
Abstract
Background
The lignification of branches can promote the accumulation of nutrients, increase plant survival and resistance to biotic and abiotic stresses. As an important carbon source for plants, glucose is also the carbon skeleton for lignin synthesis. Grapevine is a perennial cash crop, and highly lignified branches are essential to ensure the growth of the grapevine plant and the development of the fruit.
Methods
Here, ‘Red Globe’ grape (Vitis vinifera L.) plantlets were selected as the material and cultured with different concentrations of glucose: 0 g/L, 20 g/L, 40 g/L and 60 g/L glucose (G0, G20, G40 and G60). Among them, G0 group as control. Lignin, anthocyanins and glucose contents, plant height and microstructure were measured at each glucose concentration after 40-, 50-, and 60-day treatments. Transcriptome and metabolome were used to analyze the difference in genes and metabolites after 50 days of growth.
Results
After 50 days of cultivation, the lignin content in G40 group was the highest. And the xylem cells number also increased. To further, transcriptome and metabolome have identified a total of 3638 differentially expressed genes (DEGs) (including 245 TFs) and 510 differently accumulated metabolites (DAMs) in three comparison groups. In-depth joint analysis revealed that phenylpropanoid biosynthesis pathway significantly respond to exogenous glucose, and 37 DEGs were identified. Therefore, the phenylpropane biosynthesis pathway may be the key to exogenous glucose to increase lignin levels in grapes, with differential expression of genes being a top priority. These findings provide a new perspective for understanding the relationship between glucose and lignin in grape.
Graphical Abstract

Similar content being viewed by others


Introduction
In the process of growth and development, plants are regulated by both external environmental and internal genetic factors, which involves multi-level and multi-channel synergies. At this time, plants can synthesize a variety of special substances through various metabolic processes to respond to exogenous signals and regulate their adaptability [1]. Plant metabolism is mainly divided into two categories: primary metabolism and secondary metabolism [2]. Sugars, typical primary metabolites, are essential substances for maintaining plant life activities and growth and development. Different types of sugars are recognized by different response proteins in the plant to carry out their functions. Studies have found that sucrose is the main form of assimilate transport in plants. The glucose produced after hydrolysis is the main sugar signaling molecule, and can control gene and protein expression, cell cycle, primary and secondary metabolism, as well as growth and development processes [3, 4]. Secondary metabolites, including phenols and terpenes, are mainly involved in plant response to environmental stresses such as disease resistance and stress resistance. In Arabidopsis, it has been found that phenylpropane metabolism is one of the important secondary metabolic pathways that lead the synthesis of these secondary metabolites. This process produces phenylalanine, tryptophan and tyrosine through shikimic acid and glycolysis, and then leads to the two main branches of lignin and flavonoid synthesis, respectively, through specific enzymatic reactions [5, 6].
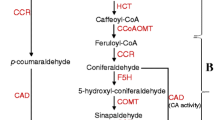
As an important signaling molecule, glucose can induce the production of lignin contents by up-regulating the expression of genes related to phenylpropanoid pathway in Arabidopsis [7, 8]. The lignin synthesis pathway mainly includes three processes. First, cinnamic acid, p-hydroxycinnamic acid and p-coumaryl-CoA were produced from phenylalanine from glucose, a product of previous photosynthesis, by phenylalanine lyase (PAL), cinnamate-4-hydroxylase (C4H) and 4-coumarine-CoA ligase (4CL), respectively [6, 9, 10]. These metabolites have been used as substrates for the next reaction and then catalyzed by cinnamyl CoA reductase (CCR) and cinnamyl alcohol dehydrogenase (CAD) to produce lignin monomer. Finally, the lignin monomer has been polymerized by laccase (LAC) and peroxidase (POX) to form lignin polymer [11,12,13].
Lignin deposition is the main manifestation of lignification in plants. Normally, this process mainly occurs in the secondary thickened cells of stems and roots, which increases the hardness of cell walls and the mechanical support force. At the same time, the insolubility and complex polymer form of lignin also increase the resistance of plants to biotic and abiotic stresses, such as hormone induction, pathogen attack, metabolic stress and injury [14]. Study had found that the increase of lignin content enhanced the stem strength and lodging resistance, and increased the yield of maize under high-density cultivation [15]. Research on lignin in fruit trees mainly focuses on the ripening and storage process of fruit, as well as many transcription factors can up-regulate or down-regulate lignin content by binding to gene promoters on the lignin synthesis pathway. PbrMYB169 can activate the promoter activity of lignin metabolic structure genes 4CL, C3H, CCR, CAD, CCoAOMT and LAC, thus participating in the formation of pear stone cells [16]. In apple, MdMYB93 induced the expression of Md4CL to regulate lignin accumulation [17]. Besides, over-expressed EgNAC141 gene increased the lignin content and lignified area of the in transgenic Arabidopsis [18]. However, there are few studies on the regulation of lignin biosynthesis by sugar, especially glucose, and the expression of biosynthesis-related genes. Research has found that the lignin deposition occurred after the peak of sugar accumulation, and the amount of sugar accumulation directly affected the lignin biosynthesis in peach fruit [19]. Chapelle et al. investigated the impact of the absence of stem-specific beta-glucosidases (BGLU45, BGLU46 and BGLU47) on lignin and monolignols and found that knockout mutants for BGLU45 or BGLU46 do not have a lignin-deficient phenotype [20]. But the results of Lin et al. suggested that UGT72B1 working as a glycosyltransferase may be a key component in modulating cell wall lignification of plants functioning specifically in stems [21]. It is shown that the presence of carbohydrates has a direct bearing on the extent of lignification, and presents evidence suggests that sugar mediated control of lignin deposition may occur through ahexokinase-independent sugar signaling pathway [22]. Staining with phloroglucinol-HCl showed that dark-grown Arabidopsis thaliana seedlings require exogenous sucrose in order to accumulate lignins [23].
Grapes, one of the most popular fruit trees, are cultivated all over the world. The lignification of branches during plant growth can improve plant stress resistance and defense capabilities [24, 25]. Buchanan et al. [26] found that the carbon skeleton of lignin was mainly provided by glucose metabolites. It was found that Hexokinase1 (HXK1), the first glucoreceptor identified, acts as a rate-limiting enzyme for glycolysis, catalyzing the phosphorylation of hexose, ensuring that glucose molecules enter the cell and are phosphorylated instead of spreading from the cell membrane to the outside [27]. In addition, there is also a glucose signal transduction system in plants, namely SnRK1 and TOR signal transduction systems indirectly respond to glucose signals by sensing energy levels in the body [28]. However, how exogenous glucose regulates grapevine lignification remains unclear.
In this study, ‘Red Globe’ grape (Vitis vinifera L.) plantlets were cultured with different concentrations of glucose (0, 20, 40 and 60 g/L). Further, lignin, anthocyanins, glucose and microstructure were assayed of grape stem. Finally, we integrated transcriptome and metabolome techniques to analysis the difference in genes and metabolites after 50 days of growth so that to explore the relationship between lignin and glucose in grape stem. Newly identified genes and regulators provide new insights into further network elucidation, and finally, providing theoretical reference for glucose regulation of grape stem lignification.
Materials and methods
Plant materials
‘Red globe’ (Vitis. vinifera L.) test-tube seedlings, as treat materials, were cultured in chamber. The growth condition was 16 h/8 h of light/dark at 23 °C. After 40 days, a single bud stem from consistent growth and pollution-free plantlets, which underwent single-factor (glucose) multiple concentration treatment, was transferred to a new triangular flask with 50 mL modified GS solid medium containing 0.2 mg/L IAA, where the four treatments are as follows: G0 (0 g/L glucose) as a CK, G20 (20 g/L glucose), G40 (40 g/L glucose), and G60 (60 g/L glucose), respectively.
