
Abstract
Background
Continuous monocropping obstacles are common in plants, especially medicinal plants, resulting in disease outbreaks and productivity reductions. Foliar disease, mainly caused by Fusarium oxysporum, results in a severe decrease in the yield of Pseudostellaria heterophylla annually. Determining an effective biomethod to alleviate this disease is urgently needed to improve its productivity and quality.
Results
This study screened thirty-two keystone bacterial genera induced by pathogens in P. heterophylla rhizosphere soil under continuous monocropping conditions. Pseudomonas, Chryseobacterium, and Flavobacterium, referred to as the beneficial microbiota, were significantly attracted by pathogen infection. The P. palleroniana strain B-BH16-1 can directly inhibit the growth and spore formation of seven primary pathogens of P. heterophylla foliar disease by disrupting fusaric acid production via the emission of volatile organic compounds (VOCs). In addition, strain B-BH16-1 enhances the disease resistance of P. heterophylla by obliterating the pathogen and assembling beneficial microbiota.
Conclusion
Pathogen-induced Pseudomonas reshaped phyllosphere microbial communities via direct antagonism of pathogens and indirect disruption of the pathogen virulence factor biosynthesis to enhance disease suppression and improve yields. These results show that inhibiting pathogen virulence biosynthesis to reshape the plant microbial community using disease-induing probiotics will be an innovative strategy for managing plant disease, especially under continuous monoculture conditions.
Similar content being viewed by others


Background
Long-term monocropping leads to an imbalance in the soil microbial community and physical-chemical properties, including the accumulation of pathogens and elimination of probiotics, causing production obstacles, and resulting in yearly productivity reduction, known as continuous monocropping obstacles. Continuous monocropping obstacles are common in plants, especially in medicinal plants where they are most severe, such as Pseudostellaria heterophylla (Miq.) Pax ex Pax et Hoffm (TaiZiShen) [1,2,3]. P. heterophylla belongs to the Caryophyllaceae family and has been a commonly used traditional Chinese medicine in Asia for more than 100 years. It is also one of a widespread food ingredient [1]. In addition, due to their medicinal and nutritional value, P. heterophylla and its derivatives are used as raw materials in the medical, food, and cosmetic industries. Folia disease, a crucial replanting disease of P. heterophylla, results in a yield reduction of 15%~60% annually, especially under continuous monocropping for more than three seasons caused by multiple pathogens (i.e., Fusarium oxysporum, Botrytis cinerea, Alternaria alternata) [4]. Hence, identifying an effective biomethod for alleviating this disease is urgently needed to improve the productivity and quality of the P. heterophylla to support the increasing its usage in the population.
The plant-associated microbiota, as the second genome of the plant, is pivotal for plant growth, development, productivity, and disease suppression. In addition, plant-associated microbial communities coevolved with plant hosts to be more efficient at helping plant growth and resistance in diverse environments, especially under biotic stress conditions, such as those caused by a range of pathogens. Plant-associated microorganisms directly or indirectly enhance plant growth and disease resistance via nitrogen fixation, hormone and antagonistic compound synthesis, nutrient activation, and immune stimulation. Moreover, they can also form symbiotic relationships with plants, helping plants resist the invasion of pathogens and absorb nutrients. Ma and colleagues provided critical evidence for the importance of Phomopsis liquidambaris in improving plant growth and disease resistance under continuous monocropping conditions [5]. However, the dysfunction of these microbial communities can hinder plant growth and development, decreasing productivity and increasing disease outbreaks [2, 6, 7]. Hence, an effective microbiome regulation strategy to manipulate plant-associated microbiota including plant disease prevention and productivity enhancement, is essential for maintaining the agroecosystem.
Applying plant-beneficial microbiotas is an effective strategy for yield enhancement, disease prevention, and ecological protection for sustainable agricultural systems. The acquisition of these beneficial microbes is based on antagonism and growth promotion, which is the ability to sustainably colonize plants, a crucial constraint on their application. Increasing evidence shows that pathogen infection of host plants mediates the enrichment of plants probiotics in plants via disease resistance [1]. For instance, a previous study showed that the pathogen mediated the assembly of plant-beneficial bacteria on P. heterophylla, alleviating Fusarium wilt [1]. The application of pathogen-driven beneficial microbiota may be an innovative strategy for plant disease prevention in the future. Although the phenomenon of plant probiotics being mediated by pathogen infection has been well demonstrated, far less is known about their ability to manage disease and how they reshape plant-associated microbiomes to enhance disease-suppressive capabilities.
