulations within the TME of CRC tissues by integrating ST and RNA-seq. Subsets of fibroblasts were identified to reveal potential crosstalk among diverse cells. This comprehensive landscape and cellular architecture may provide novel insights into the advancement of CRC management in the future.
Methods
Patients and sample collection
In this study, spatial gene expression data were acquired by visiting the 10 × Genomics Datasets (https://www.10xgenomics.com). Spatial imaging data and feature/barcode matrix HDF5 (filtered) documents were investigated from “Space Ranger 1.2.0: Human Colorectal Cancer: Whole Transcriptome Analysis (Visium Spatial Targeted Demonstration (v1 Chemistry))”. Freshly frozen invasive adenocarcinoma of the large intestine tissue was obtained from BioIVT Asterand. The tissue was classified as T4aN0M0 (stage IIB) according to the 8th edition of the staging system issued by the American Joint Committee on Cancer (AJCC) and Union for International Cancer Control (UICC). The available 10 × genomics data used for validation study was 5 µm section from Human Intestinal Cancer named Human Intestine Cancer (FPPE). The FFPE tissue was purchased from BioIVT Asterand Human Tissue Specimens. All data analyzed in this study were available from the aforementioned open-access website; therefore, ethics approval and informed consent were waived by the National Cancer Center in China.
Tissue image preparation
The tissue was embedded and cryosectioned as described in the Visium Spatial Protocols Tissue Preparation Guide (Demonstrated Protocol CG000240). Tissue sections of 10 µm were placed on Visium Gene Expression slides, fixed, and stained following methanol fixation, hematoxylin and eosin (H&E) staining, and imaging for visible spatial protocols (CG000160). H&E images were acquired using a Nikon Eclipse Ti2-E microscope with the following settings: (a) color camera, (b) 10× objective; (c) Numerical Aperture:0.45, and (d) exposure:20 ms. The detailed description of the data process in this section was applied in the user guide (https://cdn.10xgenomics.com/image/upload/v1660261286/support-documents/CG000238_VisiumSpatialTissueOptimizationUserGuide_RevE.pdf).
Gene expression library preparation and sequencing
The Visium Gene Expression library (T1T2-E8) was prepared as described in the Visium Spatial Reagent Kit User Guide (CG000239 Rev D). Sequencing data were processed using Space Ranger. The specific parameters were as follows: (a) sequencing instrument: Illumina NovaSeq 6000, flow cell HHYWHDSXY (lanes 1–4); (b) sequencing depth: 112,228 mean reads per cell; (c) sequencing configuration: paired-end (28 × 90), Dual-Indexed Sequencing. Read 1: 28 cycles (16 bp barcode, 12 bp UMI); i7 index: 10 cycles; i5 index: 10 cycles; Read 2: 90 cycles (transcript); (d) Slide: V10A13-206; (e) Area: C1. The key metrics were as follows: (a) spots detected,3,138; (b) median genes per spot,3,538; (c) Median UMI counts per spot:8,906. The detailed description of the data process in this section was applied in the user guide (https://cdn.10xgenomics.com/image/upload/v1660261286/support-documents/CG000239_Visium_Spatial_Gene_Expression_User_Guide_Rev_F.pdf).
Processing of RNA sequencing data
The RNA-seq data were processed and visualized using the R packages Seurat, ggplot2, cowplot, dplyr, and hdf5r [24]. The SCTransform function was used to normalize the data (assay = “Spatial”). Dimensionality reduction clustering was then performed using RunPCA, RunUMAP, and RunTSNE functions (dims = 1:30). The cell types were annotated according to the previously reported cell type gene markers as follows: EPCAM-Epithelial, PECAM1-Endothelial, COL3A1-Fibroblasts, AIF1-Macrophage, CD79A-B cell, JCHAIN-Plasma cell, CD4-T cell, AKT3 and AXL-NK cells, and PTPRC-Immune cells. Fibroblasts were further extracted and classified into new clusters of c0, c1, c2, c3, c4, and c5, using the FindClusters function (resolution = 0.4). It should be claimed that the new c0-c4 were different from c0-c17 in the beginning of the study. Based on the results of the differential expression analysis, we classified fibroblasts into F1 (c1) and F2 (c0, c2, c3, and c4). The heatmap of differentially expressed genes (DEGs) were visualized using the DoHeatmap function. The FindAllMarkers function was performed to identify the top five DEGs between F1 and F2 clusters (min.pct = 0.25, logfc.threshold = 0.25, test.use = "wilcox"). The R packages irGSEA and UCell were applied to conduct Gene Set Enrichment Analysis (GSEA) of RNA-seq data from patients with colorectal cancer[25]. The gene set enrichment score was calculated using the irGSEA score function (assay = “Spatial”, seeds = 123, ncores = 1, min.cells = 3, min.feature = 0, msigdb = T, species = “Homo sapiens”, category = “H”, kcdf = “Gaussian”).