A total of 720 plantlets were used in this study (360 bottles, 2 plantlets per bottle). In order to determine the period when glucose can cause the most obvious change in lignin content, the five stems segments (begin with the second segment from the bottom up) were taken after 40, 50, and 60 days of treatment, respectively. Each treatment consisted of three biological replicates and each replicate sample was obtained from 10 bottles. All samples were frozen in liquid nitrogen immediately after collection and stored at − 80 °C for future use.
Growth index determination and tissue structure analysis
The plant height of test-tube seedlings treated with different concentrations of glucose at 40 days, 50 days and 60 days was measured using a straightedge, and three plantlets were randomly selected for each treatment in each period for determination.
Tissues staining were performed by safranin O-fast green staining [29, 30]. First, the second fresh stem segment was counted from bottom to top under four treatments in each period, fixed with Formalin-Aceto-Alcohol (FAA) fixative solution and then slice them. After that dewaxed gradiently: xylene I, 8 min; xylene II, 8 min; ethanol I, 6 min; ethanol II, 6 min; 95% ethanol 6 min; 85% ethanol 6 min; 75% alcohol 5 min. Rinsing twice with running water and then stain with safranin at room temperature for 45 min. Using water to rinse to remove excess dye and then drop fast green stain for 10–30 s. Anhydrous ethanol was used to wash off excess dye, and then xylene/ethanol solution (1/1, v/v) for 2 min until the slide was clear. Later, placing the slides into xylene I, 5 min, xylene II, 5 min and xylene III, 5 min in turn. Observing and photographing with a digital microscope (Yongxin iDM2300, Nanjing).
Determination of lignin and anthocyanin
The fresh samples were deoxidized at 105 ℃ for 5 min, then dried at 80 ℃ until the sample weight reaches a constant weight, after that 0.01 g samples dried were weighed accurately to determine lignin content according to the instructions of M1711A kit (Michy Biomedical Technology Co., Ltd, Suzhou, China).
Fresh samples were dried at 65 ℃ until the sample weight reaches a constant weight. Then, 0.1 g dried samples were weighed accurately to determine anthocyanin content according to the instructions of M0126A kit (Michy Biomedical Technology Co., Ltd, Suzhou, China).
Determination of glucose content
The glucose content was determined by HPLC (high-performance liquid chromatograph) [31]. After grinding with liquid nitrogen, 0.5 g samples were to be tested accurately and ultrasonically extracted at 35 °C with 80% chromatographic ethanol for 20 min, centrifuged at 12,000 rpm/min for 15 min, and the supernatant was taken, repeated twice and combined with the supernatant, and to set volume to 10 mL with 80% chromatographic ethanol. 2 mL was evaporated in a vacuum centrifugal concentrator (60 °C) until completely dry, redissolved with ultra-pure water and acetonitrile (v:v = 1:1), filtered through 0.22 μm organic phase microporous filter membrane, and the filtrate was added to the sample bottle to be measured. HPLC was performed by a Waters Acquity Arc system with a 2998 PDA detector (Waters Co., Milford, MA, USA) and chromatographic conditions were chromatographic column: 4.6 × 150 mm 2.5 μm mobile phase: 75% acetonitrile + 0.2% triethylamine + 24.8% ultra-pure water, flow rate: 0.8 mL/min, sample size: 10 μL, detection wavelength: 254 nm, column temperature: 40 ℃, 0.22 μm organic filter membrane filtration before use.
RNA quantification and qualification, and library construction
Total RNA was extracted from the stem segments of grape plantlets grown for 50 days under G0, G20, G40 and G60 treatments. 1% agarose gels were used to monitor RNA degradation and contamination. The NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) was used for detecting RNA purity. RNA concentration and integrity were measured using Qubit® RNA Assay Kit in Qubit®2.0 Flurometer (Life Technologies, CA, USA) and RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA), respectively [32].
Sequencing libraries were generated using NEBNext®UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following specification and index codes were added to boost to attribute sequences to samples. About 200 bp cDNA was selected, with AMPure XP Beads, for PCR amplification and then the PCR products were purified again with AMPure XP system (Beckman Coulter, Beverly, USA) to get the library. Qubit2.0 Fluorometer was used for the initial quantification (1.5 ng/μL) and validation. Whereafter, the libraries were sequenced 150 bp paired-end reads on an Illumina platform.
Transcriptome data analysis
The clean reads gained from the original data were filtered with fastp v 0.19.3 [33], standard as follows: the N content in any sequencing reads exceeds 10% of the base number of the reads; the number of low-quality (Q ≤ 20) bases contained in any sequencing reads exceeds 50% of the bases of the reads, remove the paired reads. All subsequent analyses are based on high-quality clean reads [34].
Using HISAT v2.1.0 [35] compared clean reads with reference genome and its annotation files downloaded from the V. vinifera L reference genome (GCA_000003745.2, NCBI). StringTie v1.3.4d [36] was used for predicating new gene. Using featureCounts v1.6.2 to calculate the gene alignment and fragments per kilobase of transcript per million fragments mapped (FPKM) to estimate gene expression levels. Finally, according to the differential expression screening threshold, the p value adjusted (FDR) < 0.05 and FC ≥ 2, to confirm the differentially expression genes (DEGs).
Identification and functional enrichment analysis of degs
Differential expression analyses among the G0, G20, G40 and G60 treatments were conducted with the DESeq R package (1.10.1) [37]. p value corrected using the Benjamini and Hochberg method and |log2foldchange| were used as the threshold for significant difference expression. Diamond (v0.9.24.125) [38] was used to compare the new gene with Kyoto Encyclopedia of Genes and Genomes (KEGG, https://www.genome.jp/kegg, http://www.genome.jp/kegg/), Gene Ontology (GO, http://www.geneontology.org/), non-redundant protein sequence database (NR, ftp://ftp.ncbi.nih.gov/blast/db/), Swiss-Prot protein sequence database (Swiss-Prot, http://www.uniprot.org/), Translation of European Molecular Biology Laboratory (TrEMBL, http://asia.ensembl.org/index.html), Kyoto Orthologous Groups (KOG, http://www.ncbi.nlm.nih.gov/KOG/) database sequences to obtain annotation results, and the comparison condition was E value 1e−5. Plant transcription factor was predicted using iTAK (plant transcription factor and protein kinase identifier and classifier, http://itak.feilab.net/cgi-bin/itak/index.cgiz) [39]
Extraction, qualification and quantification of metabolites
The freeze-dried sample, obtained from vacuum freeze-dryer (Scientz-100F), was smashed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz. 50 mg of lyophilized powder was dissolved in 1.2 mL 70% methanol solution, then mixed by vortex for 30 s every 30 min for 6 times in total. Following centrifugation at 12,000 rpm for 3 min, the extracts were filtrated (SCAA-104, 0.22 μm pore size; ANPEL, Shanghai, China, http://www.anpel.com.cn/). After that, sample extracts were analyzed by UPLC-ESI-MS/MS system (UPLC, SHIMADZU Nexera X2, https://www.shimadzu.com.cn/; MS, Applied Biosystems 4500 Q TRAP, https://www.thermofisher.cn/cn/zh/home/brands/applied-biosystems.html), and the analysis conditions were as follows: UPLC: column, Agilent SB-C18 (1.8 µm, 2.1 mm × 100 mm). The mobile phase consisted of solvent A (pure water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). Sample measurements gradient program were 5%:95% A/B within 9 min, 5%:95% A/B within 1 min, 95%:5% A/B was adjusted within 1.1 min and kept for 2.9 min, flow velocity, 0.35 mL/min; the column oven, 40 ℃; injection volume was 4 μL. The effluent was alternatively connected to an ESI-triple quadrupole-linear ion trap (Q TRAP)-MS. Linear Ion Trap (LIT) and triple quadrupole (QQQ) scans were acquired on the API 6500 Q TRAP LC/MS/MS System, including an ESI Trubo Ion-Spray interface. The ESI source operation parameters were as follows: source temperature 550 ℃; ion spray voltage (IS) 5500 V (positive ion mode)/-4500 V (negative ion mode); ion source gas I (GSI), gas II (GSII), and curtain gas (CUR) were set at 50, 60, and 25 psi, respectively; the collision-activated dissociation (CAD) was high. Instrument tuning and mass calibration were performed with 10 and 100 μmol/L polypropylene glycol solutions in QQQ and LIT modes, respectively. QQQ scans were acquired as MRM experiments with collision gas (nitrogen) set to medium. DP (declustering potential) and CE (collision energy) for individual MRM transitions was done with further DP and CE optimization. A specific set of MRM transitions were monitored for each period according to the metabolites eluted within this period [40].