Increasing evidence has shown that Pseudomonas, a beneficial microbiota, is widely applied to enhance plant disease resistance and growth, especially to resist biotic and abiotic stresses. For instance, Prabhukarthikeyan and colleagues demonstrated that P. fluorescens promoted plant growth and mediated rhizome rot disease resistance in turmeric plants [8]. It was also found that P. chlororaphis enhanced disease suppression and growth promotion in maize [9]. In addition, coinoculation of six Pseudomonas strains strongly promoted garlic growth [10]. A previous study showed that Pseudomonas strains were able to alleviate Fusarium wilt in P. heterophylla [1]. However, Pseudomonas can directly inhibit pathogens by producing antifungal metabolites, which has been confirmed; far less is known about how Pseudomonas reshapes phyllosphere microbial communities in P. heterophylla to suppress the disease.
In the present study, we hypothesized that persistent pathogen-induced Pseudomonas colonization could reshape the phyllosphere microbial communities in P. heterophylla, which would benefit plant fitness and confer disease suppression. Therefore, this study aims to (i) screen and identify pathogen-induced probiotics in P. heterophylla under continuous monocropping conditions, (ii) explore broad-spectrum antipathogen violating metabolites from pathogen-induced Pseudomonas, and (iii) reveal the microecological mechanism of disease resistance in this species.
Materials and methods
Field site description, experimental design, and soil sampling
The field site description, experimental design, and soil sampling procedure were described in our previous study [1]. A long-term field trial was conducted in Shibing County (27°4’21"N, 108°8’0"E, and 759 m a.s.l.) in Guizhou Province, China. The continuous monoculture experiment started in December 2015; P. heterophylla was continuously planted. In the third season, viz. in 2019, plants with different disease severities were selected, and their rhizosphere soils were collected. The rhizosphere soil microbiome was sequenced in our previous study [1].
Analysis of the dominant bacteria associated with disease
The microbial community assembly processes were assessed by the neutral model with the R code and the weighted abundance based on the Raup–Crick metric (RCbray) according to the description of Xun et al. [11] and Wen et al. [12]. The null modeling approach was used to evaluate phylogenetic patterns of the rhizosphere microbiome by calculating the β-nearest taxon index (βNTI) between pairs of samples as described in Stegen et al. Sample pairs with |βNTI| > 2 are expected to result from deterministic processes, while |βNTI| < 2 values indicate that selection pressure is weak and that stochastic processes likely govern community assembly. Values derived from βNTI analyses reflect the driving force of factors that influence community assembly processes as phylogenetic turnover correlates with environmental dissimilarity. Mantel tests were performed to evaluate whether βNTI values differed significantly using the “vegan” package. Furthermore, RCbray was used to examine the role of dispersal in community assembly by estimating OTU turnover. We combined the results from the βNTI and RCbray analyses to determine the relative proportion of the overall community assembly governed by deterministic and stochastic processes within each sample. The presence of sample pairs with a |βNTI| < 2 and a |RCbray| > 0.95 indicated that bacterial community turnover was dominated by dispersal. More specifically, limited dispersal was dominant when RCbray > 0.95, while RCbray < − 0.95 indicated homogenizing dispersal [12]. In addition, machine learning was used to distinguish rhizosphere bacteria associated with diseased and healthy rhizosphere soils. The “important” bacteria were selected by cross-tabulations in R with “randomForest.” Wilcoxon tests (the “stat” package) were used to detect differences in rhizosphere bacteria between the diseased and healthy samples. The dominant bacteria that may be associated with microbial community assembly were selected for further confirmation.
Isolation of rhizosphere culturable bacteria
The isolation of culturable bacteria from rhizosphere soil of P. heterophylla with different disease severities was conducted using a dilution plate method on Luria-Bertani (LB) agar media (5 g NaCl, 10 g tryptone, and 5 g yeast extract per liter). Briefly, all collected rhizosphere soil samples were sonicated in 20 ml of PBS solution for 30 min, and an aliquot (2 ml) of the suspension was diluted ten times. An aliquot (100 µl) of the dilution suspension was coated onto LB medium and cultured at 25℃ for two days, after which individual colonies were isolated and stored at -80℃ in 20% glycerol. The 16 S rRNA genes of the isolated bacterial strains was amplified with universal primers 27 F (AGAGTTTGATCCTGGCTCAG) and 1492R (TACGGYTACCTTGTTACGACTT). PCR amplicons were verified on a 1% agarose gel. The reaction products were purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States) and then sequenced on an ABI 3730XL DNA Analyzer (Applied Biosystems, CA, United States) according to standard protocols by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China). The 16 S rRNA gene sequences were aligned with the NCBI NR database by BLASTn to determine the approximate phylogenetic affiliation of the strains. Taxonomy was confirmed by phylogenetic analysis using neighbor-joining methods, which was bootstrapped 1 000 times.