Spatial transctiptomic analysis
The R packages Seurat, dplyr, and hdf5r were used for spatial transcriptomic (ST) analysis [24]. Tissue images were loaded using the Read10X_h5 function of Seurat. We visualized the count and features of gene expression using the VlnPlot function of R packages ggplot2 and cowplot. The structure of the tissue section was divided into cancer and stromal regions after being read by pathology experts. The spatial locations of epithelial cells, plasma cells, fibroblasts, B cells, T cells, endothelial cells, and NK cells were analyzed and visualized using the DimPlot function. We conducted ST analysis based on the method of multimodal intersection, which made a combination of gene expression modal and spatial architecture modal of colorectal cancer tissue sections. After identifying the top five DEGs (tDEGs), the spatial location of expression of these genes was determined using the SpatialFeaturePlot function (alpha = c (0.5, 1)). In addition, the immunohistochemical (IHC) and fluorescent staining images of these genes, which were available in the Human Protein Atlas (HPA) database (https://www.proteinatlas.org/), were also displayed to demonstrate the intracellular expression sites of these genes.
Immunological, methylation, function enrichment and survival related analyses
In this section, mRNA transcriptomic data of colorectal cancer were investigated from cohorts within The Cancer Genome Atlas (TCGA) database. The standardized pan-cancer dataset TCGA TARGET GTEx (PANCAN, N = 19,131, G = 60,499) was downloaded from the UCSC (https://xenabrowser.net/). Gene expression data for colon adenocarcinoma/rectum adenocarcinoma esophageal carcinoma (TCGA-COAD/READ) were selected for subsequent analysis. Furthermore, we extracted the expression data of the tDEGs in each sample and transformed each expression value into log2 (x + 0.001). The mcpcounter, XCELL, EPIC, and ESTIMATE methods in the R package IOBR was used to estimate the population assumption of tumor-infiltrating immune and spatial cell populations [26]. Additionally, gene markers of immunoregulatory pathways, including chemokines, receptors, MHC, immunoinhibitors, and immunostimulators, were extracted to calculate Pearson’s correlation with tDEGs. We performed a similar correlation analysis using the expression values of the tDEGs and RNA-moderated genes (m1A, m5C, and m6A). Mutation data (MuTect2) were processed and visualized using the R packages, maftools and ComplexHeatmap [27, 28]. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted using the R packages clusterProfilerorg and org.Hs.eg.db [29]. The least absolute shrinkage and selection operator (LASSO) regression algorithm and multivariate Cox regression analysis were used to construct a prognostic model of survival using the R package glmnet [30]. The log-rank test was used to compare differences in survival between groups. Time-ROC analysis was used to compare the predictive accuracy of the risk score. The Spearman correlation between the model and immune cells was analyzed using the R package ggstatsplot.
Statistical analysis
All analyses in this study were conducted using R software for Windows. In the immune infiltrating analysis, we extracted the expression data of the tDEGs in each sample and transformed each expression value into log2 (x + 0.001) after filtering all normal samples. Then, Spearman’s correlation was calculated in each subgroup [31]. In the similar method, Pearson correlation was performed for methylation and immune regulation analysis [32]. The chi square test was utilized to evaluate the difference in gene mutation frequency in each group of samples [33]. Considering the survival analysis, we took the first step to convert counts data to TPM and normalize the data log2 (TPM + 1), keeping samples with clinical information at the same time. For Kaplan–Meier curves, p-values and hazard ratio (HR) with 95% confidence interval (CI) were generated by log-rank tests and univariate cox proportional hazards regression, which were used to compare differences in survival between the groups [34]. The timeROC analysis was conducted to further compare the predictive accuracy. LASSO with tenfold cross-validation was used for feature selection [35]. Statistical analyses with two sides were performed, and all the difference with P < 0.05 was considered statistically significant.
Results
Analysis of identity and density of gene expression distribution
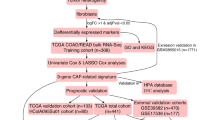
The design of this study is briefly summarized and illustrated in Fig. 1A. After the RNA-seq profiling data, 18 clusters (cluster 0 to cluster 17) were identified automatically by Seurat. The gene expression and gene number of these clusters showed a wide range of changes according to the violin plots (Fig. 1B, D). We further explored the spatial location of the density of gene expression distribution. As shown, there were more genes enriched in cancer regions with relatively high expression compared with stromal regions (Fig. 1C, E). Additionally, the mitochondrial gene was not detected in the tissue, which indicated the high quality of the RNA-seq data (Fig. S1).

Flow chart and pre-analysis of colorectal cancer (CRC). A The simple flow chart of study design. B, C Expression in various identities and spatial architecture. D, E Gene count in various identities and spatial architecture
Annotation of celltypes within RNA-seq
In this section, UMAP is applied to demonstrate 18 clusters (Fig. 2A). The spatial location of these clusters was well marked and provided a comprehensive display of the colorectal cancer tissue (Fig. 2B). According to the gene markers for certain cell clusters reported previously, we annotated the cell groups based on the expression of marker genes (MGs) as follows: EPCAM-Epithelial, PECAM1-Endothelial, COL3A1-Fibroblasts, AIF1-Macrophage, CD79A-B cells, JCHAIN-Plasma cells, CD4-T cells, AKT3 and AXL-NK cells, and PTPRC-Immune cells. Subsequently, we revealed the TSNE map of the corresponding cell types and genes in proper order (Fig. 2C–L). A bubble plot was then used to demonstrate the average expression and expressed percentage of MGs in clusters of 0 to 17 (Fig. 2M). The level of MGs in these clusters was also displayed using a violin plot (Fig. S2). The top three most prominently expressed genes in each cluster were investigated and displayed using a heatmap plot. From this analysis, we could clearly distinguish the various gene modules expressed in different clusters (Fig. 2N).