Metabolomic data analysis
UHPLC-MS/MS (ultra-high-performance liquid chromatography, tandem mass spectrometry and high-resolution mass spectrometry) was used to analyze the changes of metabolites and metabolic pathways involved in grape stems under G0, G20, G40 and G60 treatments. Based on metabolic database, qualitative and quantitative analyses of the metabolites of the sample were carried out, and the software Analyst (1.6.3) was used to process the mass spectrum data. Multivariate statistical analysis was conducted with principal component analysis (PCA) to understand the overall metabolite difference between each group of samples and the variation degree between samples within the group. The metabolite content data were treated with UV (unit variance scaling), and the R software, Complex Heatmap package, was used to draw the heat map. Hierarchical cluster analysis (HCA) was conducted to investigate the accumulation patterns of metabolites among different samples. Pearson’s correlation coefficient (r) was used as an evaluation index of biological repeated correlation, which was calculated by built-in cor function of R software. The closer the | r | to 1, the stronger the correlation between the two duplicates sample is.
Orthogonal partial least-squares discriminant analysis (OPLS-DA) combined the Orthogonal signal correction (OSC) and PLS-DA methods to screen differential variables by removing uncorrelated differences, and the MetaboAnalystR package in R software was used for analysis [41, 42]. For comparison of two groups, the screening criteria for differential metabolites were VIP ≥ 1 and FC ≥ 2 as well as FC ≤ 0.5. VIP value represents the influence intensity of the difference between groups of corresponding metabolites in sample classification and discrimination of each group in the model. According to the results of differential metabolites, enrichment analysis of KEGG pathway was performed. Identified metabolites were annotated using KEGG compound database (http://www.kegg.jp/kegg/compound/), annotated metabolites were then mapped to KEGG pathway database.
Conjoint analysis of transcriptome and metabolome
In order to investigate the biological relationship between DEGs and DAMs, the correlation between DEGs and DAMs under different treatments was analyzed according to Pearson correlation coefficient (R2 > 0.8) and p value (< 0.5). Then, DEGs and DAMs in the same group were mapped to the KEGG database to determine the biological regulatory relationship between them [43].
Quantitative real-time PCR (qrt-PCR) analysis
12 DEGs that were randomly selected for quantitative real-time PCR analysis were used to verify the accuracy of transcriptome data. The primers were designed and synthesized in Sangon Biotech in Shanghai (Table S1). cDNA was synthesized using the Reverse Transcriptase M-MLV (RNaseH-) kit (TaKaRa Biotechnology. Lanzhou, China). Light Cycler® 96 Real-Time PCR System (Roche, Basel, Switzerland) was used to perform qRT-PCR of 12 DEGs and VvActin (LOC100232866) as reference gene. The relative expression levels of the DEGs were calculated using a 2−△△CT method [44, 45]. The PCR amplification system was 20 μL, including upstream and downstream primers 1 μL, cDNA 1.5 μL, ddH2O 9 μL, 12.5 μL TaKaRa SYBR Premix Ex Taq.II (TaKaRa Biotechnology. Beijing, China). The reaction procedure was denaturation at 95 ℃ for 30 s; denatured at 95 ℃, for 10 s; annealing at 60 ℃ for 30 s; extend for 30 s at 72 ℃; there were 50 cycles in total, with each treatment consisting of three biological repeats.
Data analysis
All data were collected from three replicates (each replicate being 10 plantlets) and were expressed as mean ± SD. The statistical analysis was performed using SPSS 20.0 software. Duncan’s test was used to describe the level of difference between the treatments. Data were visualized using Adobe illustrator 2020 software, Adobe Photoshop CS6 and TBtools (2.029) [46].
Results
Physiological and biochemical analysis of grape stem treated with glucose
In this study, the endogenous glucose content of grape stem showed obvious difference after different concentrations of glucose were applied. When the plantlets grew to 40 days, the plant height of G0 and G60 was significantly lower than that of G20 and G40 treatments. With the increase of growth time, the stem segments of grapes under the four treatments gradually extended, but the plant height under the G60 treatment was always the lowest (Fig. 1A–B). In addition, it was found that with the increase of glucose concentration, grape stem gradually turned red, and the color gradually deepened (Fig. 1A). The glucose content in stem segment showed that when the plants grew to 40 days and 50 days, the glucose content gradually increased with the increase of treatment concentration. At the 50th day, the glucose content in stem segment was the highest, 26.20 mg/g under G60 treatment. It showed a trend of first increasing and then decreasing at the 60th day (Fig. 1C).

Changes in physiological and biochemical analysis of grape stem treated with glucose. A Plant phenotypes under 4 treatments at 40 days, 50 days and 60 days, the line segment indicates a length of 2 cm. B Plant height. C Glucose contents. D Lignin contents. E Anthocyanin contents. F Safranin-O/fast green staining under 4 treatments at for 40 days, 50 days, and 60 days. magnification 40 ×. The 4 treatments were G0 (without glucose) as a CK, G20 (with 20 g/L glucose), G40 (with 40 g/L glucose), G60 (with 60 g/L glucose). Three biological replicates (each replicate was collected randomly from 10 bottles of ‘Red Globe’) were performed for each treatment. The values (means ± SD) are the averages of three biological replicates. Bars having different lowercase letters indicate statistically significant differences by Duncan's multiple range test (p < 0.05)
Further, lignin content showed a trend of first increase and then decrease in each period, and the highest content was 261.25 mg/g under G40 treatment after 50-day treatment (Fig. 1D). At the same time, the anthocyanin content indicated which gradually increased with the increase of glucose concentration at the 40th day. At the 50th day of growth, the anthocyanin content showed an opposite trend to that of lignin at G20 and G40, while the two contents showed the same trend at the 60 days (Fig. 1D–E). It was speculated that the endogenous substances were in the process of continuous synthesis and metabolism at the 40 days of growth in test-tube seedlings. At day 50, the lignin and anthocyanin synthesis precursors were consistent, and the synthesis process competed with the substrate, so it showed the opposite trend, while at day 60, the test-tube seedlings were already in a stable state with the same trend. At the same time, saffron solid green stain analysis declared that as the concentration of glucose treatment increased, the stem secondary cell wall thickened and xylem cells rose gradually, which was consistent with the above results (Fig. 1F).
The transcriptome distinction of grape stem under glucose treatments
Based on the differential changes of lignin content, we selected grape test-tube seedlings stem grown for 50 days for transcriptome and metabolome analysis. The transcriptome analysis results indicate that a total of 91.02 Gb of clean data was obtained, and the clean data of each sample was up to 6 Gb. The percentage of Q30 bases was 91% or above. The GC content of clean reads ranged from 45.32% to 45.85% (Table 1). In addition, compared with the reference genome sequence, it was found that the mapped reads of the 12 samples in this experiment accounted for 89.93–91.93% of clean reads, indicating that the transcriptome data were of high quality and there was no contamination in the experiment (Table 1). Pearson’s correlation coefficient (r) was used as an evaluation index of biological repeated correlation. The closer R2 is to 1, the stronger the correlation between the two samples is. In this study, R2 were all greater than 0.8, indicating high repeatability between the two samples (Fig. 2A). Unsupervised PCA showed that grape stem under four glucose concentrations could be significantly divided into four groups on the score map, indicating that there were significant differences in the transcription of the four groups samples (Fig. 2B). According to the differential expression screening threshold, fold change ≥ 2 and FDR < 0.05. A total of 22,018 genes were differentially expressed in at least one comparison. G0 vs G20 group has 521 different expression genes (DEGs), of which 310 were up-regulated and 211 were down-regulated. G0 vs G40 had 1815 DEGs, among which 1254 were up-regulated and 561 were down-regulated. There were 2119 DEGs in G0 vs G60, including 1217 up-regulated DEGs and 902 down-regulated DEGs (Fig. 2C). Otherwise, Venn diagram could show the overlap of different genes between different comparison combinations. As shown in Fig. 2D–F, there were 235 DEGs in G0 vs G20, G0 vs G40 and G0 vs G60, among which 88 were up-regulated and 146 down-regulated DEGs.

