
Abstract
Prenatal ethanol exposure (PEE) (mainly through maternal alcohol consumption) has become widespread. However, studies suggest that it can cause intrauterine growth retardation (IUGR) and multi-organ developmental toxicity in offspring, and susceptibility to various chronic diseases (such as neuropsychiatric diseases, metabolic syndrome, and related diseases) in adults. Through ethanol’s direct effects and its indirect effects mediated by maternal-derived glucocorticoids, PEE alters epigenetic modifications and organ developmental programming during fetal development, which damages the offspring health and increases susceptibility to various chronic diseases after birth. Ethanol directly leads to the developmental toxicity of multiple tissues and organs in many ways. Regarding maternal-derived glucocorticoid-mediated IUGR, developmental programming, and susceptibility to multiple conditions after birth, ethanol induces programmed changes in the neuroendocrine axes of offspring, such as the hypothalamus-pituitary-adrenal (HPA) and glucocorticoid-insulin-like growth factor 1 (GC-IGF1) axes. In addition, the differences in ethanol metabolic enzymes, placental glucocorticoid barrier function, and the sensitivity to glucocorticoids in various tissues and organs mediate the severity and sex differences in the developmental toxicity of ethanol exposure during pregnancy. Offspring exposed to ethanol during pregnancy have a “thrifty phenotype” in the fetal period, and show “catch-up growth” in the case of abundant nutrition after birth; when encountering adverse environments, these offspring are more likely to develop diseases. Here, we review the developmental toxicity, functional alterations in multiple organs, and neuroendocrine metabolic programming mechanisms induced by PEE based on our research and that of other investigators. This should provide new perspectives for the effective prevention and treatment of ethanol developmental toxicity and the early prevention of related fetal-originated diseases.
Similar content being viewed by others


Introduction
Drinking can be harmful to individuals [1,2,3,4]. Epidemiological investigation shows that an estimated 10% of pregnant women consume alcohol globally, and prenatal ethanol exposure (PEE) has further increased in recent years [5, 6]. Many studies have shown that PEE has a more obvious damage to the fetus than the mother due to immature development and incompletely established homeostasis, which not only induces fetal developmental toxicity (including intrauterine growth retardation (IUGR) and fetal alcohol syndrome (FAS)), but also can lead to a series of long-term development-related health problems and susceptibility to conditions in adulthood (such as neuropsychiatric disorders and metabolic syndrome (MS)) [1,2,3,4, 7,8,9]. In the 1990s, Dr. David Barker performed a large-scale epidemiological investigation and found that the incidence of coronary heart disease, hypertension, hyperlipidemia and obesity was increased in adults with IUGR; he thus proposed the hypothesis of the intrauterine origin of adult diseases [10,11,12,13]. Over the past three decades, numerous scholars have continued studies on the relationship between adverse environments during pregnancy, birth weight and the development of diseases in adults, and put forward a new concept — the Developmental Origins of Health and Disease (DOHaD). Upon further studies regarding DOHaD, the concept of intrauterine programming of diseases was proposed and improved.
Moisiadis and Matthews studied the role of endogenous glucocorticoids (GCs) (cortisol in humans and corticosterone in rodents) in fetal programming in 2014, believing that maternal GC overexposure may be the main cause of fetal programming and susceptibility to diseases in offspring [14]. Studies conducted in our laboratory also found that PEE increased fetal exposure to maternal GCs by stimulating the maternal hypothalamus-pituitary-adrenal (HPA) axis and opening the placental GC barrier, increasing susceptibility to a variety of conditions such as MS, non-alcoholic fatty liver disease (NAFLD), and depression in adults [2, 15]. It was further found that the expression level of insulin-like growth factor 1 (IGF1) in the fetal liver determined the birth weight, organ structure, and functional development of the fetus [16,17,18]. Furthermore, PEE affected the level of GCs to regulate the expression of IGF1 [19, 20]. A cohort from Weeks et al. also reported that adults (22.2 to 44.4 years old) with fetal alcohol spectrum disorders (FASDs) caused by PEE had an increased incidence of metabolic abnormalities, including type 2 diabetes, low HDL, high triglycerides, and female-specific overweight and obesity [21]. Therefore, changes in intrauterine neuroendocrine metabolic programming may be the key mechanism for a variety of neuropsychiatric and metabolic diseases in IUGR offspring caused by PEE [22]. However, there is no systematic experiment to support this claim or hypothesis to explain it.
Currently, research on the developmental toxicity of PEE has made significant progress, but there is a lack of full understanding of its characteristics on offspring impacts, susceptibility to multiple diseases in utero programming, and mechanisms of disease occurrence in PEE-induced adulthood. According to the work of our laboratory and that of other researchers, this review outlines the characteristics of offspring developmental toxicity caused by PEE, the role of neuroendocrine programming and epigenetics in it, sex differences and possible mechanisms, suggesting that alterations in the HPA axis and GC-IGF1 axis programming may be the core mechanisms for the susceptibility to metabolic-related diseases in offspring caused by PEE, and proposing “dual programing” in utero and the “two strikes” theory of disease occurrence in adulthood caused by PEE. We wish to promote understanding of the toxicity caused by PEE and the mechanisms involved, to provide new perspectives on the early prevention and treatment of ethanol developmental toxicity-related fetal-derived diseases.
Short- and long-term effects of PEE
Many products contain ethanol, such as all baijiu, wine products, some skin-care products, dyes, paint removers, gasoline, disinfectants, detergents, and pesticide residue on food. Pregnant women often consciously or unconsciously contact the above items. Among these, maternal drinking has the most obvious damage. In this section, according to different occurrence times of effects on the fetus, the developmental toxicity of ethanol is divided into short-term toxicity and long-term toxicity. Adverse effects on embryo and fetus are defined as “short-term toxicity,“ and, on postnatal offspring, they are called “long-term toxicity.“
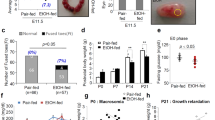
Drinking alcohol during development of the embryo or fetus may cause miscarriage. The more drinks that are consumed and the longer the drinking occurs, the greater the possibility of spontaneous abortion. Drinking more than three days a week was associated with a higher risk of miscarriage [23,24,25,26,27]. IUGR is defined as the limitation on the growth and development of embryos or fetuses, which is characterized by multiple organ dysfunction, growth retardation, and low birth weight [22, 28]. IUGR is also one type of short-term toxicity caused by PEE [29,30,31,32]. An epidemiological investigation showed that the birth weights of offspring from mothers who drank were significantly lower than those of the control group [33]. Mouse studies found that acute ethanol exposure that occurred twice during pregnancy might inhibited cell proliferation during the trimester one, cell migration and differentiation during trimester two, and cellular communication and neurotransmission during trimester three in offspring [34]. Studies in our laboratory also suggested that ethanol exposure during middle and late pregnancy significantly reduced the birth weight in mice, and the IUGR rate was as high as 80% [35]; moreover, ethanol exposure led to abnormal development or function in many types of tissues and organs in the fetus, such as the hippocampus, hypothalamus, adrenal gland, cartilage and bone (Table 1) [36,37,38,39].
In addition to short-term toxicity, PEE can also lead to long-term toxicity (Table 1). “Catch-up growth” is defined as the phenomenon by which longitudinal growth velocity transiently stands above the statistical limits of normality for age and/or maturity after the removal of a growth-inhibiting condition, which has been shown to be associated with increased susceptibility to various conditions including osteoarthritis, MS, insulin resistance and other metabolic diseases such as diabetes and obesity [37,38,39,40,41]. PEE induced low birth weights, while mounting epidemiological and experimental studies have shown that low-birth-weight individuals frequently present IGF1-induced “catch-up growth” after birth [37, 40], causing physical and mental abnormalities and increased susceptibility to MS and various metabolic diseases in adulthood [42, 43]. Studies showed that PEE increased apoptosis and inhibited proliferation of islet β cells and decreased insulin secretion in rat (at 13 weeks of age) and guinea pig (on postnatal day 150–200) before and after birth [8, 44, 45]. Our studies also showed that the adult offspring of rats exposed to ethanol during pregnancy had increased blood glucose levels when fed a normal diet and elevated blood glucose levels, insulin levels, and insulin resistance index when fed a high-fat diet, leading to decreased glucose tolerance and diabetes at postnatal week 17 and 24 [2, 41]. The mechanism of these effects is related to abnormal development of the pancreas mediated by programming alteration of the GC-IGF1 axis [46]. In rats, offspring with IUGR induced by PEE also showed “catch-up growth” in body weight, elevated blood triglyceride levels, increased liver lipid and glycogen synthesis, and inhibited lipid output, and these rats were more likely to have typical NAFLD phenotypes than the control rats [2]. In addition, we found that PEE of rats caused retardation of bone growth, inhibition of endochondral ossification, delayed development of primary and secondary ossification centers [36], and poor cartilage quality in offspring before and after birth [39]; these findings were consistent with those of Simpson et al. [47]. Moreover, the persistent poor cartilage quality before and after birth (at postnatal week 24) caused by PEE in rats was related to the low functional programming of local IGF1 signaling, while high-fat diet after birth increased blood cholesterol content and deposition in cartilage, which further induced osteoarthritis [38].
Characteristics of developmental toxicity induced by PEE
Epidemiological investigations showed that fetuses suffered from FAS when pregnant women were exposed to ethanol at 3.0–4.3 g/kg·d [54], or even at doses as low as 0.35 g/kg·d [55]. Simpson et al. found that when pregnant rats were fed diets of equal calories but different ethanol proportions, the fetal offspring in the high-dose group (36% ethanol-derived calories) showed significant reductions in body weight and length and inhibition of ossification, while only a delay in radius was observed in the low-dose group (15% ethanol-derived calories) [47]. It has been suggested that the higher the ethanol dose during pregnancy, the more developmental toxicity to the fetus. However, there are reported differences in the toxicity of low-dose ethanol exposure to the fetus. The results of the epidemiological investigations of Kelly et al. and Robinson et al. indicated that the toxicity of light alcohol (1–2 drinks per week or per occasion in Kelly’s study; 2–6 standard drinks per week in Robinson’s study) consumption during pregnancy was minimal or could not be observed in the offspring [56, 57]. However, Gray et al. believed that these negative results have arisen from, for example, the lack of rigorous experimental design, short observation times, and incomplete detection indicators. They suggested that due to the different metabolic rates of ethanol among individuals, peak blood alcohol levels are higher and residual time is longer in pregnant women with slow metabolism; therefore, low-dose ethanol consumption can seriously impair fetuses in this subpopulation [58]. The findings of Lewis et al. corroborate those of Gray et al. in a study of 15,000 pregnant women, Lewis et al. found that despite the same amount of ethanol consumed during pregnancy, the intelligence quotient (IQ) scores of the 8-year-old children of women with ethanol metabolic enzyme deficiency were lower than those of women with normal ethanol metabolic enzyme levels [9]. Moreover, Lewis et al. also suggested that the genetic variation of both mother and fetus is an important moderator of the fetal effects of alcohol [9]. This suggests that the fetal toxicity induced by PEE is not only related to the amount of ethanol consumed but also to the ethanol metabolic capacity and genetic variation of the mother and fetus. Therefore, Gray indicated that the determination of a ‘safe’ level which can be uniformly recommended for all pregnant women seems unrealistic [58].