Annotation of celltypes within RNA-seq. A UMAP visualization of RNA-seq. B Spatial location of identities within CRC. C–L TSNE plots of diverse gene markers. M Average expression levels of gene markers in identities. N Top 3 genes with significantly different expression in each identity
The integration of rank-based gene set enrichment analysis
Pathology experts were consulted to determine the cancerous and stromal regions of the tissue. As shown in Fig. 3A, the relatively dark areas surrounded by red dotted lines were identified as regions of colon cancer, whereas the lighter areas without red dotted lines were stromal regions. The distribution of annotated cell clusters was represented comprehensively, including epithelial cells, plasma cells, fibroblasts, B cells, T cells, endothelial cells, and NK cells within the tissue architecture (Fig. 3B). In combination with tissue imaging, a large number of epithelial cells were located in the cancer region, while fibroblasts were mainly located in the stroma, which was in line with our expectations. We then determined the DEGs with increased or decreased expression or no significant difference within the aforementioned clusters using AUCell, UCell, singscore, and ssgsea (Fig. 3C). Furthermore, we investigated the correlation between these clusters and the inflammatory response using UCell. The density distribution of UCell within the aforementioned clusters is shown in Fig. 3D. In addition, PCA, tSEN, and UMAP methods were applied to show the results of the dimension reduction analysis (Fig. 3E–G). The UCell scores of hallmark-inflammatory-response in these clusters were calculated and are shown in Fig. 3H, I. Subsequently, we explored the correlation between clusters and a variety of HALLMARK pathways using the robust rank aggregation (RRA) method, which can comprehensively evaluate the results of difference analysis and screen out gene sets that are significantly enriched in most gene set enrichment analysis methods. According to the results, the top 3 relevant pathways were hallmark-oxidative-phosphorylation, hallmark-e2f-targets and hallmark-unfolded-protein-response (Fig. 3J).

The integration of rank-based gene set enrichment analysis. A Distribution of cancer and stromal regions. B Spatial architecture of celltypes within CRC. C Analysis of genes with significantly different expression. D UCell’s distribution map of celltypes in inflammatory pathway. E–G PCA, tSEN and UMAP maps of inflammatory pathway. H, I Ucell score maps of inflammatory pathway. J Correlation analysis of pathway in clusters
Analysis of fibroblasts subpopulation in CRC tissue
Fibroblasts are important components of RNA sequencing results. The UMAP plot is shown and the fibroblasts are circled by a dotted line (Fig. 4A). From the visualization of the PCA and UMAP results, fibroblasts were classified into subpopulations F1 and F2 (Fig. 4B, C). To verify if immune cells were mixed in the F1 and F2, we demonstrated the expression of the gene makers of each cell type in extracted fibroblasts (c0-c4), indicating no mixture of immune cells was involved (Fig S3). We then explored the spatial location of the two fibroblast subtypes F1 and F2 within CRC tissue. The F1 subtype was mainly located in the stromal region, whereas a portion of the F2 subtype was located in the cancer region (Fig. 4D). To investigate the differences and heterogeneity in biological genetics, we analyzed differentially expressed genes (DEGs). According to the results of DEGs, there were five top genes with the most differential expression between F1 and F2 after weighted analysis, with the top five differentially expressed genes (tDEGs) identified, including SPP1, CXCL10, APOE, APOC1, and LYZ (Fig. 4E). The expression of tDEGs in the 18 identities mentioned above was visualized (Fig. S4A–E). tDEGs were enriched in identity 13 (Fig S4F). We further showed the spatial landscapes of the expression of these five genes and compared them with those of the original CRC tissue. We found that the tDEGs were mainly expressed in the stromal region of the tissue (Fig. 4F–K). Additionally, the UMAP plots of tDEGs are shown (Fig. 4L–P). To further validate these findings, we applied another sample of large intestine CRC named Human Intestine Cancer (FPPE) from 10 × genomics database. The information of CRC samples and datasets used in the study was summarized in Table S1. The gene expression and gene number of the clusters of validation sample showed a wide range of changes according to the violin plots (Fig. S5A, C). We further explored the spatial location of the density of gene expression distribution. As shown, there were more genes enriched in cancer regions with relatively high expression compared with stromal regions (Fig. S5B, D). In further study of validation, we demonstrated the spatial distribution of tDEGs expression. It revealed that tDEGs were mainly enriched in the stromal area of the CRC tissue, which was consistent with previous findings (Fig. S5E–J).