RNA-seq analysis of grape stems after adding glucose. A Genes’ correlation analysis. B Principal component analysis (PCA) of transcriptome sequencing data, each point represents an independent biological replicate. C Number of differentially expressed genes (DEGs) in G0 vs G20, G0 vs G40, and G0 vs G60 group (FC > 2 and FDR < 0.05). D Venn diagram of all DEGs in G0 vs G20, G0 vs G40, and G0 vs G60 group. E Venn diagram of up-regulated DEGs in G0 vs G20, G0 vs G40, and G0 vs G60 group. F Venn diagram of down-regulated DEGs in G0 vs G20, G0 vs G40, and G0 vs G60 group
Degs functional enrichment analysis
In order to further clarify the effect of exogenous glucose on the lignification of grape stem and better analyze the function of DEGs, GO annotation was carried out for all comparison groups. It was found that 1397 GO terms were enriched in G0 vs G20, of which 927, 134 and 366 were involved in biological processes (BP), cell components (CC) and molecular functions (MF), respectively. A total of 2515 GO terms were enriched in G0 vs G40, among which 1646, 217 and 651 were involved in BP, CC and MF, respectively. Similarly, 2755 GO terms were enriched in G0 vs G60, among which 1834, 248 and 366 were related to BP, CC and MF, respectively (Table S2). The top 20 BP terms according to a threshold of p < 0.05 in G0 vs G20, G0 vs G40 and G0 vs G60 groups were screened, which showed that flavonoid biosynthetic process (GO:0009813), flavonoid metabolic process (GO:0009812) and systemic acquired resistance (GO:0009627) enriched in all 3 comparable groups. Only G0 vs G40 and G0 vs G60 groups enriched cellular response to hypoxia (GO:0071456), cellular response to decreased oxygen levels (GO:0036294), cellular response to oxygen levels (GO:0071453), response to hypoxia (GO:0001666), response to decreased oxygen levels (GO:0036293) and response to oxygen levels (GO:0070482). In addition, compared with G0 vs G20 and G0 vs G40, phenylpropanoid biosynthetic process (GO:0009699) and phenylpropanoid metabolic process (GO:0009698) were just enriched in the top 20 BP terms of G0 vs G60. This indicated that secondary metabolic reactions are the main response processes under the different concentration of exogenous glucose sugars (Fig. 3A; Table S3).

DEGs functional enrichment analysis. A The top 20 BP terms in G0 vs G20, G0 vs G40 and G0 vs G60 groups (p < 0.05). B Top 20 KEGG pathway in G0 vs G20. C Top 20 KEGG pathway in G0 vs G40. D Top 20 KEGG pathway in G0 vs G60
In depth, KEGG pathway enrichment analysis revealed that 80, 121 and 125 KEGG metabolic pathways were enriched in G0 vs G20, G0 vs G40 and G0 vs G60 comparison groups, respectively. The top 20 KEGG pathways had contained biosynthesis of secondary metabolites (ko01110), flavonoid biosynthesis (ko00941), zeatin biosynthesis (ko00908), metabolic pathways (ko01100), flavone and flavonol biosynthesis (ko00944), plant hormone signal transduction (ko04075), galactose metabolism (ko00052), pentose and glucuronate interconversions (ko00040) in the three comparison groups (Fig. 3B–D). Furthermore, phenylpropanoid biosynthesis (ko00940) only existed in G0 vs G20 and phenylalanine metabolism (ko00360) was enriched in G0 vs G40 and G0 vs G60.
Transcription factors (tfs) enrichment analysis
A total of 3639 transcription factors (TFs) were identified from all genes, among which 245 TFs were differentially enriched, and the top 10 TF families were MYBs (38), AP2/ERF-ERFs (34), NACs (25), bHLHs (17), WRKYs (16), LOBs (9), bZIPs (8), GRASs (8), C2H2s (7), and HSFs (7) (Fig. 4A). Furthermore, the expression heatmap of the top 5 TFs family members showed that the expression of genes framed in red under the four treatments increased first and then decreased, and the expression level was the highest under the G40 treatment, including VIT14s0068g01490 (NAC68-like), VIT15s0048g02270 (NTM1-like9) (Fig. 4B), VIT11s0052g00100 (bHLH35) (Fig. 4C), VIT02s0025g01280 (WRKY41), VIT04s0008g05760 (WRKY40-like), VIT06s0004g07500 (WRKY24), VIT09s0018g00240 (WRKY40-like), VIT15s0046g01140 (WRKY46) (Fig. 4D), VIT18s0001g09850 (MYB44) (Fig. 4E), VIT03s0063g00460 (ERF109), VIT07s0005g00820 (ERF073), and VIT12s0028g03270 (ERF4), respectively (Fig. 4F). Meanwhile, the expression of TFs framed in blue, VIT15s0021g02690 (UNE10) (Fig. 4C) and VIT11s0016g02410 (MYBS3) (Fig. 4E), was gradually reduced under four treatments. These differential expression transcription factors (TF) may be key in linking glucose and lignin.

Transcription factors (TFs) enrichment analysis. A The top 10 TF families. B NAC TF family. C bHLH TF family. D WRKY TF family. E MYB TF family. F ERF TF family. The red box indicated that the expression level was first up-regulated and then down-regulated, and was the highest under G40 treatment. Blue boxes indicate a gradual decline in expression. The expression quantity is FPKM value
The metabolome distinction of grape stem under glucose treatments
In order to confirm whether there are differences between the samples, we conducted PCA, and the results showed that grape stem under four glucose concentrations could be significantly divided into four groups on the score map, indicating that there were significant differences in the metabolites of the four groups samples (Fig. 5A). Metabolomic analyses showed that a total of 849 metabolites were identified in all samples and classified into 11 categories, among which were flavonoids (213), phenolic acids (189), lipids (97), amino acids and derivatives (47), alkaloids (43), organic acids (43), nucleotides and derivatives (36), terpenoids (33), lignans and coumarins (23), tannins (22), and others (103), respectively (Fig. 5B; Table S4). Besides, to investigate the accumulation dynamics of identified metabolites accumulated at different glucose concentration, orthogonal partial least-squares discriminant analysis (OPLS-DA) was performed, resulting in 510 differentially accumulated metabolites (DAMs) being found (Table S5). VIP > 1.0, fold change (FC) ≥ 2 as well as ≤ 0.5 as the threshold values were used to identify all metabolites, and there 203 DAMs were found in G0 vs G20, among which 160 DAMS were up-regulated and 43 DAMS were down-regulated (Fig. 5C). In G0 vs G40 group, 280 DAMs were identified, of which 219 were up-regulated and 61 were down-regulate. G0 vs G60 group had 369 DAMs, among which 257 were up-regulated and 112 were down-regulated (Fig. 5C). It is found that 134 DAMs expressed in all comparison groups throughout Veen analysis of all DAMs (Fig. 5D).