Time is another important factor in the toxicity induced by PEE. Short-term exposure to high doses of ethanol directly damages fetal tissue, especially during stages of fetal development, resulting in miscarriage, stillbirth, and malformation [59, 60]. Fish et al. found that after acute exposure to 2.8 g/kg ethanol twice in early pregnancy, the morphology of the cerebellum, hippocampus, striatum, and corpus callosum changed in fetal mice. In the offspring of male mice (on postnatal day 28–45), activity increased in maze tests and the preference for a rich environment decreased, and in the female offspring, exploratory behavior increased and cage running decreased [61]. Diaz et al. also found that acute ethanol exposure in mid-pregnancy (12 or 15 days of pregnancy) was associated with neurobehavioral abnormalities in offspring of early and late adolescent or young adult Sprague Dawley rats, but differed with exposure time, species (although there are few reports), and sex [62]. Typically, these abnormalities were greater in Long Evans males and Sprague Dawley female rats [62]. Chronic ethanol exposure often causes persistent damage in the middle and late stages of pregnancy and leads to serious fetal developmental toxicity (such as IUGR [35]). A study by Dettmer et al. showed that chronic ethanol exposure during pregnancy caused long-term inhibitory effects on N-methyl-d-aspartate receptor subtype 2B (NR2B) (a glutamate-gated ion channel) in the cerebral cortex of guinea pig offspring and upregulated the expression of glutamate receptor subunits (GluR2/3); these changes may be related to the neurobehavioral changes observed in guinea pig offspring on postnatal day 61 [63]. A study by Iqbal et al. showed that PEE led to changes in the protein level of the γ-aminobutyric acid type A (GABA-A) receptor β-2/3 subunit in the adult offspring of guinea pigs and damaged their spatial learning ability, which was potentially related to FAS and long-term harm [51]. Previous studies in our laboratory found that ethanol exposure during middle and late pregnancy in rats caused the dysfunction of glucose lipid metabolism in liver and HPA axis, therefore increasing the susceptibility of adult offspring to NAFLD, MS, and cholesterol-accumulation-related osteoarthritis, and there were differences between males and females [2, 35, 38, 39, 41, 64].
In summary, PEE, whether acute or chronic exposure (at least at high doses), causes a variety of adverse effects in offspring. Ethanol exposure in early pregnancy leads to structural abnormalities of the nervous system and adverse pregnancy outcomes, while in middle and late pregnancy, it is more likely to lead to IUGR, functional abnormalities, and susceptibility to multiple diseases in adulthood.
Developmental toxicity induced by the direct action of ethanol
In adults, 90–98% of absorbed ethanol is metabolized in the liver, while only 2–10% is directly excreted from the kidneys and exhaled from the lungs. Ethanol and its main metabolite, acetaldehyde, are small hydrophilic compounds that can directly enter the fetus through the placental barrier. After drinking three to five bottles of alcoholic drinks (approximately 150 mg/dl), the average human blood alcohol concentration is 33 mM, and the blood ethanol concentration of alcoholics is approximately 20–170 mM [65]. Previous work in our lab indicated that at a maternal ethanol consumption of 4 g/kg·d in rats, the serum ethanol concentration in the mother and fetus was 87 mM and 58 mM, respectively [2], indicating that ethanol can easily cross the placenta into the fetus. Cumming et al. indicated that the activity of liver alcohol dehydrogenase (ADH) in a fetus during late pregnancy or in newborn sheep was only 7% that of adults, and ADH activity in the placenta was even lower [66]. Pikkarainen and Räihä measured the activity of ADH in humans, and found that although fetal ADH activity in the second month of pregnancy was detectable, it was just 3–4% of the adult level. With an increase in gestational age, the activity of ADH gradually increases, but is always lower than that of adults [67]. Because the capacity of the fetal liver to metabolize ethanol is not mature, the actual metabolic burden in the developing fetus may be much higher than that in adults under similar conditions. In other words, the fetus is more prone to the direct toxic effects of ethanol. Chen et al. found that ethanol directly inhibited the Wnt/β-catenin pathway through oxidative stress to delay the differentiation of bone mesenchymal stem cells (BMSCs) and bone dysplasia, or directly upregulated IGF1 signaling pathway to cause ovarian cell apoptosis and premature ovarian insufficiency in female Sprague-Dawley rats [68]. Sun et al. also found that PEE can affect individual metabolism of endogenous and exogenous substances in early adulthood due to the direct effect of ethanol on impairment of protein levels and enzyme activities of cytochrome P450s (CYPs) in rats [69]. These results indicate that ethanol causes direct toxicity to multiple fetal tissues and organs via many mechanisms.
Developmental toxicity mediated by fetal exposure to excessive maternal glucocorticoids induced by PEE
In most previous studies, the developmental toxicity of ethanol was believed to be related to the direct action of ethanol on a fetus. However, in recent years, the influence of elevated levels of maternal-derived GCs induced by PEE has attracted more and more attention. Although current studies have shown that PEE increases maternal and fetal blood ethanol levels in a dose-dependent manner [2, 35, 70,71,72], there has been no direct comparison of the effects of different doses of prenatal ethanol on fetal serum GC levels in humans. The reports are different in animal models, but the existing literature suggests that the higher the dose of prenatal ethanol and the longer the duration, the higher the maternal and fetal GC levels [2, 35, 70,71,72].
Maternal glucocorticoid overexposure in offspring induced by PEE
The placenta is crucial for normal fetal development. Placental 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) and P-glycoprotein (P-gp) are key mediators of maternal GC inactivation and transport [73,74,75,76]. 11β-HSD2, the key enzyme catalyzing GC oxidation, is believed to be the “gate-keeper” of the placental barrier against GCs [77, 78]. Because adrenal gland function is immature during the fetal period, fetal GC levels mainly originate from the mother through the placental GC barrier [79, 80]. Population and rodent studies showed that placental 11β-HSD2 activity was easily affected by various adverse conditions during pregnancy, resulting in exposure of the developing fetus to excessive maternal GCs [81, 82]. The concentration of cortisol in the serum and urine of drinkers was significantly higher than that in non-drinkers [83,84,85,86]. We also confirmed in rats that PEE increased maternal GC levels via a stress response and opened the placental GC barrier (ethanol directly downregulated 11β-HSD2 expression through cAMP/PKA/EGR1 signaling), resulting in fetal exposure to excessive maternal-derived GCs and inducing IUGR [35].
Placental ATP binding cassette (ABC) transporters protect placental and fetal tissues by excreting exogenous and endogenous metabolites. ABCB1/MDR1, namely P-gp, is the most abundant drug efflux transporter expressed in the membrane on the maternal side of the placental syncytiotrophoblast, representing another GC barrier on the placenta. P-gp transports GCs back to the maternal side against the concentration gradient, which limits the entry of GCs into placental cells and fetuses, thus reducing fetal exposure to maternal GCs [87]. Studies have suggested that the expression of P-gp decreased in the intestinal epidermal cells of mice exposed to ethanol [88]. Our previous study also showed that the expression of P-gp in rat placenta decreased after exposure to ethanol alone [89], the mechanism of which is related to expression and activity changes in the JNK/YB-1/P300 pathway [90]. In addition, P-gp inducers can decrease maternal-derived GCs and IUGRs of offspring caused by adverse environmental conditions (including ethanol exposure) during pregnancy [90]. It is suggested that the expression and functional inhibition of placental P-gp are also involved in the opening of the placental GC barrier caused by PEE. In conclusion, changes in the placental barrier caused by PEE caused fetal overexposure to maternal-derived GCs.
Excessive exposure of fetuses to maternal glucocorticoids mediates offspring IUGR and postnatal susceptibility to multiple conditions
Basal levels of GCs play an important role in regulating the biosynthesis and metabolism of saccharides, fat, and protein and the proliferation and differentiation of cells. GCs are not only important factors in regulating fetal development and maturity, but they are also critical in determining the fate of the fetus after birth. Maternal GCs are the main source of fetal GC levels [35, 80]. A large number of reports suggest that high maternal GCs program the susceptibility of offspring to multiple conditions after birth [22, 79]. Human offspring with IUGR were found to have increased cortisol concentrations of umbilical cord blood at birth [91]. A variety of adverse factors during pregnancy can lead to increased fetal GCs and abnormal fetal development [81, 82, 92, 93]. Our previous studies also confirmed that the blood corticosterone levels of IUGR fetal rats exposed to ethanol during pregnancy were significantly higher than those of the control group; under these conditions, activity of the HPA axis in the maternal rats increased, while the synthesis of fetal adrenal steroids was inhibited, suggesting that maternal-derived GC overexposure occurred in the fetal rats [2, 35]. Furthermore, the increased expression of fetal liver triglyceride (TG) synthase, decreased neuronal activity in the paraventricular nucleus (PVN) region of the fetal hypothalamus, decreased activity of the fetal adrenal IGF1 signaling pathway, and inhibition of osteoclast differentiation at the osteo-cartilage interface were found to be mainly related to high maternal GCs induced by PEE [2, 35,36,37]. These studies all indicate that overexposure to maternal GCs may be another important mechanism of offspring IUGR and the susceptibility to multiple diseases in adulthood caused by PEE.
Mechanism of maternal-derived glucocorticoid-mediate intrauterine neuroendocrine metabolic programming induced by PEE
Intrauterine programming refers to the process of permanent changes in morphology and function of embryonic or fetal tissues and organs [22]. Under physiological conditions, a variety of neuroendocrine hormones are involved in programming the development and function of multiple fetal organs. As a core component of the “DOHaD”, intrauterine programming is also responsible for pathologies (susceptibility to multiple conditions). However, to date, the intrauterine programming mechanism of IUGR in offspring has not been systematically clarified. Recently, a series of studies by us and other researchers may have confirmed that alterations in intrauterine neuroendocrine metabolic programming induced by maternal GC overexposure were associated with susceptibility to multiple chronic diseases in adults [22, 94, 95].
Intrauterine maternal glucocorticoid overexposure and developmental programming of the HPA axis
The HPA axis, a neuroendocrine axis, plays an important role in prenatal and postnatal stress and defense responses. Mounting studies from human or animals have indicated that PEE causes changes in an offspring’s HPA axis, which are manifested as low basal activities from the intrauterine to early postnatal periods and high stress sensitivity throughout life [14, 96,97,98].
Low basal activity programming of the HPA axis
Although conclusions were not entirely consistent, accumulating studies have shown that the HPA axis has low basal activity in IUGR rats exposed prenatally to ethanol [96,97,98,99]. Previous studies from our group also found that the blood adrenocorticotropic hormone (ACTH) and corticosterone levels in IUGR rats induced by PEE were lower after birth than those of the control rats [41, 64]; it was further confirmed that fetal rats exposed to alcohol during pregnancy had low adrenal function, mainly manifested by reduced expression of steroidogenic acute regulatory protein (StAR) and cytochrome P450 family 11 subfamily A member 1 (P450scc) [35, 37], suggesting that the low basal activity of the HPA axis caused by PEE originated from the fetus.