Analysis of fibroblasts subpopulation in CRC tissue. A Fibroblasts in UMAP visualization. B, C PCA and UMAP of fibroblasts clustered into F1 and F2. D The spatial location of F1 and F2. E Top five differentially expressed genes (tDEGs) identified by differential analysis F–J Spatial expression of SPP1, CXCL10, APOE, APOC1 and LYZ. K Original CRC tissue. L–P UMAP plots of SPP1, CXCL10, APOE, APOC1 and LYZ
Immunoassay and methylation analyses of tDEGs
Immune infiltration analysis was performed to investigate the relationship between the tDEGs and various types of immune cells. Based on the COAD and COADREAD cohorts, we found that tDEGs were closely related to the immune infiltration of the tumor microenvironment (TME) within CRC (all P < 0.05). In addition to the fibroblasts, tDEGs were mainly related to cytotoxic lymphocytes and monocytic lineage according to the results (all P < 0.05) (Fig. 5A). Additionally, 150 gene markers of immunoregulatory pathways, including chemokines (41 gene markers), receptors (18 gene markers), MHC (21 gene markers), immunoinhibitors (24 gene markers), and immunostimulators (46 gene markers) were extracted to calculate the Pearson correlation with the tDEGs. The results showed that tDEGs play important roles in the immunoregulatory pathways of the TME in CRC. There was a strong correlation between tDEGs and immunoregulation of the TME (all P < 0.05) (Fig. 5B). Considering the important regulatory role of methylation on cell function and pathways, we extracted the expression of tDEGs and gene markers of three types of RNA modification methods, including m1A (10 gene markers), m5C (13 gene markers), and m6A (21 gene markers). We further classified the gene markers of methylation into writers, readers, and erasers, according to their functions. The correlation between tDEGs and methylation regulation was comprehensively demonstrated in this analysis (Fig. 5C). Then, Xcell, EPIC, and ESTIMATE methods were performed to investigate immune infiltration. All these methods revealed that tDEGs were closely related to the TME of colorectal cancer tissue architecture. The detail data was provided as Tables S1, S2, S3.

Immunoassay and methylation analyses of tDEGs. A Correlation analysis between tDEGs and TME components. B Correlation analysis between tDEGs and regulatory factors of TME. C Correlation analysis between tDEGs and modification including m1A, m5C and m6A
Function enrichment analysis of the DEGs between F1 and F2
Gene Ontology (GO) analysis was conducted to elucidate the roles of the DEGs in cellular components (CC), molecular functions (MF), and biological processes (BP). According to the analysis, DEGs were mainly involved in extracellular structure organization of BP, collagen-containing extracellular matrix of CC, and extracellular matrix structural constituents of MF (Fig. 6A). Gene Set Variation Analysis (GSVA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis were performed. DEGs were mainly enriched in antigen processing and presentation, Epstein-Barr virus, phagosome, human T-cell leukemia virus 1 infection, and metabolic pathways (Figs. 6B, C). The results of the GO and KEGG analyses are summarized in Table S2. Furthermore, we integrated the results of functional enrichment analysis and visualized the connection between them in one plot (Fig. 6D).

Function enrichment analysis of the DEGs between F1 and F2. A Gene Ontology (GO) analysis of the DEGs. B, C Gene Set Variation Analysis (GSVA) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis. D Connection plot of function enrichment analysis
Potential survival prediction module and immunologic correlation analysis
To investigate the impact of tDEGs on the survival of patients with CRC, we established a risk score based on the expression of tDEGs using the Least Absolute Shrinkage and Selection Operator (LASSO). The overall survival (OS) trend that changed with the increase in the risk score is shown (Fig. 7A). The Kaplan–Meier survival analysis of CRC patients with high- and low-risk scores was performed, indicating that the OS of patients with high-risk scores was poorer (HR = 1.896, 95%CI:1.266–2.839, P < 0.01) (Fig. 7B). Then, a receiver operating characteristic (ROC) curve was established to evaluate the accuracy and specificity of the prediction for 1-year, 3-year, and 5-year OS (Fig. 7C). In addition, the trends of progression-free survival (PFS) and disease-specific survival (DSS) change with risk score (PFS panel and DSS panel) were also demonstrated (Fig. S6A, D). Similar Kaplan–Meier and ROC curves for PFS and DSS were obtained (PFS: HR = 2.385, 95%CI:1.637–3.477, P < 0.01; DSS: HR = 2.717, 95%CI:1.575–4.687, P < 0.01) (Fig. S6B, C; E–F). Furthermore, we investigated the relationship between genes in the OS panel (gene panel: APOE, CXCL10, and SPP1) and eight gene markers related to immune checkpoints. It showed that CRC patients with low OS-panel expression were more correlated with immune checkpoints (Fig. 7D–F). The waterfall diagram of tumor mutational burden (TMB) was used to explore the heterogeneity of genes within the OS panel (Fig. S7A–C). Spearman analysis was applied to explore the correlation between the OS panel and immune infiltration within the TME, including B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and myeloid dendritic cells. Consequently, there was a significant positive correlation between the OS panel and CD4+ T cells (P = 4.9e−05, Spearman = 0.19, 95%CI: [0.10, 0.28]), macrophages (P = 7.41e−11, Spearman = 0.30, 95%CI: [0.21, 0.38]), and myeloid dendritic cells (P = 9e−3, Spearman = 0.12, 95%CI: [0.03, 0.21]), while B cells had a significant negative correlation (P = 3e−2, Spearman = − 0.10, 95%CI: [− 0.19, − 0.01]) (Fig. 7G–L).