Metabolome analysis of grape stems after adding glucose. A Principal component analysis (PCA) of metabolome sequencing data. B Category of differentially accumulated metabolites (DAMs) (VIP > 1.0, FC > 1). C Number of DAMs in G0 vs G20, G0 vs G40, and G0 vs G60 group. D Venn diagram of total DAMs in G0 vs G20, G0 vs G40, and G0 vs G60 group. Each point represents an independent biological replicate
KEGG pathway enrichment analysis was performed for differential metabolites of the three comparison groups (Fig. 6). It was found that 58, 68 and 72 KEGG metabolic pathways were enriched in G0 vs G20, G0 vs G40 and G0 vs G60, respectively (Table S6). The analysis of top 20 KEGG metabolic pathways showed that carbon metabolism, vitamin B6 metabolism, flavonoid biosynthesis and pyrimidine metabolism pathways were all enriched. Compared with G0 vs G20, pentose phosphate pathway, glutathione metabolism, carbapenem biosynthesis and sulfur relay system were detected both in G0 vs G40 and G0 vs G60; Compared with G0 vs G20 and G0 vs G40, when the concentration of exogenous glucose reaches 60 g/L, flavone and flavonol biosynthesis, pentose and glucuronate interconversions and other metabolic pathways were enriched under G0 vs G60 (Fig. 6A–C).

Metabolome KEGG analysis. Top 20 KEGG pathway in G0 vs G20 (A), G0 vs G40 (B) and G0 vs G60 (C). The red font indicates the KEGG pathway enriched in the three groups
Combined transcriptome and metabolome analysis
Pearson correlation coefficient R2 > 0.8 (or < − 0.8) and p < 0.05 were used for conjoint analysis of transcriptome and metabolome. DEGs and DAMs in G0 vs G20, G0 vs G40 and G0 vs G60 were mapped to the corresponding KEGG to clarify the relationship between key genes and metabolites associated with grape plantlets stem under different concentration glucose. The results showed that the metabolic pathways, biosynthesis of secondary metabolites, flavonoid biosynthesis, flavone and flavonol biosynthesis, pentose and glucuronate interconversions, carbon fixation in photosynthetic organisms, starch and sucrose, phenylpropanoid biosynthesis phenylalanine metabolism pathways were enriched in all the three comparison groups, which may be the key pathways to connecting glucose and lignin (Fig. 7A–C).

Combined transcriptome and metabolome KEGG analysis. Top 20 KEGG pathway in G0 vs G20 (A), G0 vs G40 (B) and G0 vs G60 (C). The red font indicates the KEGG pathway enriched in the three groups
In this study, transcriptome and metabolome were combined to explore the relationship between glucose and lignin in grape stem. It was found that there were 37 DEGs and 8 DAMs during the emergence of lignification caused by glucose, which showed different expression level (Fig. 8). First, glucose passed through the carbon metabolism and shikimate acid pathway to phenylalanine, and then entered the phenylpropanoid biosynthesis to form lignin. It was found that there were significant differences in gene and expression under different concentrations of glucose treatments, including phenylalanine amylase (PAL, 7), 4-coumaric acid CoA ligase (4CL, 3), cinnamyl CoA reductase (CCR, 3), cinnamyl alcohol dehydrogenase (CAD, 4), coniferol acyltransferase (CFAT, 5), caffeine-O-methyltransferase (COMT, 3), and peroxidase (POD, 12). However, DEGs belonged to the same gene family also showed different expression patterns. The expression of PALs was first up-regulated, reaching the highest under G40, and after decreased, among which the expression of PAL (VIT_06s0004g02620) was the highest under four treatments, whereafter p-coumaric acid went through certain ways to produce caffeic acid on the lateral pathway, and then caffeic acid and 5-Hydroxyferulic were converted into ferulic acid and sinapic acid by COMT, respectively. On the other side, p-coumaric acid, caffeic acid, sinapic acid and ferulic acid could be converted into the corresponding -CoA esters by 4CL. CCR, the first enzyme that catalyzes the synthesis of lignin monomer, catalyzed -CoA esters to transform into aldehydes material, which generate alcohols by CAD. PODs were mainly involved in the monomer polymerization process of lignin synthesis and responsible for the oxidation of monolignin together, including p-coumarol, coniferol, and sinapol. The expression of POD64 (VIT_18s0001g01140) was higher under G0, G20 and G40 than G60. Finally, four kinds of monomer lignin were obtained, such as H-, G- and S-lignin.

The pathway related to lignin biosynthesis and the heat map of changes in metabolites and transcripts. Red and blue boxes indicated up-regulated and down-regulated genes (square) and metabolites (round), respectively. Color bars represent the expression fold changes (log2FC) of DEGs and the relative content of DAMs. White font represents key gene family based on KEGG database. Black font represents key metabolite based on KEGG database. PAL phenylalanine amylase, 4CL 4-coumaric acid CoA ligase, CCR cinnamyl CoA reductase, CAD cinnamyl alcohol dehydrogenase, CFAT coniferol acyltransferase, COMT caffeine-O-methyltransferase, POD peroxidase
To validate the reliability of RNA-Seq results, 12 DEGs (including TFs) were selected randomly to perform qRT-PCR analysis. As shown in Fig. 9A–L, these genes were 4CL9, PALY, CCR1, CAD, POD64, UNE10, and TFs were MYB4-like, bHLH35, C3H29-like, ERF4, MYB44, and NAC68-like. Their relative expression level exhibited a similar tendency with RNA-seq FPKM value, confirming that transcriptome data were reliable for further analysis.

Quantitative real-time PCR (qRT-PCR) validation. qRT-PCR validation of the structural genes involved in the lignin biosynthesis and the candidate transcript factors under adding glucose. Error bars indicated the standard deviation of three independent replicates. Asterisks (*, ** or ***) indicate a significant difference at p < 0.05, p < 0.01 or p < 0.001, respectively
Discussion
Glucose regulates plant growth and development by facilitating cell division and acting as energy sources, osmotic regulatory substances as well as signal molecule [47]. Interestingly, glucose is also the carbon skeleton of lignin [26]. In this study, the grape plantlets showed different growth states after applying different concentrations of glucose (Fig. 1). In our study, we investigated the relationship between exogenous glucose and lignin in grape stem. The results indicated that with the increase of growth time, the grapes stem under the four treatments gradually extended, but the plant height under the G60 treatment was always the lowest, as well as the lignin content was lower (Fig. 1A, B). This is similar to the findings in Arabidopsis Thaliana, which was AtLAC4/11/17 mutant showed severe dwarfization of plants and almost zero lignin content in stems [12]. In addition, it was found that with the increase of glucose concentration, grape stem gradually turned red and deepened as well as the lignin content increased (Fig. 1A). Equally, lignin synthesis genes, COMT and C4H, have increased expression, underlying the lignin accumulation that leads to russet formation in pear and apple [48, 49]. This may be due to the high concentration of glucose under G60, which caused osmotic stress on plants, resulting in the decrease of lignin synthesis ability. The results of Saffron solid green stain showed that the secondary cell wall of grape stem thickened with higher sugar concentration, which also indicated that the lignification degree enhanced. This maybe the result of increase of lignin contents. Besides, the first three steps of the phenylpropyl pathway provides precursor substances for metabolites of all downstream branches, in which the lignin and flavonoid (including anthocyanin synthesis) pathways are the two main branches [10]. In this study, the lignin content showed an opposite trend to that of anthocyanin under G20 and G40 after 50-day treatment (Fig. 1D, E). Similarly, Hu et al. [50] found that overexpressing miR7125 or inhibiting CCR transiently in apple fruit promoted anthocyanin biosynthesis but reduced lignin production under light-induced conditions, which suggested that there is a competition between the synthesis of lignin and anthocyanin. Therefore, the opposite trend of lignin and anthocyanin content in this study may be due to substrate competition during their synthesis.