The concentration of basal GCs during the fetal period and normal development of the fetal adrenal gland determine fetal maturity and postnatal fate [14, 100, 101]. It was found that development of adrenal function in IUGR offspring was delayed, resulting in low-activity programming of the HPA axis, which is consistent with the low activity of the HPA axis observed in offspring exposed prenatally to ethanol [97]. Our previous studies found that ethanol exposure during middle and late pregnancy inhibited the functional development of the HPA axis in fetal rats, which was manifested through fetal overexposure to maternal GCs, decreased corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP) levels in the hypothalamus, and decreased StAR and P450scc (the key genes of adrenal steroid hormone synthesis) expression [35, 37, 102]. Moreover, fetal maternal GC overexposure inhibited the steroid synthesis function of fetal adrenal cortical cells in a concentration-dependent manner, the mechanism of which was mainly related to the hypofunctional programming of adrenal corticosteroid synthesis induced by the GC activation system, including 11β-HSD1/2, GC receptor (GR), and CCAAT/enhancer binding protein α (C/EBPα). These effects continued after birth and even into adulthood [37]. Therefore, the changes in functional programming of the adrenal gland caused by maternal-derived GC overexposure induced by prenatal ethanol are an important reason for the intrauterine origin of the low basal activity of the HPA axis.
Stress hypersensitivity programming of the HPA axis
A number of studies from humans and animals have confirmed that stress hypersensitivity of the HPA axis is one of the key mechanisms in MS (e.g., diabetes and NAFLD) and mental disorders (e.g., depression and schizophrenia) susceptibility originating from fetuses [97, 103,104,105,106]. PEE programmably alters the stress sensitivity of the HPA axis in human and animal offspring [35, 97], but its specific mechanism has not been fully clarified. Our previous studies found that PEE in rats reduced serum ACTH and CORT levels by 65% and 60%, respectively, but there were no significant differences between PEE and control groups after chronic stress. Moreover, the increased rates of ACTH and CORT concentrations in offspring after PEE were significantly higher than those in offspring without exposure [41], suggesting that the offspring exposed prenatally to ethanol had stress hypersensitivity of the HPA axis, which was consistent with other basic research and clinical reports [97, 98, 107, 108]. Furthermore, we found that, in the offspring of rats exposed to ethanol during pregnancy, excitatory potential increased in the PVN of the hypothalamus whether examining fetal or adult offspring and this effect was characterized by invariable expression of an excitatory glutamate transporter (vesicular glutamate transporter 2 (VGluT2)) and decreased expression of inhibitory GABA synthase (65 kDa glutamic acid decarboxylase (GAD65)), resulting in a significant increase in the VGluT2/GAD65 ratio [109]. It is suggested that PEE permanently changes the setting point and sensitivity of hypothalamic PVN in offspring, resulting in stress hypersensitivity of the HPA axis.
As an advanced regulatory center, the hippocampus plays an important role in the HPA axis. The affinity of hippocampal mineralocorticoid receptor (MR) for GC is more than 10 times that of the GR [110]. When the GC level is low, almost all the GCs bind to the MR to regulate basal activity of the HPA axis; when GC levels increase, saturating the MR, GC then binds to the GR, resulting in glutamate release [111] and attenuation of the overactive HPA axis to the basal state through the Glu-GABA synaptic connection [112, 113]. Therefore, the balance of hippocampal MR/GR expression is critical to regulate HPA-axis activity, which directly determines the ratio of hippocampal VGluT2/GAD65 [104, 114]. It has been found that hippocampal GR participates in perinatal programming of the HPA axis by negatively regulating the expression of CRH [115]. After birth, the balance of MR and GR is maintained [116, 117]. However, once they are imbalanced, dysfunction of the HPA axis occurs, which is a typical response of the hippocampus to chronic stress or depression. Our studies found that PEE increased GR expression in fetal rats, leading to decreased MR/GR and increased VGluT2/GAD65 expression ratios. When the offspring of female rats exposed prenatally to ethanol were subjected to chronic stress (cool water swimming), the expression of GR in the hippocampus was higher than that of the control rats, the expression ratio of MR/GR was further reduced, and the VGluT2/GAD65 ratio was further increased. At the same time, pathological changes such as cell arrangement disorder, deep nuclear staining and shrinkage in the dentate gyrus (DG) and CA3 areas of the hippocampus were aggravated [102, 109]. These results suggest that PEE reduces negative regulatory effects on the hypothalamus to enhance its excitatory potential. The above changes can continue into adulthood. After chronic stress, the hippocampus damage is aggravated and the regulatory ability to HPA axis is further reduced, leading to hyperexcitation of the hypothalamus and finally to stress hypersensitivity of the HPA axis.
Intrauterine maternal glucocorticoid overexposure and developmental programming of the GC-IGF1 axis
IGF1 and its downstream signals are involved in regulating the development, differentiation, and metabolism of tissues and organs during the intrauterine period [16, 18, 118]. IGF1 regulates cell proliferation and apoptosis, and glucose and lipid metabolism by binding to the IGF1 receptor (IGF1R) [119]. Although IGF1 is expressed in almost all embryonic tissues [17], it is mainly produced in the liver during the fetal period. Hepatic IGF1 or IGF1R knockout or mutation significantly reduces fetal birth weight and length [120,121,122]. Studies confirmed that blood IGF1 levels decreased in fetuses with IUGR in mammals, and these levels were significantly higher in IUGR offspring exhibiting “catch-up growth” than in those not exhibiting “catch-up growth” [123,124,125], which is related to adult metabolic diseases [2, 126]. A study in our laboratory also showed that the expression of liver and serum IGF1 in fetuses of rats exposed to ethanol during pregnancy was lower than that of controls [2], while they also increased significantly after birth, and the increased rate was higher than that of the controls, which may be one of the intrauterine programming mechanisms of NAFLD [2].
GCs promote fetal maturation, while this process is theoretically accompanied by growth inhibition. Increasing numbers of studies have shown that high levels of GCs inhibit the expression of IGF1 in various tissues and cells of human and sheep [19, 20]. A series of studies confirmed that PEE increases the level of serum corticosterone in fetal rats, but decreases the levels of serum and liver IGF1. After birth, the levels of corticosterone decrease, while those of IGF1 increase. After a high-fat diet, corticosterone levels in the offspring further decrease, but IGF1 levels further increase and body weight shows “catch-up growth” [2]. Similarly, when fetal blood corticosterone levels in male IUGR offspring rats exposed to ethanol prenatally increase, the expression of IGF1 and downstream steroid synthase enzymes in the adrenal gland decreases. After birth, when corticosterone levels decrease in the offspring of rats, the expression of adrenal IGF1 and downstream steroid synthase enzymes shows a compensatory increase [37]. This negative change suggests that there may be an axial relationship between GC and IGF1 in multiple tissues (such as the liver and adrenal gland) (Fig. 1), which may be a physiological axis of fetal development and maturity, mediating adaptive changes and compensatory effects under the adverse intrauterine environment.
To confirm how maternal GC overexposure mediates programming alterations of the GC-IGF1 axis in fetuses, researchers systematically observed changes in corticosterone levels, the adrenal GC activation system, IGF1 signaling, and steroid synthesis before and after birth in IUGR rats induced by PEE in a rat model [37]. The results showed that maternal-derived GCs increased the expression of 11β-HSD1; decreased the expression of 11β-HSD2; increased the expression of MR, GR, and C/EBPα; and inhibited C/EBPβ and IGF1 signaling in fetal adrenal glands. Moreover, endogenous corticosterone content and the expression of corticosteroid synthase system decreased in the offspring exposed prenatally to ethanol. In the early postnatal period, these offspring showed low basal activity of the HPA axis and suppression of the adrenal GC activation system, but enhanced IGF1 signaling; after a postnatal high-fat diet, the HPA axis showed stress hypersensitivity, accompanied by local GC-IGF1 axis-mediated changes in organs and tissues (such as the adrenal gland and liver) [2, 64, 127]. This suggests that GC-IGF1-axis programming plays an important role in maintaining activity of the HPA axis and regulating organ and tissue functions before and after birth.

GC-IGF1 axis mediates programming changes in the offspring exposed prenatally to ethanol. HPA, hypothalamic-pituitary-adrenal; GCs, glucocorticoids; IGF1, insulin like growth factor 1; IUGR, intrauterine growth retardation
Epigenetic modifications in intrauterine programing and genetic toxicity induced by PEE
Epigenetics refers to heritable regulation of gene expression that does not involve gene sequence changes and is a reflection of environmental stimuli on genetic factors. Examples include DNA methylation, histone modification, and non-coding RNAs [128, 129]. It is believed that epigenetic changes are involved in the intrauterine origin of adult-onset diseases and are responsible for the continuation of fetal programming that is induced by adverse intrauterine environments into the postnatal period and even the next generation of offspring [79]. Studies have suggested that PEE induces epigenetic changes in various organs and cells (Table 2) [130, 131]. As early as the 1990s, Garro et al. found that acute ethanol (3 g/kg twice a day) exposure from the ninth to the eleventh day of gestation caused extensive DNA hypomethylation in fetal mouse [132]. Recently, News et al. reported that pregnant mouse exposure to alcohol (2 g/kg) on embryonic 18.5 could result in incorporation of alcohol into gestating fetal brains and contribute to rapid histone acetylation in the brain in part by direct deposition of alcohol-derived acetyl groups onto histones in an ACSS2-dependent manner [133]. Based on the whole embryo culture technique, Liu et al. also found that alcohol exposure (88 mM) during mouse embryonic neurodevelopment induced alterations in the DNA methylation of the fetal brain, mainly occurring on chromosomes 7, 10, and X, which further caused delayed closure of the neural tube [134].
In addition, PEE in mice can alter acetylation of key genes and the expression of non-coding RNAs (lncRNA1354, miR-467b-5p, and miR-302c) in the hippocampus, liver and sperm cells of offspring to affect the development of tissues and organs in adults (postnatal day 87 or 70) [135, 136]. Moisiadis and Matthews reviewed multiple publications and found that excessive maternal GCs in fetuses can induce universal DNA methylation, histone acetylation, and miRNA expression, which might participate in fetal programing and intergenerational inheritance of fetal-derived neurological, cardiovascular and metabolic diseases [79]. Moreover, studies from our group indicated that PEE decreased H3K9 and H3K14 histone acetylation within the IGF1 promoter and increased IGF1 gene expression in multiple fetal organs of rats (e.g., the liver and bones), triggering altered GC-IGF1 axis programming (high GC and low IGF1 in intrauterine; low GC and high IGF1 after birth); these epigenetic changes were inherited by the F2 generation [127]. Stouder et al. also found that PEE in mice caused hypomethylation of the imprinted gene H19 in the somatic and spermatogonial cells of offspring, which was transferred to F2 generation [137]. These epigenetic changes in imprinted genes and the GC-IGF1 axis partly explain the intrauterine origin, long-term effect and multigenerational inheritance of developmental toxicity in offspring induced by PEE [136]. PEE can also lead to expression and promoter methylation changes in common imprinted genes such as IGF2 [44, 138, 139]. However, it mediates developmental toxicity and multigenerational inheritance effects in PEE offspring and whether there are other mechanisms involved in PEE-induced multigenerational inheritance needs to be further studied.