Potential survival prediction module and immunologic correlation analysis. A Correlation plot of risk score and OS-panel. B Kaplan–Meier survival analysis of OS-panel. C ROC curve of OS-panel. D–F Heatmaps of correlation between immune check points and APOE, CXCL10 and SPP1. G–L Spearman analyses of correlation between OS-panel and B cell, T cell CD4+, T cell CD8+, neutrophil, macrophage and myeloid dendritic cell. *P < 0.05, **P < 0.01, **P < 0.001, ***P < 0.0001
Histocyte level analysis of tDEGs
For a deeper exploration of tDEGs, we searched the Human Protein Atlas (HPA) database (https://www.proteinatlas.org) for histological and subcellular-level information which was available. Immunohistochemical staining images demonstrated the spatial location of SPP1, APOE, and LYZ in CRC tissue (Fig. 8A–C). According to the HPA database, SPP1 was detected in the Golgi apparatus and predicted to be secreted (Fig. 8D). The cell atlas showing fluorescent staining of subcellular structures demonstrated the spatial distribution of the SPP1 protein, nucleus, and microtubules (Fig. 8E). APOE was detected in vesicles and predicted to be secreted (Fig. 8F). Fluorescent staining of the APOE protein, nucleus, and microtubules is shown (Fig. 8G). LYZ was detected in the Golgi apparatus, actin filaments, and nucleoplasms (Fig. 8H). Subcellular fluorescent staining also demonstrated the distribution characteristics of the LYZ protein and microtubules (Fig. 8I).

Histocyte level analysis of tDEGs. A–C Immunohistochemical staining (IHC) of SPP1, APOE and LYZ. D–E Intracellular distribution and fluorescence staining of SPP1. F–G Intracellular distribution and fluorescence staining of APOE. H–I Intracellular distribution and fluorescence staining of LYZ
Discussions
In this study, we revealed the tissue architecture of stage IIB CRC and thoroughly characterized the heterogeneous spatial landscape of diverse subpopulations within the TME by integrating ST and RNA-seq. The expression landscape of all or selected genes at various spatial locations in the cancer and stromal regions, obtained through consultation with pathologists, was visualized. The identification of different fibroblast subtypes (F1 and F2) may address the potential interactions within the TME underlying CRC proliferation, progression, and metastasis. Immunoassay, methylation, and functional enrichment analyses of tDEGs further characterized the biological heterogeneity between F1 and F2, elucidating the roles of tDEGs in the TME. These findings reveal the tissue architecture and provide novel insights into the management of CRC.
Carcinomas are intricate heterocellular structures comprising epithelial cancer cells, stromal fibroblasts, and diverse immune cell populations. Interactions between these TME and cells facilitate cancer progression and influence the effectiveness of the existing therapies. The TME contributes to systemic inflammation, increases oxidative stress and fibrosis, and affects the cachectic state of CRC patients through inflammatory factors, including tumor necrosis factor alpha (TNFα) and certain chemokines such as interleukin IL-1 and IL-6 [36, 37]. In this study, we annotated epithelial cells and related cells within the TME and illustrated the spatial distribution of diverse cell types. Our analysis revealed that the TME in CRC tissues is predominantly located in the stromal region. An important aspect of the interaction between various components is the inflammatory response pathway that is activated within the TME. We investigated the intricate interplay between epithelial cells and various components of the TME and the inflammatory response pathways. A significant number of genes exhibited differential expressions in response to these factors. According to the analysis of rank-based gene set enrichment, several genes that showed differential expression in various components were functionally enriched in the oxidative phosphorylation pathway. It has been shown that inhibition of oxidative phosphorylation (OXPHOS) can resist the hypoxic state in TME, thereby reducing the inhibitory effect on immune effector cells, increasing oxygenation of hypoxic tumor areas and reactivating the immune response. The prognostic improvement of OXPHOS inhibitors has been observed in cancer immunotherapy and radiation therapy [38]. Meanwhile, a previous study investigated the immune subtypes (C1–C6) of CRC and identified novel TME profiles. Among these, C2 exhibits greater activation of pathways associated with immune system function, apoptosis, DNA repair, mTOR signaling, and oxidative phosphorylation [39]. These findings contribute to our understanding of the interplay between various pathways and components within the TME of CRC.