The lignin biosynthetic genes are generally regulated by the same suite of transcriptional regulators [51]. There were 245 TFs differentially enriched after adding glucose, and the top 3 TF families were MYB (n = 38), AP2/ERF-ERF (34), NAC (25), and many members showed a trend increasing first then decreasing, which under the G40 treatment was the highest. Some MYBs can positively or negatively regulate lignin synthesis, for example, CmMYB8 inhibited lignin accumulation in chrysanthemum [52]. The expression of MYB44 (VIT18s0001g09850) under G40 was the highest (Fig. 4E), which may be involved in the conversion from glucose to lignin. Also, there has been lots of studies about MYB44 function in plant growth and development, including regulates abiotic and biotic stress, which includes drought tolerance, salt tolerance, cold tolerance, responding to phosphate and nitrogen starvation, and disease resistance [53]. AP2/ERF-ERF, a class of plant-specific transcription factors, could regulate various processes of plant development and play a very important role in hormone regulation and stress response, such as MsERF8, AtERF98, and JERF011 could enhance tolerance to salt [54,55,56]. In addition, a novel AP2/ERF gene named Ii049 was isolated and characterized from Isatis indigotica by Ma et al. [57], and they found that overexpressing Ii049 make of high lignan/lignin biosynthesis gene and SA content, resulting in a large amount of lignan/lignin accumulation from Isatidis hairy root. In this study, we found the expression of ERF109 (VIT03s0063g00460), ERF073 (VIT07s0005g00820) and ERF4 (VIT12s0028g03270) were highest under G40 (Fig. 4F), which was consistent with the trend of lignin content (Fig. 1D), so we suspected that 3 ERF genes are also involved in lignin synthesis. NAC transcription factors are considered “master switches” in lignin formation process [58]. In Arabidopsis thaliana, overexpression of AtSND2 increased the wall thickness of xylem fibers [59]. In poplar, overexpression of SND2 homolog PopNAC154 led to an increase in bark to xylic ratio without affecting SCW thickness [60]. In the same vein, NAC68-like (VIT14s0068g01490) and NTM1-like9 (VIT15s0048g02270) showed the expression pattern of first up-regulation and then down-regulation, we also suspected that they are involved in the synthesis of lignin.
There is a close relationship between sugar metabolism and lignin biosynthesis in plants. To be specific, lignin synthesis is related to carbohydrate metabolism through the shikimic acid pathway and phenylpropane metabolic pathway [61, 62]. Some research showed that carbon source of lignin biosynthesis was provided by glucose. After d-glucose labeled with C14 was introduced into a spruce twig by infusion, lignin in the cambium was radioactive, indicating the ultimate organic source of lignin was d-glucose [26, 63]. In this process, a large number of secondary metabolites related to lignin synthesis were produced, and their contents and types have important effects on lignin synthesis [64]. Similarly, in our study, the conjoint analysis of transcriptome and metabolome KEGG enrichment indicated that both phenylpropanoid biosynthesis and phenylalanine metabolism pathways were enriched in all the three groups and 37 DEGs and 8 DAMs significantly respond to glucose treatment (Fig. 8), which may also be key to deciphering the relationship between glucose and lignin.
A number of metabolic pathways are usually involved in glycolysis, including pentose phosphate pathway, shikimic pathway and phenylpropanoid pathway and glucose is also produced by glycolytic metabolism. HXK is an important enzyme in glycolysis and plays an important role in glucose sensing and signal transduction [27]. However, our result of RNA-seq showed no difference in the expression of HXK, consistent with the result in pear callus [65] which explicated that HXK may not play a significant role in the lignin biosynthesis caused by glucose. Besides, glucose can induce phenylpropanoid compounds by up-regulating the expressions of genes related to the phenylpropanoid pathway [8]. As the first rate-limiting pathway in lignin synthesis, the level of PAL activity affects lignin directly the whole process of lignin synthesis [66]. There were 7 PALs be identified, among which PAL (VIT_06s0004g02620) had the same trend as lignin content (Figs. 1D, 8). In Lotus japonicus, it was found that the PAL enzyme activity was significantly increased, the root lignin content was increased by 46%, and the root cell wall was 50% thicker in over-expressed of LjPAL1 than that of the wild type, while inhibiting the expression of LjPAL1 reduced the lignin content by 11% [67]. The level of gene expression in the lignin synthesis pathway effects with each other, resulting in the lignin content. In Manihot esculenta C, MePAL6 was silenced by VGIS, resulting in decreased expression of 4CL, HCT, CCR and other lignin synthesis-related genes and decreased lignin content [68]. In our study, under G60 treatment, the expression of most genes in the lignin synthesis pathway decreased, resulting in dwarfing of grape plantlets than other treatments. In Arabidopsis thaliana, inhibiting the expression of CCR1, CADC and CADD, participated in lignin synthesis, can reduce the lignin content in stem tissues of transgenic lines by 50% compared with the wild type, and cause serious dwarfing and male sterility, which affects the normal growth and development of the transgenic lines [69]. We speculated that the decrease of lignin synthesis might lead to plant dwarfing. It can be seen that the expression of key genes in the lignin synthesis pathway is the key to changes in content. However, in-depth functional verification is also necessary.
Conclusion
In this paper, the secondary metabolites and microstructure of ‘Red globe’ grape seedlings stem grew to 40 days, 50 days and 60 days after four concentrations of glucose treatment were studied, and further analyzed the 50-day grape stem by transcriptomics and metabolomics under four concentrations of glucose. The results showed that the application of a certain concentration of glucose could increase the lignin content in grape stem, especially under 40 g/L glucose. In addition, glucose increases the number of xylem cells. The combined analysis of transcriptome and metabolomics indicates that the secondary metabolite synthesis pathway is significantly influenced by glucose. In particular, phenylpropanoid biosynthesis pathway prominently respond to glucose, and further, 37 genes differentially express.
This study provides valuable genetic resources for the subsequent metabolic engineering of lignin, and provides a certain reference for the study of glucose regulation of lignin synthesis as well as the relationship between lignin and anthocyanin.
Availability of data and materials
No datasets were generated or analyzed during the current study.
Abbreviations
- PAL:
-
Phenylalanine lyase
- C4H:
-
Cinnamate-4-hydroxylase
- 4CL:
-
4-Coumarine-CoA ligase
- CCR:
-
Cinnamyl CoA reductase
- CAD:
-
Cinnamyl alcohol dehydrogenase
- LAC:
-
Laccase
- POX:
-
Peroxidase
- HXK1:
-
Hexokinase1
- FAA:
-
Formalin-aceto-alcohol
- HPLC:
-
High-performance liquid chromatograph
- OPLS-DA:
-
Orthogonal partial least-squares discriminant analysis
- OSC:
-
Orthogonal signal correction
- qPCR:
-
Quantitative real-time PCR
- DEGs:
-
Different expression genes
- GO:
-
Gene ontology
- BP:
-
Biological processes
- CC:
-
Cell components
- MF:
-
Molecular functions
- TFs:
-
Transcription factors
- DAMs:
-
Differentially accumulated metabolites
- PAL:
-
Phenylalanine amylase
- 4CL:
-
4-Coumaric acid CoA ligase
- CFAT:
-
Coniferol acyltransferase
- COMT:
-
Caffeine-O-methyltransferase
- POD:
-
Peroxidase
- FPKM:
-
Fragments per kilobase of transcript per million fragments mapped
References
Fang C, Fernie AR, Luo J. Exploring the diversity of plant metabolism. Trends Plant Sci. 2019;24(1):83–98. https://doi.org/10.1016/j.tplants.2018.09.006.
Fengxia Z, Dong W. The applications of metabolomics in plant biology current status and prospective. Rev China Agric Sci Technol. 2013;15(2):5.
Moore B, Sheen J. Plant sugar sensing and signalling—a complex reality. Trends Plant Sci. 1999;4(7):250. https://doi.org/10.1016/s1360-1385(99)01433-8.
Rolland F, Moore B, Sheen J. Sugar sensing and signaling in plants. Plant Cell. 2002;14(Suppl):S185-205. https://doi.org/10.1105/tpc.010455.
Feng X, Le H, Zhenjie W, Jin J, Yutao S. Advances in understanding how branches of the phenylpropanoid pathway respond to biotic stress. China Plant Protection. 2023;43:8.
Fraser CM, Chapple C. The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book. 2011;9: e0152. https://doi.org/10.1199/tab.0152.