“Dual programing” and “two strikes” mechanisms mediate multiple fetal-originated Diseases caused by PEE
“Thrifty phenotypes” are fetal adaptive changes, along with nutrition and energy redistribution, which maintain the normal development and function of crucial organs when fetuses were exposed to adverse environments during pregnancy. However, such individuals become susceptible to diseases in adulthood [146]. Thus, we hold that when a mother is exposed to ethanol during pregnancy, on the one hand, ethanol directly enters the circulation of the fetus to injure fetal tissues; on the other hand, under conditions of high maternal GCs, the expression of 11β-HSD2 and P-gp decreases, and the placental GC barrier opens. Then, the maternal-derived GCs injure fetal tissues and modulate IGF1 signaling in various fetal organs and tissues to trigger the redistribution of nutrition and energy to protect the development of crucial organs (such as liver and brain), while attenuating the development of non-critical organs (such as bone and cartilage) to maintain fetal survival. This makes the fetus “thrifty.“
After birth, with the withdrawal of maternal-derived GCs and low GC expression in the offspring themselves, liver IGF1 expression continues to increase [37]. Therefore, the levels of liver and serum IGF1 in the offspring exposed prenatally to ethanol were significantly higher than those in the control group for a long time after birth, along with notable postnatal “catch-up growth” [2], but the blood levels of growth hormone (GH) were consistently lower (data unpublished). In other words, the excessive increase in hepatic IGF1 expression mediated by GCs is responsible for the “catch-up growth” of IUGR offspring, rather than the traditional GH-IGF1 axis. Interestingly, although GC-IGF1 axis programming (high GC and low IGF1 in intrauterine; low GC and high IGF1 after birth) was altered in IUGR offspring subjected to PEE, the change in expression of genes involving liver glycolipid metabolism was not altered. Based on these findings, we have proposed that this process be defined as the “one programing” where the functional changes in various organs are caused by GCs under PEE and continue to adulthood. Meanwhile, the programming changes in multiple organ functions mediated by the GC-IGF1 axis were defined as “another programing,“ which mainly reflects the pre- and postnatal adaptive and compensatory changes in the multi-organ functions of offspring exposed to ethanol during pregnancy.
The HPA axis showed low basal activity and hypersensitivity to chronic stress in IUGR offspring induced by PEE, as well as the GC-dependent phenotype of glucose and lipid metabolism [41, 64]. Those findings indicated that the excessive maternal-derived GCs induced by PEE and ethanol itself caused the “first strike” to the fetuses, which disrupted fetal development of organs and further induced adaptive alterations. Outside of the adverse intrauterine environment and under conditions of adequate postnatal nutrition, the offspring demonstrated “catch-up growth” through redistribution of nutrients and energy to compensate for the organ dysplasia. However, it was confirmed by the rat and zebrafish embryo models that those changes might disturb fetal-originated metabolic programing and exacerbate the abnormal morphology of tissues and organs and dysfunction of glucose and lipid metabolism to exhaust the metabolic potential of the offspring, thus increasing susceptibility to metabolic diseases [2, 126, 147, 148]. When the offspring again suffered from adverse factors postnatally, such as high-fat diet or chronic stress [41] (these were defined as the “second strike”), various metabolic diseases developed. Therefore, “dual programing” (including the “one programing” and “another programing”) and “two strikes” (including the “first strike” and “second strike”) induced by PEE mediate the dysplasia of multiple organs and susceptibility to various diseases in the offspring.
Sex-based differences and mechanism of developmental toxicity induced by PEE
Although the developmental toxicity of ethanol has been reported in both female and male animals and the high sensitivity of the HPA axis is very consistent between sexes, numerous studies still suggest that there are sex-based differences in severity and specific manifestations [61, 62].
Sex-based differences caused by placental barrier
Studies have shown that PEE inhibits the expression and activity of 11β-HSD2 and P-gp more significantly in female placentas than males [149]. The suppressed expression and activity of 11β-HSD2 are related to the higher 11β-HSD2 DNA methylation level in female rat placenta [150]. When Sprague Dawley rats were exposed to ethanol in the middle and late stages of pregnancy, the expression of 11β-HSD2 was decreased in female placenta and increased in male placenta [149]. The difference in PEE between male and female placentas makes it easier for maternal-derived GCs to enter female fetuses, resulting in sex-based differences in developmental toxicity. Furthermore, high maternal cortisol levels have been reported to induce GC resistance in male fetuses, while female fetuses remain sensitive [151]. In addition, the GR gene is mainly composed of five subtypes: GRα, GRβ, GRγ, GRA, and GRP [152, 153]. GRα has the strongest activation effect on target genes; GRA, GRβ, and GRP cannot bind to GCs [154, 155]; GRP is related to GC resistance; and GRγ only has ~ 50% of the activation effect of GRα [156]. Human placental trophoblasts express 12 GR protein subtypes, which makes the placenta uniquely sensitive to GCs [157]. Under the high intrauterine GC levels induced by PEE, the male placenta develops GC resistance by increasing GRβ, GRA, and GRP expression, while the female placenta increases GC sensitivity by decreasing expression of GRβ and enhancing the interaction between GRαA, GRαD3, and GRαC [157]. Therefore, the differences in placental barrier and GC sensitivity are the reasons for the sex differences in developmental toxicity caused by PEE. However, whether the difference in GC sensitivity of other tissues and organs mediates the sex difference in developmental toxicity of PEE needs to be further studied.
Sex-based differences caused by neuroendocrine metabolic programming
The differences in HPA axis development and sensitivity might also be responsible for the sex differences in susceptibility to adult-onset diseases [158]. Studies from humans have demonstrated that the basal activity and sensitivity of the HPA axis are much higher in females than in males [159, 160]. In rats, as the basal serum corticosterone levels, corticosterone secretion rate, and reactivities to ACTH and corticosterone in female are higher than those in males in physiology, the HPA axis is also activated more easily in females than in males, especially during proestrus when serum estrogen reaches its peak, which further increases the activity and sensitivity of the HPA axis [161, 162]. Our previous studies observed that PEE increased blood corticosterone concentration in rat offspring, upregulated GR expression in the hippocampus, decreased or did not change the expression of IGF1 and GAD67 from the intrauterine to postnatal phase in females, but increased the expression of GR, IGF1, and GAD67 in the male offspring hippocampus prenatally and in adulthood [102, 109]. Additional studies from both mice and rats indicated that PEE disrupted development of the hypothalamic-pituitary-gonadal (HPG) axis and delayed its maturation [163, 164], which may also be one of the reasons for sex differences. PEE caused postponed adolescence, abnormal vaginal development, abnormal estrogen synthesis and secretion, and sexual behavior changes in female animals [1, 165,166,167]; however, abnormal testicular development (including a decreased number of interstitial glands), vesicles in the seminiferous tubules, suppressed testosterone synthesis and secretion, hyposensitivity to luteinizing hormone (LH), feminization, hyposensitivity of the HPG axis and sexual debility were observed in the male offspring of rats [7, 168,169,170,171,172].
Furthermore, the HPG axis and HPA axis can mutually regulate each other’s functions at various tissue levels [97]. For instance, estradiol can promote ACTH release during stress, thereby increasing GC levels, while progesterone competes with estradiol to inhibit HPA axis activity; only when progesterone is at extremely low levels does the regulatory effect of estrogen on the HPA axis become evident [161, 162]. Testosterone can inhibit CRH mRNA levels and reduce the body’s responsiveness to stress [162, 173]. Moreover, the CRH gene promoter region contains estrogen and androgen response elements, and estrogen can promote CRH transcription in the hypothalamus, while androgens inhibit CRH transcription; therefore, testosterone can reduce corticosterone production, while estrogen has the opposite effect [97, 162, 173]. This may be one of the reasons why females are more sensitive to stress compared to males and why the basal activity and sensitivity of the HPA axis in female offspring induced by PEE are higher than in males [159, 160]. In addition, an increase in GC levels inhibited the activity of 17β-hydroxysteroid dehydrogenase (an important enzyme in testosterone synthesis) in the testes, leading to a reduction in serum testosterone levels in males [174], which can also inhibit the release of gonadotropin-releasing hormone (GnRH) from the hypothalamus and interfere with the release of follicle-stimulating hormone (FSH) and LH induced by GnRH, thereby reducing estrogen levels [175]. IGF1 is also important for the development of the HPG axis, including testis and ovaries [175,176,177]. Studies in our lab also found that PEE caused elevated blood corticosterone levels and decreased expression of IGF1 in the liver, testes and ovaries of the fetal rats [2, 37, 49], which resulted in ovarian and testicular dysplasia, therefore decreasing estrogen and testosterone levels in female and male offspring, respectively [48, 49]. In summary, all HPA, HPG and GC-IGF1 axes may be involved in the sex difference of developmental toxicity caused by PEE.
Summary and prospects
The mechanism of dysplasia of fetal tissues and organs and susceptibility to fetal-originated diseases in offspring caused by PEE is not only related to the direct actions of ethanol or its metabolites, but also related to the fetal neuroendocrine metabolic programming changes (HPA and GC-IGF1 axis programming). The “dual programing” and “two strikes” mechanisms induced by the direct effects of ethanol and neuroendocrine programming lead to the occurrence of various metabolic diseases in adult offspring (Fig. 2). Epigenetic modifications, placental barrier and neuroendocrine metabolic programming and HPG axis participated in sex-based differences and intergenerational inheritance induced by PEE, but some key points of these are still absolutely unclear.

The intrauterine programing mechanism of adult metabolic diseases induced by PEE. GC, glucocorticoid; IGF1, insulin like growth factor 1; IUGR, intrauterine growth retardation; 11β-HSD2, 11β-hydroxysteroid dehydrogenase 2; P-gp, P-glycoprotein; HPA axis, hypothalamic-pituitary-adrenal axis
Additionally, there are other common neuroendocrine axes (systems) with important roles in the human including the hypothalamic pituitary thyroid axis (HPT) and renin angiotensin system (RAS) except for HPA, GC-IGF1, and HPG axes. Hannigan and Cudd et al. showed that PEE led to a decrease in triiodothyronine (T3) and T4 levels in Long-Evans rats and goat offspring, both prenatally and after birth, while having no significant effect on free T4 [178, 179]. Wilcoxon et al. also indicated that PEE could result in permanent programming changes in the HPT axis in offspring rats and behavioral and cognitive dysfunction in offspring rats, and decreased expression of GAP-43 and GR in hippocampus, while supplementing with T4 can reverse these changes [180, 181]. Moreover, studies by Fidalgo and us also suggested that PEE inhibited the binding of angiotensin (Ang) IV to Ang II type 1α receptor (AT1R), increased the expression of angiotensin converting enzyme (ACE), Ang II, and AT1R in the serum and kidney, while decreasing the expression of ACE2, AT2R, and Mas receptor (MasR), thereby eliminating the memory consolidation effect of exogenous Ang IV on male offspring aged 3–6 months, and causing kidney and bone dysplasia, as well as adult (24 weeks after birth) nephrotic syndrome and osteoporosis [182,183,184]. All of these indicate that PEE may also lead to changes in other neuroendocrine axes such as HPT and RAS. However, current related studies are still limited, mostly at the level of animals and observational studies, lacking in-depth programming mechanisms and epidemiological investigations. There have also been no reports on whether maternal glucocorticoid overexposure caused by PEE is involved in changes in the HPT axis and RAS system. At the same time, due to differences in exposure time and dose, there are still many inconsistencies in the existing literature [52]. Therefore, more systematic research is urgently needed.