To investigate the transformation of stromal cells in the TME by CRC cells, a study conducted single-cell sequencing and discovered that somatic cell copy number alterations (SCNAs) are widespread in immune cells, fibroblasts, and endothelial cells in both TME and normal tissues within each individual. Moreover, the percentage of fibroblasts with SCNAs was considerably greater in tumors (11.1–47.7%) than in adjacent normal tissues (1.1–10.6%) [40]. This led to our interest in fibroblasts in the TME of CRC. Fibroblasts can be divided into two parts, F1 and F2, according to the FindClusters function. Analysis of spatial transcriptomics revealed that F1 was mainly expressed in the stroma, whereas a portion of F2 was expressed in the cancer region of CRC. We identified a large number of genes that exhibited significantly different expression levels between F1 and F2 plants. Among these genes, SPP1, APOE, CXCL10, APOC1, and LYZ had the highest weights in the weighted analysis and were the top 5 differentially expressed genes (tDEGs). Spatial transcriptomics was performed to reveal the genetic and spatial heterogeneity of these two groups, demonstrating that tDEGs were mainly expressed in the stromal region of CRC tissue. An emerging study revealed a positive correlation between tumor-specific FAP (+) fibroblasts and SPP1 (+) macrophages, which were tightly localized, as demonstrated by immunofluorescence staining and spatial transcriptomics [41]. This interaction may be regulated by TGF-β and interleukin-1, which promote the formation of bridging protein structures that facilitate immunological rejection and restrict T-cell infiltration. Interestingly, they observed that patients with high FAP or SPP1 expression levels exhibited limited therapeutic benefits from anti-PD-L1 therapy. These findings suggest that disrupting the interaction between FAP (+) fibroblasts and SPP1 (+) macrophages may represent a promising therapeutic strategy to improve immunotherapy. APOE was found to be associated with lipid homeostasis and inflammation in the normal brain and could be a therapeutic target for Alzheimer's disease [42, 43]. According to previous studies, CXCL10 is a novel therapeutic target for autoimmune diseases including inflammatory intestinal disease, multiple sclerosis, and rheumatoid arthritis [44]. In addition, CXCL10 is involved in the metastasis of colon cancer through activation of the PI3K/Akt pathway by CXCR3, leading to inhibition of GSK-3β phosphorylation and upregulation of Snail expression, thereby regulating epithelial mesenchymal transition in colon cancer cells [45]. APOC1 is considered a biomarker that indicates the prognosis of various cancer types [46,47,48,49]. A series of studies have revealed an association between LYZ and diseases, including temporal lobe epilepsy and ulcerative colitis [50, 51].
RNA methylation modification is a key mechanism of epigenetic regulation in the immune response and tumorigenesis, and research on RNA methylation has become a hot topic in recent years. The four major RNA adenosine modifications include m(6)A, m(1)A, alternative polyadenylation, and adenosine-to-inosine RNA editing [52]. In this study, we examined the correlation between tDEGs and RNA methylation modifications including m1A, m5C, and m6A. tDEGs were shown to be strongly associated with changes in RNA methylation, suggesting a crucial role in regulation within the TME of CRC. Furthermore, we present landscapes that illustrate the immunomodulatory regulation and components within the TME, demonstrating the intricate immune interactions between tDEGs and the TME. Survival analysis, immunohistochemistry, and fluorescence staining further enriched the clinical translational significance and basic researching insight of the tDEGs and provided a deeper understanding at the molecular level. In the survival analysis, the receiver operating characteristic (ROC) value was observed to be lower than 0.7, indicating the predictive performance was not strong, nevertheless, this was one attempt at the clinical translational application of tDEGs and predicting survival status was not the key point of the study. In the Kaplan–Meier analysis, it revealed a significant different survival of High- and Low-score groups with P < 0.002, demonstrating a strong performance in predicting long-term survival. In general, more translational applications based on tDEGs in various areas are warranted in the feature.
Despite the use of relatively advanced ST analysis techniques in this study, which integrated multiple analytical methods to illustrate the spatial architecture of CRC and reveal the potential interactions of several TME components, there are still some limitations. First, due to database resource limitations, the sample included in this study was small, which may have led to an underrepresentation of the study. We believe that more patients will be enrolled in the study based on these findings. Second, immune-related cellular interactions within the TME were derived by cell sequencing, thus obtaining expression for correlation analysis as well as pathway analysis, lacking direct demonstration from pathway experiments. Third, although ST arrays can provide spatial transcriptomic data, their coverage and resolution are limited. Owing to the size of the ST array, it may not be possible to cover the entire tissue of interest. Additionally, the resolution may vary around single-cell level (1–10 cell) across different spots on the array. Furthermore, transcriptomic data are only accessible within each spot and information may be lost between adjacent spots. However, the development of ST technology offers the potential for higher resolutions and shorter intervals between spots in the future. This may enable researchers to capture the transcriptional profiles of individual cells within complex tissues more accurately.
Conclusions
This study characterized the tissue architecture of CRC by integrating RNA-seq and ST analyses, demonstrating spatial heterogeneity and potential cross-talk within the TME. Various landscapes have revealed a strong association between tDEGs, immunological regulation, and RNA methylation modification. The findings showing a comprehensive tissue structure and cellular landscape provide novel insights into the mechanism of CRC progression and the discovery of novel therapeutic targets.
Availability of data and materials
The datasets used in the current study are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Sinicrope FA. Increasing incidence of early-onset colorectal cancer. N Engl J Med. 2022;386:1547–58.
Jin J. Screening for colorectal cancer. JAMA. 2021;325:2026–2026.
Osumi H, Shinozaki E, Yamaguchi K, Zembutsu H. Clinical utility of circulating tumor DNA for colorectal cancer. Cancer Sci. 2019;110:1148–55.
Li J, Ma X, Chakravarti D, Shalapour S, DePinho RA. Genetic and biological hallmarks of colorectal cancer. Genes Dev. 2021;35:787–820.