Rook F, Gerrits N, Kortstee A, van Kampen M, Borrias M, Weisbeek P, et al. Sucrose-specific signalling represses translation of the Arabidopsis ATB2 bZIP transcription factor gene. Plant J. 1998;15(2):253–63. https://doi.org/10.1046/j.1365-313x.1998.00205.x.
Solfanelli C, Poggi A, Loreti E, Alpi A, Perata P. Sucrose-specific induction of the anthocyanin biosynthetic pathway in Arabidopsis. Plant Physiol. 2006;140(2):637–46. https://doi.org/10.1104/pp.105.072579.
Douglas CJ. Phenylpropanoid metabolism and lignin biosynthesisfrom weeds to trees. Trends Plant Sci. 1996;1(6):8.
Dong NQ, Lin HX. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J Integr Plant Biol. 2021;63(1):180–209. https://doi.org/10.1111/jipb.13054.
Muro-Villanueva F, Mao XY, Chapple C. Linking phenylpropanoid metabolism, lignin deposition, and plant growth inhibition. Curr Opin Biotech. 2019;56:202–8. https://doi.org/10.1016/j.copbio.2018.12.008.
Schuetz M, Benske A, Smith RA, Watanabe Y, Tobimatsu Y, Ralph J, et al. Laccases direct lignification in the discrete secondary cell wall domains of protoxylem. Plant Physiol. 2014;166(2):798-U489. https://doi.org/10.1104/pp.114.245597.
Shigeto J, Tsutsumi Y. Diverse functions and reactions of class III peroxidases. New Phytol. 2016;209(4):1395–402. https://doi.org/10.1111/nph.13738.
Moura JCMS, Bonine CAV, Viana JDF, Dornelas MC, Mazzafera P. Abiotic and biotic stresses and changes in the lignin content and composition in plants. J Integr Plant Biol. 2010;52(4):360–76. https://doi.org/10.1111/j.1744-7909.2010.00892.x.
Zhihai Z, Xuan Z, Zhelong L, Jian W, Hangqin L, Leina Z, et al. A Large transposon insertion in the stiff1 promoter increases stalk strength in maize. Plant Cell. 2020;32:13. https://doi.org/10.1105/tpc.19.00486.
Xue C, Yao JL, Xue YS, Su GQ, Wang L, Lin LK, et al. PbrMYB169 positively regulates lignification of stone cells in pear fruit. J Exp Bot. 2019;70(6):1801–14. https://doi.org/10.1093/jxb/erz039.
Legay S, Guerriero G, Andre C, Guignard C, Cocco E, Charton S, et al. MdMyb93 is a regulator of suberin deposition in russeted apple fruit skins. New Phytol. 2016;212(4):977–91. https://doi.org/10.1111/nph.14170.
Sun Y, Jiang C, Jiang R, Wang F, Zhang Z, Zeng J. A Novel NAC transcription factor from eucalyptus, EgNAC141, positively regulates lignin biosynthesis and increases lignin deposition. Front Plant Sci. 2021;12: 642090. https://doi.org/10.3389/fpls.2021.642090.
Yang AiZhen YA, Zhang ZhiYi ZZ, Cao AiJuan CA, Meng HaiLing MH, Wang YouNian WY. Studies of changes in sugar accumulation and lignin deposition during peach fruit endocarp development. Acta Horticulturae Sinica. 2009;36(8):6.
Chapelle A, Morreel K, Vanholme R, Le-Bris P, Morin H, Lapierre C, et al. Impact of the absence of stem-specific beta-glucosidases on lignin and monolignols. Plant Physiol. 2012;160(3):1204–17. https://doi.org/10.1104/pp.112.203364.
Lin JS, Huang XX, Li Q, Cao YP, Bao Y, Meng XF, et al. UDP-glycosyltransferase 72B1 catalyzes the glucose conjugation of monolignols and is essential for the normal cell wall lignification in. Plant J. 2016;88(1):26–42. https://doi.org/10.1111/tpj.13229.
Rogers LA, Dubos C, Cullis IF, Surman C, Poole M, Willment J, et al. Light, the circadian clock, and sugar perception in the control of lignin biosynthesis. J Exp Bot. 2005;56(416):1651–63. https://doi.org/10.1093/jxb/eri162.
Newman LJ, Perazza DE, Juda L, Campbell MM. Involvement of the R2R3-MYB, AtMYB61, in the ectopic lignification and dark-photomorphogenic components of the det3 mutant phenotype. Plant J. 2004;37(2):239–50. https://doi.org/10.1046/j.1365-313x.2003.01953.x.
Mazid M, Khan T, Mohammad F. Role of secondary metabolites in defense mechanisms of plants. Biol Med. 2011;3(2):18.
Yan Y, Wang P, Lu Y, Bai YJ, Wei YX, Liu GY, et al. MeRAV5 promotes drought stress resistance in cassava by modulating hydrogen peroxide and lignin accumulation. Plant J. 2021;107(3):847–60. https://doi.org/10.1111/tpj.15350.
Buchanan BB, Gruissem W, Jones RL. Biochemistry and molecular biology of plants. John Wiley & Sons; 2015.
Moore B, Zhou L, Rolland F, Hall Q, Cheng WH, Liu YX, et al. Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science. 2003;300(5617):332–6. https://doi.org/10.1126/science.1080585.
Lastdrager J, Hanson J, Smeekens S. Sugar signals and the control of plant growth and development. J Exp Bot. 2014;65(3):799–807. https://doi.org/10.1093/jxb/ert474.
Liao K, Han L, Yang Z, Huang Y, Du S, Lyu Q, et al. A novel in-situ quantitative profiling approach for visualizing changes in lignin and cellulose by stained micrographs. Carbohydr Polym. 2022;297: 119997. https://doi.org/10.1016/j.carbpol.2022.119997.
Baldacci-Cresp F, Spriet C, Twyffels L, Blervacq AS, Neutelings G, Baucher M, et al. A rapid and quantitative safranin-based fluorescent microscopy method to evaluate cell wall lignification. Plant J. 2020;102(5):1074–89. https://doi.org/10.1111/tpj.14675.
Panagopoulou EA, Chiou A, Nikolidaki EK, Christea M, Karathanos VT. Corinthian raisins (Vitis vinifera L., var. Apyrena) antioxidant and sugar content as affected by the drying process: a 3-year study. J Sci Food Agric. 2019;99(2):915–22. https://doi.org/10.1002/jsfa.9263.
Zhang Z, Long Y, Yin X, Wang W, Li W, Chen T, et al. Metabolome and transcriptome analysis of sulfur-induced kiwifruit stem laccase gene involved in syringyl lignin synthesis against bacterial canker. J Agric Food Chem. 2023;71(36):13566–76. https://doi.org/10.1021/acs.jafc.3c02653.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Oshlack NMDA. Corset: enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014;15:410.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. https://doi.org/10.1038/nbt.3122.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60. https://doi.org/10.1038/nmeth.3176.
Zheng Y, Jiao C, Sun H, Rosli HG, Pombo MA, Zhang P, et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant. 2016;9(12):1667–70. https://doi.org/10.1016/j.molp.2016.09.014.
Fraga CG, Clowers BH, Moore RJ, Zink EM. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal Chem. 2010;82(10):4165.
Thevenot EA, Roux A, Xu Y, Ezan E, Junot C. Analysis of the human adult urinary metabolome variations with age, body mass index, and gender by implementing a comprehensive workflow for univariate and OPLS statistical analyses. J Proteome Res. 2015;14(8):3322–35. https://doi.org/10.1021/acs.jproteome.5b00354.
Chong J, Xia J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics. 2018;34(24):4313–4. https://doi.org/10.1093/bioinformatics/bty528.
Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1. https://doi.org/10.1093/bioinformatics/btt285.
Zhang L, Zhao T, Sun X, Wang Y, Du C, Zhu Z, et al. Overexpression of VaWRKY12, a transcription factor from Vitis amurensis with increased nuclear localization under low temperature, enhances cold tolerance of plants. Plant Mol Biol. 2019;100(1–2):95–110. https://doi.org/10.1007/s11103-019-00846-6.