Finally, with accumulating research on fetal-originated diseases, translational medicine is continuing to bring basic research to clinical practice or clinical applications. Though avoiding ethanol exposure during pregnancy could be the best option, in cases where exposure has already occurred, based on the temporal characteristics of ethanol-induced developmental toxicity, sex differences, epigenetic programming and “dual programing” and “two strikes” mechanisms, interventions of ethanol exposure during early pregnancy primarily should focus on fetal structural abnormalities and abnormal pregnancy outcomes, while ethanol exposure during mid to late pregnancy primarily should aim to prevent long-term effects and increased susceptibility to multiple diseases in adult; moreover, specific interventions can be targeted towards male or female offspring based on the reasons for sex differences, respectively, while epigenetics may serve as diagnostic and warning markers or intervention targets; meanwhile, voiding a second strike after birth is also one of the effective strategies to prevent fetal-originated diseases caused by PEE. However, due to the some unclear molecular mechanism of the developmental toxicity and susceptibility to various diseases caused by PEE, the established targets at present are greatly limited. Therefore, based on neuroendocrine metabolic programming, the primary and secondary prevention of birth defects induced by prenatal adverse environments (e.g., ethanol exposure) needs to be further studied.
Data Availability
All data are included in the manuscript.
Abbreviations
- PEE:
-
Prenatal ethanol exposure
- IUGR:
-
Intrauterine growth retardation
- FAS:
-
Fetal alcohol syndrome
- MS:
-
Metabolic syndrome
- DOHaD:
-
Developmental Origins of Health and Disease
- HPA:
-
Hypothalamus-pituitary-adrenal
- NAFLD:
-
Non-alcoholic fatty liver disease
- IGF1:
-
Insulin-like growth factor 1
- FASDs:
-
Fetal alcohol spectrum disorders
- GC:
-
Glucocorticoid
- SGA:
-
Small for gestational age
- GD:
-
Gestational day
- PD:
-
Postanal day
- PW:
-
Postanal week
- CYP:
-
Cytochrome P450
- CORT:
-
Corticosteron
- IQ:
-
Intelligence quotient
- NR2B:
-
N-methyl-d-aspartate receptor subtype 2B
- GluR2/3:
-
Glutamate receptor subunits 2/3
- GABA-A:
-
γ-aminobutyric acid type A
- ADH:
-
Alcohol dehydrogenase
- BMSCs:
-
Bone mesenchymal stem cells
- 11β-HSD2:
-
11β-hydroxysteroid dehydrogenase type 2
- P-gp:
-
P-glycoprotein
- ABC:
-
ATP binding cassette
- TG:
-
Liver triglyceride
- PVN:
-
Paraventricular nucleus
- ACTH:
-
Adrenocorticotropic hormone
- StAR:
-
Steroidogenic acute regulatory protein
- P450scc:
-
P450 family 11 subfamily A member 1
- CRH:
-
Corticotrophin-releasing hormone
- AVP:
-
Arginine vasopressin
- GR:
-
Glucocorticoid receptor
- C/EBPα:
-
CCAAT/enhancer binding proteinα
- VGluT2:
-
Vesicular glutamate transporter 2
- GAD65:
-
65 kDa glutamic acid decarboxylase
- MR:
-
Mineralocorticoid receptor
- DG:
-
Dentate gyrus
- IGF1R:
-
Insulin-like growth factor 1 receptor
- GABRD:
-
δsubunit GABAA receptor
- Dnmt1:
-
DNA methyltransferase 1
- Dnmt3a:
-
DNA methyltransferase 3α
- MeCP2:
-
Methyl-CpG binding protein 2
- H19:
-
H19 imprinted maternally expressed transcript
- Nlgn3:
-
Neuroligin-3
- Elavl2:
-
ELAV-like RNA binding protein 2
- Sim1:
-
SIM BHLH transcription factor 1
- Cyp4f13:
-
Cytochrome P450 family 4 subfamily f polypeptide 13
- GH:
-
Growth hormone
- HPG:
-
Hypothalamic-pituitary-gonadal
- GnRH:
-
Gonadotropin-releasing hormone
- FSH:
-
Follicle-stimulating hormone
- HPT:
-
Hypothalamic pituitary thyroid axis
- RAS:
-
Renin angiotensin system
- T3:
-
triiodothyronine
- T4:
-
Thyroxine
- Ang:
-
Angiotensin
- AT1R:
-
Angiotensin II type 1α receptor
- ACE:
-
Angiotensin converting enzyme
- MasR:
-
Mas receptor
References
Esquifino AI, Sanchis R, Guerri C. Effect of prenatal alcohol exposure on sexual maturation of female rat offspring. Neuroendocrinology. 1986;44(4):483–7.
Shen L, Liu Z, Gong J, Zhang L, Wang L, Magdalou J, et al. Prenatal ethanol exposure programs an increased susceptibility of non-alcoholic fatty Liver Disease in female adult offspring rats. Toxicol Appl Pharmacol. 2014;274(2):263–73.
Fraser A, Ebrahim S, Ben-Shlomo Y, Davey Smith G, Lawlor DA. Intrauterine growth retardation, insulin resistance, and nonalcoholic fatty Liver Disease in children: response to Nobili et al. Diabetes Care. 2007;30(11):e124. author reply e5.
Pruett D, Waterman EH, Caughey AB. Fetal alcohol exposure: consequences, diagnosis, and treatment. Obstet Gynecol Surv. 2013;68(1):62–9.
Popova S, Lange S, Probst C, Gmel G, Rehm J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: a systematic review and meta-analysis. Lancet Glob Health. 2017;5(3):e290–e9.
Popova S, Lange S, Probst C, Gmel G, Rehm J. Global prevalence of alcohol use and binge drinking during pregnancy, and fetal alcohol spectrum disorder. Biochem Cell Biol. 2018;96(2):237–40.
Chen JJ, Smith ER. Effects of perinatal alcohol on sexual differentiation and open-field behavior in rats. Horm Behav. 1979;13(3):219–31.
Chen L, Nyomba BL. Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism. 2003;52(4):454–62.
Lewis SJ, Zuccolo L, Davey Smith G, Macleod J, Rodriguez S, Draper ES, et al. Fetal alcohol exposure and IQ at age 8: evidence from a population-based birth-cohort study. PLoS ONE. 2012;7(11):e49407.
Barker DJ. Fetal origins of coronary Heart Disease. Br Heart J. 1993;69(3):195–6.
Barker DJ. The fetal origins of coronary Heart Disease. Eur Heart J. 1997;18(6):883–4.
Barker DJ. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci. 2004;359(1449):1359–66.
Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261(5):412–7.
Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol. 2014;10(7):391–402.
Yu Y, Shi Z, Xu D, Li Y, Qin J, Zhang Z, et al. Prenatal ethanol exposure increases susceptibility to depression- and anxiety-like behavior in adult female offspring and its underlying mechanism. Reprod Toxicol. 2020;96:36–46.
Roberts CT, Owens JA, Sferruzzi-Perri AN. Distinct actions of insulin-like growth factors (IGFs) on placental development and fetal growth: lessons from mice and guinea pigs. Placenta. 2008;29(Suppl A):42–7.
Agrogiannis GD, Sifakis S, Patsouris ES, Konstantinidou AE. Insulin-like growth factors in embryonic and fetal growth and skeletal development (review). Mol Med Rep. 2014;10(2):579–84.
Netchine I, Azzi S, Houang M, Seurin D, Perin L, Ricort JM, et al. Partial primary deficiency of insulin-like growth factor (IGF)-I activity associated with IGF1 mutation demonstrates its critical role in growth and brain development. J Clin Endocrinol Metab. 2009;94(10):3913–21.
Hyatt MA, Budge H, Walker D, Stephenson T, Symonds ME. Ontogeny and nutritional programming of the hepatic growth hormone-insulin-like growth factor-prolactin axis in the sheep. Endocrinology. 2007;148(10):4754–60.
Inder WJ, Jang C, Obeyesekere VR, Alford FP. Dexamethasone administration inhibits skeletal muscle expression of the androgen receptor and IGF-1–implications for steroid-induced myopathy. Clin Endocrinol (Oxf). 2010;73(1):126–32.
Weeks O, Bossé GD, Oderberg IM, Akle S, Houvras Y, Wrighton PJ, et al. Fetal alcohol spectrum disorder predisposes to metabolic abnormalities in adulthood. J Clin Invest. 2020;130(5):2252–69.
Zhang C, Xu D, Luo H, Lu J, Liu L, Ping J, et al. Prenatal xenobiotic exposure and intrauterine hypothalamus-pituitary-adrenal axis programming alteration. Toxicology. 2014;325:74–84.
Armstrong BG, McDonald AD, Sloan M. Cigarette, alcohol, and coffee consumption and spontaneous abortion. Am J Public Health. 1992;82(1):85–7.
Rasch V. Cigarette, alcohol, and caffeine consumption: risk factors for spontaneous abortion. Acta Obstet Gynecol Scand. 2003;82(2):182–8.
Henriksen TB, Hjollund NH, Jensen TK, Bonde JP, Andersson AM, Kolstad H, et al. Alcohol consumption at the time of conception and spontaneous abortion. Am J Epidemiol. 2004;160(7):661–7.
Ammon Avalos L, Kaskutas LA, Block G, Li DK. Do multivitamin supplements modify the relationship between prenatal alcohol intake and miscarriage? Am J Obstet Gynecol. 2009;201(6):563e1–9.
Avalos LA, Roberts SC, Kaskutas LA, Block G, Li DK. Volume and type of alcohol during early pregnancy and the risk of miscarriage. Subst Use Misuse. 2014;49(11):1437–45.
Fowden AL, Forhead AJ. Endocrine mechanisms of intrauterine programming. Reproduction. 2004;127(5):515–26.
Peacock JL, Bland JM, Anderson HR. Effects on birthweight of alcohol and caffeine consumption in smoking women. J Epidemiol Community Health. 1991;45(2):159–63.
Larroque B, Kaminski M, Lelong N, Subtil D, Dehaene P. Effects of birth weight of alcohol and caffeine consumption during pregnancy. Am J Epidemiol. 1993;137(9):941–50.
Shu XO, Hatch MC, Mills J, Clemens J, Susser M. Maternal Smoking, alcohol drinking, caffeine consumption, and fetal growth: results from a prospective study. Epidemiology. 1995;6(2):115–20.
Lundsberg LS, Bracken MB, Saftlas AF. Low-to-moderate gestational alcohol use and intrauterine growth retardation, low birthweight, and preterm delivery. Ann Epidemiol. 1997;7(7):498–508.
Aliyu MH, Wilson RE, Zoorob R, Brown K, Alio AP, Clayton H, et al. Prenatal alcohol consumption and fetal growth restriction: potentiation effect by concomitant Smoking. Nicotine Tob Res. 2009;11(1):36–43.
Kleiber ML, Mantha K, Stringer RL, Singh SM. Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J Neurodev Disord. 2013;5(1):6.