Arneth B. Tumor microenvironment. Medicina (Kaunas, Lithuania) 2019; 56.
Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221: 107753.
Laplane L, Duluc D, Bikfalvi A, Larmonier N, Pradeu T. Beyond the tumour microenvironment. Int J Cancer. 2019;145:2611–8.
Bilotta MT, Antignani A, Fitzgerald DJ. Managing the TME to improve the efficacy of cancer therapy. Front Immunol. 2022;13: 954992.
Rao A, Barkley D, França GS, Yanai I. Exploring tissue architecture using spatial transcriptomics. Nature. 2021;596:211–20.
Longo SK, Guo MG, Ji AL, Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet. 2021;22:627–44.
Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, Hajdu CH, Simeone DM, Yanai I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. 2020;38:333–42.
Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, Nombela-Arrieta C, Steinmetz LM, Trumpp A, Haas S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22:38–48.
Saviano A, Henderson NC, Baumert TF. Single-cell genomics and spatial transcriptomics: discovery of novel cell states and cellular interactions in liver physiology and disease biology. J Hepatol. 2020;73:1219–30.
Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstråhle J, Larsson L, Bai Y, Zhu B, Bhaduri A, Meyers JM, Rovira-Clavé X, Hollmig ST, Aasi SZ, Nolan GP, Lundeberg J, Khavari PA. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell. 2020;182:497-514.e22.
Chen H, Murray E, Sinha A, Laumas A, Li J, Lesman D, Nie X, Hotaling J, Guo J, Cairns BR, Macosko EZ, Cheng CY, Chen F. Dissecting mammalian spermatogenesis using spatial transcriptomics. Cell Rep. 2021;37: 109915.
Andersson A, Larsson L, Stenbeck L, Salmén F, Ehinger A, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Borg Å, Frisén J, Engblom C, Lundeberg J. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun. 2021;12:6012.
Larroquette M, Guegan JP, Besse B, Cousin S, Brunet M, Le Moulec S, Le Loarer F, Rey C, Soria JC, Barlesi F, Bessede A, Scoazec JY, Soubeyran I, Italiano A. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J Immunothera Cancer. 2022;10:e003890.
Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstråhle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L, Salmén F, Ogris C, Wallenborg K, Lagergren J, Ståhl P, Sonnhammer E, Helleday T, Lundeberg J. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun. 2018;9:2419.
Tavares-Ferreira D, Shiers S, Ray PR, Wangzhou A, Jeevakumar V, Sankaranarayanan I, Cervantes AM, Reese JC, Chamessian A, Copits BA, Dougherty PM, Gereau RWT, Burton MD, Dussor G, Price TJ. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med. 2022;14:e8186.
Ou Z, Lin S, Qiu J, Ding W, Ren P, Chen D, Wang J, Tong Y, Wu D, Chen A, Deng Y, Cheng M, Peng T, Lu H, Yang H, Wang J, Jin X, Ma D, Xu X, Wang Y, Li J, Wu P. Single-nucleus RNA sequencing and spatial transcriptomics reveal the immunological microenvironment of cervical squamous cell carcinoma. Adv Sci. 2022;9:e2203040.
Guo W, Zhou B, Yang Z, Liu X, Huai Q, Guo L, Xue X, Tan F, Li Y, Xue Q, Gao S, He J. Integrating microarray-based spatial transcriptomics and single-cell RNA-sequencing reveals tissue architecture in esophageal squamous cell carcinoma. EBioMedicine. 2022;84: 104281.
Peng Z, Ye M, Ding H, Feng Z, Hu K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J Transl Med. 2022;20:302.
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573-3587.e29.
Andreatta M, Carmona SJ. UCell: robust and scalable single-cell gene signature scoring. Comput Struct Biotechnol J. 2021;19:3796–8.
Zeng D, Ye Z, Shen R, Yu G, Wu J, Xiong Y, Zhou R, Qiu W, Huang N, Sun L, Li X, Bin J, Liao Y, Shi M, Liao W. IOBR: multi-omics immuno-oncology biological research to decode tumor microenvironment and signatures. Front Immunol. 2021;12: 687975.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (Oxford, England). 2016;32:2847–9.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Engebretsen S, Bohlin J. Statistical predictions with glmnet. Clin Epigenet. 2019;11:123.
Sedgwick P. Spearman’s rank correlation coefficient. BMJ (Clin Res Ed). 2014;349: g7327.
Amdisen A. Pearson’s correlation coefficient, p-value, and lithium therapy. Biol Psychiat. 1987;22:926–8.
Cohen J. A power primer. Psychol Bull. 1992;112:155–9.
Ranstam J, Cook JA. Kaplan–Meier curve. Br J Surg. 2017;104:442.
Kang J, Choi YJ, Kim IK, Lee HS, Kim H, Baik SH, Kim NK, Lee KY. LASSO-based machine learning algorithm for prediction of lymph node metastasis in T1 colorectal cancer. Cancer Res Treat. 2021;53:773–83.
AlMusawi S, Ahmed M, Nateri AS. Understanding cell–cell communication and signaling in the colorectal cancer microenvironment. Clin Transl Med. 2021;11: e308.