Yang H, Liu J, Huang S, Guo T, Deng L, Hua W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene. 2014;538(1):113–22. https://doi.org/10.1016/j.gene.2013.12.057.
Chen C, Wu Y, Li J, Wang X, Zeng Z, Xu J, et al. TBtools-II: a “one for all, all for one” bioinformatics platform for biological big-data mining. Mol Plant. 2023;16(11):1733–42. https://doi.org/10.1016/j.molp.2023.09.010.
Guo WJ, Pommerrenig B, Neuhaus HE, Keller I. Interaction between sugar transport and plant development. J Plant Physiol. 2023. https://doi.org/10.1016/j.jplph.2023.154073.
Shi CH, Qi BX, Wang XQ, Shen LY, Luo J, Zhang YX. Proteomic analysis of the key mechanism of exocarp russet pigmentation of semi-russet pear under rainwater condition. Sci Hortic-Amsterdam. 2019;254:178–86. https://doi.org/10.1016/j.scienta.2019.04.086.
Wang YZ, Zhang SJ, Dai MS, Shi ZB. Pigmentation in sand pear (Pyrus pyrifolia) fruit: biochemical characterization, gene discovery and expression analysis with exocarp pigmentation mutant. Plant Mol Biol. 2014;85(1–2):123–34. https://doi.org/10.1007/s11103-014-0173-1.
Hu YJ, Cheng H, Zhang Y, Zhang J, Niu SQ, Wang XS, et al. The MdMYB16/MdMYB1-miR7125-MdCCR module regulates the homeostasis between anthocyanin and lignin biosynthesis during light induction in apple. New Phytol. 2021;231(3):1105–22. https://doi.org/10.1111/nph.17431.
Zhao Q, Dixon RA. Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci. 2011;16(4):227–33. https://doi.org/10.1016/j.tplants.2010.12.005.
Zhu L, Guan YX, Zhang ZH, Song AP, Chen SM, Jiang JF, et al. CmMYB8 encodes an R2R3 MYB transcription factor which represses lignin and flavonoid synthesis in chrysanthemum. Plant Physiol Bioch. 2020;149:217–24. https://doi.org/10.1016/j.plaphy.2020.02.010.
Wang FH, Yang F, Zhu DF, Saniboere B, Zhou B, Peng D. MYB44 plays key roles in regulating plant responses to abiotic and biotic stress, metabolism, and development. J Plant Biochem Biot. 2023. https://doi.org/10.1007/s13562-023-00864-y.
Chen TT, Yang QC, Gruber M, Kang JM, Sun Y, Ding W, et al. Expression of an alfalfa (Medicago sativa L.) ethylene response factor gene MsERF8 in tobacco plants enhances resistance to salinity. Mol Biol Rep. 2012;39(5):6067–75. https://doi.org/10.1007/s11033-011-1421-y.
Zhang ZJ, Wang J, Zhang RX, Huang RF. The ethylene response factor AtERF98 enhances tolerance to salt through the transcriptional activation of ascorbic acid synthesis in Arabidopsis. Plant J. 2012;71(2):273–87. https://doi.org/10.1111/j.1365-313X.2012.04996.x.
Tang YH, Qin SS, Guo YL, Chen YB, Wu PZ, Chen YP, et al. Genome-wide analysis of the AP2/ERF gene family in physic nut and overexpression of the gene in rice increased its sensitivity to salinity stress. PLoS ONE. 2016;11(3):e0150879. https://doi.org/10.1371/journal.pone.0150879.
Ma RF, Xiao Y, Lv ZY, Tan HX, Chen RB, Li Q, et al. AP2/ERF transcription factor, positively regulates lignan biosynthesis in through activating salicylic acid signaling and lignan/lignin pathway genes. Front Plant Sci. 2017;8:1361. https://doi.org/10.3389/fpls.2017.01361.
Sun YM, Jiang CX, Jiang RQ, Wang FY, Zhang ZG, Zeng JJ. A novel NAC transcription factor from, EgNAC141, positively regulates lignin biosynthesis and increases lignin deposition. Front Plant Sci. 2021;12:642090. https://doi.org/10.3389/fpls.2021.642090.
Zhong R, Lee C, Zhou J, McCarthy RL, Ye ZH. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008;20(10):2763–82. https://doi.org/10.1105/tpc.108.061325.
Grant EH, Fujino T, Beers EP, Brunner AM. Characterization of NAC domain transcription factors implicated in control of vascular cell differentiation in Arabidopsis and Populus. Planta. 2010;232(2):337–52. https://doi.org/10.1007/s00425-010-1181-2.
Liu J, Huang QH, Kang PZ, Liang L, Chen JJ. Lignin accumulation in three pumelo cultivars in association with sucrose and energy depletion. Biomolecules. 2019;9(11):701. https://doi.org/10.3390/biom9110701.
Herrmann KM, Weaver LM. The shikimate pathway. Annu Rev Plant Physiol. 1999;50:32.
Acerbo SN, Schubert WJ, Nord FF. Investigations on lignins and lignification. XXII. 1 The conversion of D-glucose into lignin in Norway Spruce. J Am Chem Soc. 1960;82(3):5.
Xu M, Zhang MX, Shi YN, Liu XF, Li X, Grierson D, et al. EjHAT1 participates in heat alleviation of loquat fruit lignification by suppressing the promoter activity of key lignin monomer synthesis gene EjCAD5. J Agr Food Chem. 2019;67(18):5204–11. https://doi.org/10.1021/acs.jafc.9b00641.
Jiao Y, Gong X, Qi K, Xie Z, Wang Y, Yuan K, et al. Transcriptome analysis provides new ideas for studying the regulation of glucose-induced lignin biosynthesis in pear calli. Bmc Plant Biol. 2022;22(1):310. https://doi.org/10.1186/s12870-022-03658-x.
Jun SY, Sattler SA, Cortez GS, Vermerris W, Sattler SE, Kanga CH. Biochemical and structural analysis of substrate specificity of a phenylalanine ammonia-lyase. Plant Physiol. 2018;176(2):1452–68. https://doi.org/10.1104/pp.17.01608.
Chen YP, Li FJ, Tian L, Huang MC, Deng RF, Li XL, et al. The phenylalanine ammonia lyase gene is involved in plant defense responses to pathogens and plays diverse roles in rhizobium symbioses. Mol Plant Microbe In. 2017;30(9):739–53. https://doi.org/10.1094/Mpmi-04-17-0080-R.
Yao XW, Liang X, Chen Q, Liu Y, Wu CL, Wu MF, et al. regulates lignin accumulation to shape cassava resistance against two-spotted spider mite. Front Plant Sci. 2023;13:1067695. https://doi.org/10.3389/fpls.2022.1067695.
Thévenin J, Pollet B, Letarnec B, Saulnier L, Gissot L, Maia-Grondard A, et al. The simultaneous repression of CCR and CAD, two enzymes of the lignin biosynthetic pathway, results in sterility and dwarfism in. Mol Plant. 2011;4(1):70–82. https://doi.org/10.1093/mp/ssq045.
Acknowledgements
This research was financially supported by the Key Project of Gansu Provincial Natural Science Fund (22JR5RA831) and the FuXi Foundation of Gansu Agricultural University (No. Ganfx-03J02).
Author information
Authors and Affiliations
Contributions
BHC and JM designed the experiments and revised the manuscript. HW performed the analyses and wrote the manuscript. JJH, CCZ, YML and HMG participated in the determination of physiological–biochemical indicators. GPL and ZHM participated in the revision of the manuscript. All the authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, H., Huang, J., Zhang, C. et al. Glucose enhanced lignin accumulation in grapevine stems via promoting phenylpropanoid biosynthesis. Chem. Biol. Technol. Agric. 11, 152 (2024). https://doi.org/10.1186/s40538-024-00676-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40538-024-00676-9