Liang G, Chen M, Pan XL, Zheng J, Wang H. Ethanol-induced inhibition of fetal hypothalamic-pituitary-adrenal axis due to prenatal overexposure to maternal glucocorticoid in mice. Exp Toxicol Pathol. 2011;63(7–8):607–11.
Pan Z, Zhang X, Shangguan Y, Hu H, Chen L, Wang H. Suppressed osteoclast differentiation at the chondro-osseous junction mediates endochondral ossification retardation in long bones of Wistar fetal rats with prenatal ethanol exposure. Toxicol Appl Pharmacol. 2016;305:234–41.
Huang H, He Z, Zhu C, Liu L, Kou H, Shen L, et al. Prenatal ethanol exposure-induced adrenal developmental abnormality of male offspring rats and its possible intrauterine programming mechanisms. Toxicol Appl Pharmacol. 2015;288(1):84–94.
Ni Q, Wang L, Wu Y, Shen L, Qin J, Liu Y, et al. Prenatal ethanol exposure induces the osteoarthritis-like phenotype in female adult offspring rats with a post-weaning high-fat diet and its intrauterine programming mechanisms of cholesterol metabolism. Toxicol Lett. 2015;238(2):117–25.
Ni Q, Tan Y, Zhang X, Luo H, Deng Y, Magdalou J, et al. Prenatal ethanol exposure increases osteoarthritis susceptibility in female rat offspring by programming a low-functioning IGF-1 signaling pathway. Sci Rep. 2015;5:14711.
Cianfarani S, Germani D, Branca F. Low birthweight and adult insulin resistance: the catch-up growth hypothesis. Arch Dis Child Fetal Neonatal Ed. 1999;81(1):F71–3.
Xia LP, Shen L, Kou H, Zhang BJ, Zhang L, Wu Y, et al. Prenatal ethanol exposure enhances the susceptibility to metabolic syndrome in offspring rats by HPA axis-associated neuroendocrine metabolic programming. Toxicol Lett. 2014;226(1):98–105.
Painter RC, Osmond C, Gluckman P, Hanson M, Phillips DI, Roseboom TJ. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG. 2008;115(10):1243–9.
Nomura Y, Wickramaratne PJ, Pilowsky DJ, Newcorn JH, Bruder-Costello B, Davey C, et al. Low birth weight and risk of affective disorders and selected medical Illness in offspring at high and low risk for depression. Compr Psychiatry. 2007;48(5):470–8.
Dobson CC, Thevasundaram K, Mongillo DL, Winterborn A, Holloway AC, Brien JF, et al. Chronic prenatal ethanol exposure alters expression of central and peripheral insulin signaling molecules in adult guinea pig offspring. Alcohol. 2014;48(7):687–93.
Dobson CC, Mongillo DL, Brien DC, Stepita R, Poklewska-Koziell M, Winterborn A, et al. Chronic prenatal ethanol exposure increases adiposity and disrupts pancreatic morphology in adult guinea pig offspring. Nutr Diabetes. 2012;2(12):e57.
Xiao D, Kou H, Gui S, Ji Z, Guo Y, Wu Y, et al. Age-characteristic changes of glucose metabolism, pancreatic morphology and function in male offspring rats Induced by prenatal ethanol exposure. Front Endocrinol (Lausanne). 2019;10:34.
Simpson ME, Duggal S, Keiver K. Prenatal ethanol exposure has differential effects on fetal growth and skeletal ossification. Bone. 2005;36(3):521–32.
Liu M, Zhang Q, Pei L, Zou Y, Chen G, Wang H. Corticosterone rather than ethanol epigenetic programmed testicular dysplasia caused by prenatal ethanol exposure in male offspring rats. Epigenetics. 2019;14(3):245–59.
Ni Y, Xu D, Lv F, Wan Y, Fan G, Zou W et al. Prenatal ethanol exposure induces susceptibility to premature ovarian insufficiency. J Endocrinol. 2019.
Youngentob SL, Glendinning JI. Fetal ethanol exposure increases ethanol intake by making it smell and taste better. Proc Natl Acad Sci U S A. 2009;106(13):5359–64.
Iqbal U, Dringenberg HC, Brien JF, Reynolds JN. Chronic prenatal ethanol exposure alters hippocampal GABA(A) receptors and impairs spatial learning in the guinea pig. Behav Brain Res. 2004;150(1–2):117–25.
Gray SP, Kenna K, Bertram JF, Hoy WE, Yan EB, Bocking AD, et al. Repeated ethanol exposure during late gestation decreases nephron endowment in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2008;295(2):R568–74.
Sozo F, Vela M, Stokes V, Kenna K, Meikle PJ, De Matteo R, et al. Effects of prenatal ethanol exposure on the lungs of postnatal lambs. Am J Physiol Lung Cell Mol Physiol. 2011;300(1):L139–47.
May PA, Gossage JP, Brooke LE, Snell CL, Marais AS, Hendricks LS, et al. Maternal risk factors for fetal alcohol syndrome in the western cape province of South Africa: a population-based study. Am J Public Health. 2005;95(7):1190–9.
Willhite CC, Hendrickx AG, Burk DT, Book SA. Warnings and the hazards of drinking alcoholic beverages during pregnancy. Teratology. 1988;37(6):609–11.
Kelly YJ, Sacker A, Gray R, Kelly J, Wolke D, Head J, et al. Light drinking during pregnancy: still no increased risk for socioemotional difficulties or cognitive deficits at 5 years of age? J Epidemiol Community Health. 2012;66(1):41–8.
Robinson M, Oddy WH, McLean NJ, Jacoby P, Pennell CE, de Klerk NH, et al. Low-moderate prenatal alcohol exposure and risk to child behavioural development: a prospective cohort study. BJOG. 2010;117(9):1139–50.
Gray R. Low-to-moderate alcohol consumption during pregnancy and child development–moving beyond observational studies. BJOG. 2013;120(9):1039–41.
Bâ A, Alcohol. B1 vitamin deficiency-related stillbirths. J Matern Fetal Neonatal Med. 2009;22(5):452–7.
Catlin MC, Abdollah S, Brien JF. Dose-dependent effects of prenatal ethanol exposure in the guinea pig. Alcohol. 1993;10(2):109–15.
Fish EW, Holloway HT, Rumple A, Baker LK, Wieczorek LA, Moy SS, et al. Acute alcohol exposure during neurulation: behavioral and brain structural consequences in adolescent C57BL/6J mice. Behav Brain Res. 2016;311:70–80.
Diaz MR, Mooney SM, Varlinskaya EI. Acute prenatal exposure to ethanol on gestational day 12 elicits opposing deficits in social behaviors and anxiety-like behaviors in Sprague Dawley rats. Behav Brain Res. 2016;310:11–9.
Dettmer TS, Barnes A, Iqbal U, Bailey CD, Reynolds JN, Brien JF, et al. Chronic prenatal ethanol exposure alters ionotropic glutamate receptor subunit protein levels in the adult guinea pig cerebral cortex. Alcohol Clin Exp Res. 2003;27(4):677–81.
He Z, Li J, Luo H, Zhang L, Ma L, Chen L, et al. Sex-specific increase in susceptibility to metabolic syndrome in adult offspring after prenatal ethanol exposure with post-weaning high-fat diet. Sci Rep. 2015;5:17679.
Gohlke JM, Griffith WC, Faustman EM. A systems-based computational model for dose-response comparisons of two mode of action hypotheses for ethanol-induced neurodevelopmental toxicity. Toxicol Sci. 2005;86(2):470–84.
Cumming ME, Ong BY, Wade JG, Sitar DS. Ethanol disposition in newborn lambs and comparison of alcohol dehydrogenase activity in placenta and maternal sheep, fetal and neonatal lamb liver. Dev Pharmacol Ther. 1985;8(6):338–45.
Pikkarainen PH, Räihä NC. Development of alcohol dehydrogenase activity in the human liver. Pediatr Res. 1967;1(3):165–8.
Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/beta-catenin signaling. J Bone Miner Res. 2010;25(5):1117–27.
Sun X, He L, Bi H, Huang M, Xiang E, Li X, et al. Prenatal ethanol exposure induces dynamic changes of expression and activity of hepatic cytochrome P450 isoforms in male rat offspring. Reprod Toxicol. 2022;109:101–8.
Lan N, Chiu MP, Ellis L, Weinberg J. Prenatal alcohol exposure and prenatal stress differentially alter glucocorticoid signaling in the placenta and fetal brain. Neuroscience. 2017;342:167–79.
Ngai YF, Sulistyoningrum DC, O’Neill R, Innis SM, Weinberg J, Devlin AM. Prenatal alcohol exposure alters methyl metabolism and programs serotonin transporter and glucocorticoid receptor expression in brain. Am J Physiol Regul Integr Comp Physiol. 2015;309(5):R613–22.
Hewitt AJ, Dobson CC, Brien JF, Wynne-Edwards KE, Reynolds JN. Chronic ethanol exposure increases the non-dominant glucocorticoid, corticosterone, in the near-term pregnant guinea pig. Alcohol. 2014;48(5):477–81.
Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci. 2006;26(14):3840–4.
Seckl JR, Cleasby M, Nyirenda MJ. Glucocorticoids, 11beta-hydroxysteroid dehydrogenase, and fetal programming. Kidney Int. 2000;57(4):1412–7.
Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. 11 beta-hydroxysteroid dehydrogenases: key enzymes in determining tissue-specific glucocorticoid effects. Steroids. 1996;61(4):263–9.
Evseenko DA, Paxton JW, Keelan JA. Independent regulation of apical and basolateral drug transporter expression and function in placental trophoblasts by cytokines, steroids, and growth factors. Drug Metab Dispos. 2007;35(4):595–601.
Tsugita M, Iwasaki Y, Nishiyama M, Taguchi T, Shinahara M, Taniguchi Y, et al. Differential regulation of 11beta-hydroxysteroid dehydrogenase type-1 and – 2 gene transcription by proinflammatory cytokines in vascular smooth muscle cells. Life Sci. 2008;83(11–12):426–32.
McNeil CJ, Nwagwu MO, Finch AM, Page KR, Thain A, McArdle HJ, et al. Glucocorticoid exposure and tissue gene expression of 11beta HSD-1, 11beta HSD-2, and glucocorticoid receptor in a porcine model of differential fetal growth. Reproduction. 2007;133(3):653–61.
Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: mechanisms. Nat Rev Endocrinol. 2014;10(7):403–11.
Fowden AL, Li J, Forhead AJ. Glucocorticoids and the preparation for life after birth: are there long-term consequences of the life insurance? Proc Nutr Soc. 1998;57(1):113–22.
Reynolds RM. Glucocorticoid excess and the developmental origins of Disease: two decades of testing the hypothesis–2012 Curt Richter award winner. Psychoneuroendocrinology. 2013;38(1):1–11.
Morrison JL, Botting KJ, Soo PS, McGillick EV, Hiscock J, Zhang S, et al. Antenatal steroids and the IUGR fetus: are exposure and physiological effects on the lung and cardiovascular system the same as in normally grown fetuses? J Pregnancy. 2012;2012:839656.
Ylikahri RH, Huttunen MO, Härkönen M, Leino T, Helenius T, Liewendahl K, et al. Acute effects of alcohol on anterior pituitary secretion of the tropic hormones. J Clin Endocrinol Metab. 1978;46(5):715–20.