Kasprzak A. The role of tumor microenvironment cells in colorectal cancer (CRC) cachexia. Int J Mol Sci. 2021;22.
Boreel DF, Span PN, Heskamp S, Adema GJ, Bussink J. Targeting oxidative phosphorylation to increase the efficacy of radio- and immune-combination therapy. Clin Cancer Res. 2021;27:2970–8.
Soldevilla B, Carretero-Puche C, Gomez-Lopez G, Al-Shahrour F, Riesco MC, Gil-Calderon B, Alvarez-Vallina L, Espinosa-Olarte P, Gomez-Esteves G, Rubio-Cuesta B, Sarmentero J, La Salvia A, Garcia-Carbonero R. The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur J Cancer. 2019;123:118–29.
Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, Guo L, Fu W, Tang F. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell. 2020;38:818-828.e5.
Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, Ding X, Bao R, Hong L, Jia W, Fang F, Liu H, Chen L, Zhong J, Zou D, Liu L, Han L, Ginhoux F, Liu Y, Ye Y, Su B. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat Commun. 2022;13:1742.
Lanfranco MF, Ng CA, Rebeck GW. ApoE lipidation as a therapeutic target in Alzheimer’s disease. Int J Mol Sci. 2020;21:6336.
Rebeck GW. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res. 2017;58:1493–9.
Karin N, Razon H. Chemokines beyond chemo-attraction: CXCL10 and its significant role in cancer and autoimmunity. Cytokine. 2018;109:24–8.
Wang Z, Ao X, Shen Z, Ao L, Wu X, Pu C, Guo W, Xing W, He M, Yuan H, Yu J, Li L, Xu X. TNF-α augments CXCL10/CXCR3 axis activity to induce epithelial–mesenchymal transition in colon cancer cell. Int J Biol Sci. 2021;17:2683–702.
Ren H, Chen Z, Yang L, Xiong W, Yang H, Xu K, Zhai E, Ding L, He Y, Song X. Apolipoprotein C1 (APOC1) promotes tumor progression via MAPK signaling pathways in colorectal cancer. Cancer Manage Res. 2019;11:4917–30.
Guo Q, Liu XL, Jiang N, Zhang WJ, Guo SW, Yang H, Ji YM, Zhou J, Guo JL, Zhang J, Liu HS. Decreased APOC1 expression inhibited cancer progression and was associated with better prognosis and immune microenvironment in esophageal cancer. Am J Cancer Res. 2022;12:4904–29.
Hao X, Zheng Z, Liu H, Zhang Y, Kang J, Kong X, Rong D, Sun G, Sun G, Liu L, Yu H, Tang W, Wang X. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 2022;56: 102463.
Ren L, Yi J, Yang Y, Li W, Zheng X, Liu J, Li S, Yang H, Zhang Y, Ge B, Zhang S, Fu W, Dong D, Du G, Wang X, Wang J. Systematic pan-cancer analysis identifies APOC1 as an immunological biomarker which regulates macrophage polarization and promotes tumor metastasis. Pharmacol Res. 2022;183: 106376.
Chen QL, Xia L, Zhong SP, Wang Q, Ding J, Wang X. Bioinformatic analysis identifies key transcriptome signatures in temporal lobe epilepsy. CNS Neurosci Ther. 2020;26:1266–77.
Mei F, Meng K, Gu Z, Yun Y, Zhang W, Zhang C, Zhong Q, Pan F, Shen X, Xia G, Chen H. Arecanut (Areca catechu L.) seed polyphenol-ameliorated osteoporosis by altering gut microbiome via LYZ and the immune system in estrogen-deficient rats. J Agric Food Chem. 2021;69:246–58.
Chen H, Yao J, Bao R, Dong Y, Zhang T, Du Y, Wang G, Ni D, Xun Z, Niu X, Ye Y, Li HB. Cross-talk of four types of RNA modification writers defines tumor microenvironment and pharmacogenomic landscape in colorectal cancer. Mol Cancer. 2021;20:29.
Acknowledgements
Not applicable.
Funding
The National Key R&D Program of China (2018YFC1312100).
Author information
Authors and Affiliations
Contributions
(1) Guarantor of study integrity: Dongbing Zhao. (2) Study concept and design: Zheng Li, Xiaojie Zhang, and Dongbing Zhao. (3) Provision of study materials or patients: Zheng Li, Xiaojie Zhang, Chongyuan Sun, He Fei, Zefeng Li, Dongbing Zhao. (4) Data collection and assembly: Zheng Li, Xiaojie Zhang, Chongyuan Sun, He Fei, and Zefeng Li. (5) Statistical analysis: Zheng and Li. (6) Manuscript preparation: All authors. (7) Manuscript editing: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Since this was a retrospective, observational cohort study based on open-access 10 × Genomics Datasets, the requirement for ethics approval and informed consent were waived by the National Cancer Center in China.
Consent for publication
Not applicable.
Competing interests
The authors have declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Zhang, X., Sun, C. et al. Integrating microarray-based spatial transcriptomics and RNA-seq reveals tissue architecture in colorectal cancer. J Big Data 11, 132 (2024). https://doi.org/10.1186/s40537-024-00992-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40537-024-00992-9