Stalder T, Kirschbaum C, Heinze K, Steudte S, Foley P, Tietze A, et al. Use of hair cortisol analysis to detect hypercortisolism during active drinking phases in alcohol-dependent individuals. Biol Psychol. 2010;85(3):357–60.
Thayer JF, Hall M, Sollers JJ 3rd, Fischer JE. Alcohol use, urinary cortisol, and heart rate variability in apparently healthy men: evidence for impaired inhibitory control of the HPA axis in heavy drinkers. Int J Psychophysiol. 2006;59(3):244–50.
Rico H, Gomez-Castresana F, Cabranes JA, Almoguera I, Lopez Duran L, Matute JA. Increased blood cortisol in alcoholic patients with aseptic necrosis of the femoral head. Calcif Tissue Int. 1985;37(6):585–7.
Mark PJ, Waddell BJ. P-glycoprotein restricts access of cortisol and dexamethasone to the glucocorticoid receptor in placental BeWo cells. Endocrinology. 2006;147(11):5147–52.
Wang Y, Liu Y, Sidhu A, Ma Z, McClain C, Feng W. Lactobacillus rhamnosus GG culture supernatant ameliorates acute alcohol-induced intestinal permeability and liver injury. Am J Physiol Gastrointest Liver Physiol. 2012;303(1):G32–41.
Li Y, Yan YE, Wang H. Enhancement of placental antioxidative function and P-gp expression by sodium ferulate mediated its protective effect on rat IUGR induced by prenatal tobacco/alcohol exposure. Environ Toxicol Pharmacol. 2011;32(3):465–71.
Ge C, Xu D, Yu P, Fang M, Guo J, Xu D, et al. P-gp expression inhibition mediates placental glucocorticoid barrier opening and fetal weight loss. BMC Med. 2021;19(1):311.
Goland RS, Jozak S, Warren WB, Conwell IM, Stark RI, Tropper PJ. Elevated levels of umbilical cord plasma corticotropin-releasing hormone in growth-retarded fetuses. J Clin Endocrinol Metab. 1993;77(5):1174–9.
Nolan LA, Levy A. Anterior pituitary trophic responses to dexamethasone withdrawal and repeated dexamethasone exposures. J Endocrinol. 2001;169(2):263–70.
Lesage J, Blondeau B, Grino M, Bréant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology. 2001;142(5):1692–702.
Chen Y, He Z, Chen G, Liu M, Wang H. Prenatal glucocorticoids exposure and fetal adrenal developmental programming. Toxicology. 2019;428:152308.
Lu Z, Guo Y, Xu D, Xiao H, Dai Y, Liu K et al. Developmental toxicity and programming alterations of multiple organs in offspring induced by medication during pregnancy. Acta Pharm Sinica B. 2022.
Glover V, O’Connor TG, O’Donnell K. Prenatal stress and the programming of the HPA axis. Neurosci Biobehav Rev. 2010;35(1):17–22.
Weinberg J, Sliwowska JH, Lan N, Hellemans KG. Prenatal alcohol exposure: foetal programming, the hypothalamic-pituitary-adrenal axis and sex differences in outcome. J Neuroendocrinol. 2008;20(4):470–88.
Zhang X, Sliwowska JH, Weinberg J. Prenatal alcohol exposure and fetal programming: effects on neuroendocrine and immune function. Exp Biol Med (Maywood). 2005;230(6):376–88.
Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol. 2004;151(Suppl 3):U49–62.
Marciniak B, Patro-Małysza J, Poniedziałek-Czajkowska E, Kimber-Trojnar Z, Leszczyńska-Gorzelak B, Oleszczuk J. Glucocorticoids in pregnancy. Curr Pharm Biotechnol. 2011;12(5):750–7.
Mericq V, Martinez-Aguayo A, Uauy R, Iniguez G, Van der Steen M, Hokken-Koelega A. Long-term metabolic risk among children born premature or small for gestational age. Nat Rev Endocrinol. 2017;13(1):50–62.
Lu J, Li Q, Ma G, Hong C, Zhang W, Wang Y, et al. Prenatal ethanol exposure-induced hypothalamic an imbalance of glutamatergic/GABAergic projections and low functional expression in male offspring rats. Food Chem Toxicol. 2020;141:111419.
Phillips DI, Bennett FI, Wilks R, Thame M, Boyne M, Osmond C, et al. Maternal body composition, offspring blood pressure and the hypothalamic-pituitary-adrenal axis. Paediatr Perinat Epidemiol. 2005;19(4):294–302.
Bao AM, Meynen G, Swaab DF. The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res Rev. 2008;57(2):531–53.
Cohen M, Brown DR, Myers MM. Cardiorespiratory measures before and after feeding challenge in term infants are related to birth weight. Acta paediatrica (Oslo, Norway: 1992). 2009;98(7):1183-8.
Schutter DJ. The cerebello-hypothalamic-pituitary-adrenal axis dysregulation hypothesis in depressive disorder. Med Hypotheses. 2012;79(6):779–83.
Weinberg J. Neuroendocrine effects of prenatal alcohol exposure. Ann N Y Acad Sci. 1993;697:86–96.
Gangisetty O, Bekdash R, Maglakelidze G, Sarkar DK. Fetal alcohol exposure alters proopiomelanocortin gene expression and hypothalamic-pituitary-adrenal axis function via increasing MeCP2 expression in the hypothalamus. PLoS ONE. 2014;9(11):e113228.
Lu J, Jiao Z, Yu Y, Zhang C, He X, Li Q, et al. Programming for increased expression of hippocampal GAD67 mediated the hypersensitivity of the hypothalamic-pituitary-adrenal axis in male offspring rats with prenatal ethanol exposure. Cell Death Dis. 2018;9(6):659.
Kozlovsky N, Matar MA, Kaplan Z, Zohar J, Cohen H. A distinct pattern of intracellular glucocorticoid-related responses is associated with extreme behavioral response to stress in an animal model of post-traumatic stress disorder. Eur Neuropsychopharmacol. 2009;19(11):759–71.
Treccani G, Musazzi L, Perego C, Milanese M, Nava N, Bonifacino T, et al. Stress and corticosterone increase the readily releasable pool of glutamate vesicles in synaptic terminals of prefrontal and frontal cortex. Mol Psychiatry. 2014;19(4):433–43.
Sapolsky RM, Meaney MJ. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res. 1986;396(1):64–76.
Matthews SG. Early programming of the hypothalamo-pituitary-adrenal axis. Trends Endocrinol Metab. 2002;13(9):373–80.
Krishnamurthy S, Garabadu D, Joy KP. Risperidone ameliorates post-traumatic stress disorder-like symptoms in modified stress re-stress model. Neuropharmacology. 2013;75:62–77.
Meaney MJ, Szyf M, Seckl JR. Epigenetic mechanisms of perinatal programming of hypothalamic-pituitary-adrenal function and health. Trends Mol Med. 2007;13(7):269–77.
Galeeva A, Pelto-Huikko M, Pivina S, Ordyan N. Postnatal ontogeny of the glucocorticoid receptor in the hippocampus. Vitam Horm. 2010;82:367–89.
Wada H, Breuner CW. Developmental changes in neural corticosteroid receptor binding capacity in altricial nestlings. Dev Neurobiol. 2010;70(13):853–61.
Randhawa R, Cohen P. The role of the insulin-like growth factor system in prenatal growth. Mol Genet Metab. 2005;86(1–2):84–90.
Girnita L, Worrall C, Takahashi S, Seregard S, Girnita A. Something old, something new and something borrowed: emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci. 2014;71(13):2403–27.
Walenkamp MJ, van der Kamp HJ, Pereira AM, Kant SG, van Duyvenvoorde HA, Kruithof MF, et al. A variable degree of intrauterine and postnatal growth retardation in a family with a missense mutation in the insulin-like growth factor I receptor. J Clin Endocrinol Metab. 2006;91(8):3062–70.
Wallborn T, Wüller S, Klammt J, Kruis T, Kratzsch J, Schmidt G, et al. A heterozygous mutation of the insulin-like growth factor-I receptor causes retention of the nascent protein in the endoplasmic reticulum and results in intrauterine and postnatal growth retardation. J Clin Endocrinol Metab. 2010;95(5):2316–24.
Walenkamp MJ, Losekoot M, Wit JM. Molecular IGF-1 and IGF-1 receptor defects: from genetics to clinical management. Endocr Dev. 2013;24:128–37.
Gicquel C, Le Bouc Y. Hormonal regulation of fetal growth. Horm Res. 2006;65(Suppl 3):28–33.
Kenyon C. A conserved regulatory system for aging. Cell. 2001;105(2):165–8.
Mahajan SD, Aalinkeel R, Singh S, Shah P, Gupta N, Kochupillai N. Endocrine regulation in asymmetric intrauterine fetal growth retardation. J maternal-fetal Neonatal Medicine: Official J Eur Association Perinat Med Federation Asia Ocean Perinat Soc Int Soc Perinat Obstet. 2006;19(10):615–23.
Tosh DN, Fu Q, Callaway CW, McKnight RA, McMillen IC, Ross MG, et al. Epigenetics of programmed obesity: alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. Am J Physiol Gastrointest Liver Physiol. 2010;299(5):G1023–9.
Hu W, Yuan C, Luo H, Hu S, Shen L, Chen L, et al. Glucocorticoid-insulin-like growth factor 1 (GC-IGF1) axis programming mediated hepatic lipid-metabolic in offspring caused by prenatal ethanol exposure. Toxicol Lett. 2020;331:167–77.
Arney KL, Fisher AG. Epigenetic aspects of differentiation. J Cell Sci. 2004;117(Pt 19):4355–63.
McGowan PO. Epigenetic mechanisms of perinatal programming: translational approaches from rodent to human and back. Adv Neurobiol. 2015;10:363–80.
Perkins A, Lehmann C, Lawrence RC, Kelly SJ. Alcohol exposure during development: impact on the epigenome. Int J Dev Neurosci. 2013;31(6):391–7.
Ungerer M, Knezovich J, Ramsay M. In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res. 2013;35(1):37–46.
Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcohol Clin Exp Res. 1991;15(3):395–8.
Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, et al. Alcohol metabolism contributes to brain histone acetylation. Nature. 2019;574(7780):717–21.
Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics. 2009;4(7):500–11.
Zhang CR, Ho MF, Vega MC, Burne TH, Chong S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin. 2015;8:40.
Mantha K, Laufer BI, Singh SM. Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: from immediate effects to long-term adaptation. Dev Neurosci. 2014;36(1):29–43.
Stouder C, Somm E, Paoloni-Giacobino A. Prenatal exposure to ethanol: a specific effect on the H19 gene in sperm. Reprod Toxicol. 2011;31(4):507–12.
Marjonen H, Toivonen M, Lahti L, Kaminen-Ahola N. Early prenatal alcohol exposure alters imprinted gene expression in placenta and embryo in a mouse model. PLoS ONE. 2018;13(5):e0197461.
Andreu-Fernández V, Bastons-Compta A, Navarro-Tapia E, Sailer S, Garcia-Algar O. Serum concentrations of IGF-I/IGF-II as biomarkers of alcohol damage during foetal development and diagnostic markers of foetal alcohol syndrome. Sci Rep. 2019;9(1):1562.
Jarmasz JS, Stirton H, Basalah D, Davie JR, Clarren SK, Astley SJ, et al. Global DNA methylation and histone posttranslational modifications in Human and Nonhuman Primate Brain in Association with prenatal Alcohol exposure. Alcohol Clin Exp Res. 2019;43(6):1145–62.
Gatta E, Auta J, Gavin DP, Bhaumik DK, Grayson DR, Pandey SC, et al. Emerging role of one-Carbon metabolism and DNA methylation Enrichment on δ-Containing GABAA receptor expression in the cerebellum of subjects with Alcohol Use Disorders (AUD). Int J Neuropsychopharmacol. 2017;20(12):1013–26.
Otero NK, Thomas JD, Saski CA, Xia X, Kelly SJ. Choline supplementation and DNA methylation in the hippocampus and prefrontal cortex of rats exposed to alcohol during development. Alcohol Clin Exp Res. 2012;36(10):1701–9.
Cao J, Chen Y, Xia X, Qu H, Ao Y, Wang H. Intergenerational genetic programming mechanism and sex differences of the adrenal corticosterone synthesis dysfunction in offspring induced by prenatal ethanol exposure. Toxicol Lett. 2021;351:78–88.
Haycock PC, Ramsay M. Exposure of mouse embryos to ethanol during preimplantation development: effect on DNA methylation in the h19 imprinting control region. Biol Reprod. 2009;81(4):618–27.
Veazey KJ, Carnahan MN, Muller D, Miranda RC, Golding MC. Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol Clin Exp Res. 2013;37(7):1111–22.
Hales CN, Barker DJP. The thrifty phenotype hypothesis. Br Med Bull. 2001;60:5–20.
Qiu XS, Huang TT, Deng HY, Shen ZY, Ke ZY, Mei KY, et al. Effects of early nutrition intervention on IGF1, IGFBP3, intestinal development, and catch-up growth of intrauterine growth retardation rats. Chin Med Sci J. 2004;19(3):189–92.
Kamei H, Ding Y, Kajimura S, Wells M, Chiang P, Duan C. Role of IGF signaling in catch-up growth and accelerated temporal development in zebrafish embryos in response to oxygen availability. Development. 2011;138(4):777–86.
Wilcoxon JS, Schwartz J, Aird F, Redei EE. Sexually dimorphic effects of maternal alcohol intake and adrenalectomy on left ventricular hypertrophy in rat offspring. Am J Physiol Endocrinol Metab. 2003;285(1):E31–9.
Penailillo R, Guajardo A, Llanos M, Hirsch S, Ronco AM. Folic acid supplementation during pregnancy induces sex-specific changes in methylation and expression of placental 11β-hydroxysteroid dehydrogenase 2 in rats. PLoS ONE. 2015;10(3):e0121098.
Hodyl NA, Wyper H, Osei-Kumah A, Scott N, Murphy VE, Gibson P, et al. Sex-specific associations between cortisol and birth weight in pregnancies complicated by Asthma are not due to differential glucocorticoid receptor expression. Thorax. 2010;65(8):677–83.
Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and Disease. J Allergy Clin Immunol. 2013;132(5):1033–44.
Saif Z, Dyson RM, Palliser HK, Wright IM, Lu N, Clifton VL. Identification of eight different isoforms of the glucocorticoid receptor in Guinea Pig Placenta: relationship to Preterm Delivery, Sex and Betamethasone exposure. PLoS ONE. 2016;11(2):e0148226.
Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, et al. The human glucocorticoid receptor (hGR) beta isoform suppresses the transcriptional activity of hGRalpha by interfering with formation of active coactivator complexes. Mol Endocrinol. 2005;19(1):52–64.
Lewis-Tuffin LJ, Cidlowski JA. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1–9.
Ramamoorthy S, Cidlowski JA, Corticosteroids. Mechanisms of action in Health and Disease. Rheum Dis Clin North Am. 2016;42(1):15–31. vii.
Saif Z, Hodyl NA, Hobbs E, Tuck AR, Butler MS, Osei-Kumah A, et al. The human placenta expresses multiple glucocorticoid receptor isoforms that are altered by fetal sex, growth restriction and maternal Asthma. Placenta. 2014;35(4):260–8.
Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of Disease. Horm Behav. 2011;59(3):279–89.
Seeman TE, Singer B, Wilkinson CW, McEwen B. Gender differences in age-related changes in HPA axis reactivity. Psychoneuroendocrinology. 2001;26(3):225–40.
Uhart M, Chong RY, Oswald L, Lin PI, Wand GS. Gender differences in hypothalamic-pituitary-adrenal (HPA) axis reactivity. Psychoneuroendocrinology. 2006;31(5):642–52.
Young EA. The role of gonadal steroids in hypothalamic-pituitary-adrenal axis regulation. Crit Rev Neurobiol. 1995;9(4):371–81.
Young EA. Sex differences in response to exogenous corticosterone: a rat model of hypercortisolemia. Mol Psychiatry. 1996;1(4):313–9.
Boggan WO, Randall CL, Dodds HM. Delayed sexual maturation in female C57BL/6J mice prenatally exposed to alcohol. Res Commun Chem Pathol Pharmacol. 1979;23(1):117–25.
Lan N, Yamashita F, Halpert AG, Sliwowska JH, Viau V, Weinberg J. Effects of prenatal ethanol exposure on hypothalamic-pituitary-adrenal function across the estrous cycle. Alcohol Clin Exp Res. 2009;33(6):1075–88.
Creighton-Taylor JA, Rudeen PK. Fetal alcohol exposure and effects of LHRH and PMA on LH beta-mRNA expression in the female rat. Alcohol Clin Exp Res. 1991;15(6):1031–5.
McGivern RF, Yellon SM. Delayed onset of puberty and subtle alterations in GnRH neuronal morphology in female rats exposed prenatally to ethanol. Alcohol (Fayetteville NY). 1992;9(4):335–40.
McGivern RF, McGeary J, Robeck S, Cohen S, Handa RJ. Loss of reproductive competence at an earlier age in female rats exposed prenatally to ethanol. Alcohol Clin Exp Res. 1995;19(2):427–33.
Parker S, Udani M, Gavaler JS, Van Thiel DH. Adverse effects of ethanol upon the adult sexual behavior of male rats exposed in utero. Neurobehav Toxicol Teratol. 1984;6(4):289–93.
Udani M, Parker S, Gavaler J, Van Thiel DH. Effects of in utero exposure to alcohol upon male rats. Alcohol Clin Exp Res. 1985;9(4):355–9.
Rudeen PK. Reduction of the volume of the sexually dimorphic nucleus of the preoptic area by in utero ethanol exposure in male rats. Neurosci Lett. 1986;72(3):363–8.
Rudeen PK, Kappel CA, Lear K. Postnatal or in utero ethanol exposure reduction of the volume of the sexually dimorphic nucleus of the preoptic area in male rats. Drug Alcohol Depend. 1986;18(3):247–52.
McGivern RF. Influence of prenatal exposure to cimetidine and alcohol on selected morphological parameters of sexual differentiation: a preliminary report. Neurotoxicol Teratol. 1987;9(1):23–6.
Handa RJ, Nunley KM, Lorens SA, Louie JP, McGivern RF, Bollnow MR. Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol Behav. 1994;55(1):117–24.
David R, Bell JJV, Yellon DA, Gladue ME. Glucocorticoids decrease serum testosterone levels in men by inhibiting testicular 17 beta-hydroxysteroid dehydrogenase. J Clin Endocr Metab. 1989;63(6):1265–371.
Ahmed AA, Ma W, Ni Y, Wang S, Zhao R. Corticosterone in ovo modifies aggressive behaviors and reproductive performances through alterations of the hypothalamic-pituitary-gonadal axis in the chicken. Anim Reprod Sci. 2014;146(3–4):193–201.
Yuan R, Gatti DM, Krier R, Malay E, Schultz D, Peters LL, et al. Genetic regulation of female sexual maturation and longevity through circulating IGF1. J Gerontol A Biol Sci Med Sci. 2015;70(7):817–26.
Yuan R, Meng Q, Nautiyal J, Flurkey K, Tsaih SW, Krier R, et al. Genetic coregulation of age of female sexual maturation and lifespan through circulating IGF1 among inbred mouse strains. Proc Natl Acad Sci U S A. 2012;109(21):8224–9.
Hannigan JH, Bellisario RL. Lower serum thyroxine levels in rats following prenatal exposure to ethanol. Alcohol Clin Exp Res. 1990;14(3):456–60.
Cudd TA, Chen WJ, West JR. Fetal and maternal thyroid hormone responses to ethanol exposure during the third trimester equivalent of gestation in sheep. Alcohol Clin Exp Res. 2002;26(1):53–8.
Wilcoxon JS, Kuo AG, Disterhoft JF, Redei EE. Behavioral deficits associated with fetal alcohol exposure are reversed by prenatal thyroid hormone treatment: a role for maternal thyroid hormone deficiency in FAE. Mol Psychiatry. 2005;10(10):961–71.
Wilcoxon JS, Redei EE. Prenatal programming of adult thyroid function by alcohol and thyroid hormones. Am J Physiol Endocrinol Metab. 2004;287(2):E318–26.
Fidalgo S, Skipper C, Takyi A, McIver A, Tsiligkaridis T, Quadir A, et al. Low-dose chronic prenatal alcohol exposure abolishes the pro-cognitive effects of angiotensin IV. Behav Brain Res. 2017;329:140–7.
Zhu Y, Zuo N, Li B, Xiong Y, Chen H, He H, et al. The expressional disorder of the renal RAS mediates Nephrotic Syndrome of male rat offspring induced by prenatal ethanol exposure. Toxicology. 2018;400–401:9–19.
Wu Z, Pan Z, Wen Y, Xiao H, Shangguan Y, Wang H, et al. Egr1/p300/ACE signal mediates postnatal osteopenia in female rat offspring induced by prenatal ethanol exposure. Food Chem Toxicol. 2020;136:111083.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Key R&D Program of China (No. 2020YFA0803900), the National Key Research and Development Program of China (No. 2017YFC1001300), the National Natural Science Foundation of China (No. 81972036, 82030111), the Major Technological Innovation Projects of Hubei Province (No. 2019ACA140, 2020BCA071), Hubei Province’s Outstanding Medical Academic Leader program, Medical Science Advancement Program (Basic Medical Sciences) of Wuhan University (No. TFJC2018001).
Author information
Authors and Affiliations
Contributions
LL: Conceptualization, Writing–original draft, Writing–review and editing, Visualization. YW: Writing–original draft, Writing–review and editing, Formal analysis. QN: Writing–review and editing, Formal analysis. LC and HW: Conceptualization, Writing–review and editing, Supervision, Funding acquisition.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of Interest
The authors declared no conflict of interest. All authors have made a significant contribution to the paper, read and approved the version submitted, and share responsibility for it as well.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, L., Wen, Y., Ni, Q. et al. Prenatal ethanol exposure and changes in fetal neuroendocrine metabolic programming. Biol Res 56, 61 (2023). https://doi.org/10.1186/s40659-023-00473-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-023-00473-y