Antifungal activity of the dominant bacteria against pathogens
According to our previous study, the antifungal activity of volatiles from the fourteen predominant bacteria stimulated by disease was determined using the face-to-face (FTF) method [13]. Briefly, 10 µL of fresh pathogens conidia at a concentration of 5 × 105 CFU/mL was inoculated into the center of a PDA plate. The other plate contained LB medium with fresh bacteria coated on the surface (100 µL, OD600 = 1.0) or ddH2O. The two dishes were placed face-to-face with a PDA plate above. All plates were cultured at 28 ℃ in darkness. After five days of incubation, the mycelial diameter of the pathogens in each treatment was measured. The inhibition rate was calculated using the following formula:
We compared the inhibitory activity among these essential bacteria, and the broad-spectrum antifungal activity of Pseudomonas palleroniana B-BH16-1, which had the highest inhibitory activity, was determined using the FTF method described above. In addition, volatile organic compounds (VOCs) from strain B-BH16-1 were identified by GC‒MS/MS as described in our previous study [13]. The broad-spectrum antifungal activity of the VOCs was also performed using the FTF method determined above. The effects on the microscopic structure of Fusarium oxysporum cells and their sporulation behavior were examined using an OLYMPUS IX73 microscope (Olympus Corporation, Tokyo, Japan). The genes related to mycelial growth and sporulation (i.e., Smsp, iic, Fab, and PIG-L) were measured by qRT‒PCR as described in our previous study [13]. The primers used for qRT‒PCR are listed in Table S1.
Control of F. oxysporum and its fusaric acid on the tuberous roots of P. heterophylla using strain B-BH16-1
The biocontrol effect of strain B-BH16-1 against F. oxysporum on harvested tuberous roots of P. heterophylla was tested in a sealed petri dish (0.2 L airspace). Autoclaved tuberous roots of P. heterophylla (50 g) were inoculated with an F. oxysporum conidial suspension (0.1 mL, 5 × 105 CFU/mL) according to the methods of Zhou et al [13]. They were challenged with strain B-BH16-1 (the bacteria were smeared on the LB plate at the bottom of the desiccator) or with a plain LB plate as a control. The experiment was conducted independently in triplicate. The tuberous roots of P. heterophylla from the treatments were collected 10‒ and 20 days postinoculation (dpi). The fusaric acid content was detected by HPCL methods, and qRT‒qPCR was used to determine the expression of its biosynthetic genes (Fub8). The primers used for qRT‒PCR are listed in Table S1.
A pot experiment to demonstrate the disease control effect of strain B-BH16-1 on P. heterophylla
The four-leaf stage P. heterophylla seedlings were transplanted into pots and allowed to recover in a growth chamber with a 16 h light period (250 µmol/m2/s illumination)/8 h dark period at 25/18°C. A conidial suspension of F. oxysporum (5 mL, 5 × 105 CFU/mL) was inoculated into each pot. The B-BH16-1 strain (5 mL, 5 × 105 CFU/mL) or ddH2O (CK) was sprayed into the pots on the third and seventh day after inoculation of F. oxysporum. All treatments were applied under the same conditions as those described above. After fifteen days, the disease incidence was investigated.
A field experiment to demonstrate the disease control effect of strain B-BH16-1 on P. Heterophylla
The field experiment was manually conducted in September 2020 in a continuous cropping field trial located at 106°27′ E latitude, 26°11′ N longitude, and 1240 m above mean sea level in Guzhong village, Buyi and Miao township, Huaxi District, Guiyang, China. The plot size was approximately 10 m × 5 m. The region has a humid subtropical plateau monsoon climate. The plants were was divided into two groups: the control group (CK), which was treated with ddH2O; the treatment group (PP), which was treated with 2 L/m2Pseudomonas palleroniana B-BH16-1 at a concentration of 1 × 107 CFU/mL in soil for one month before planting; and the treatment group, which was sprayed with 2 L/m2 of B-BH16-1 strain at the same concentration on P. heterophylla seedings every seven days three times beginning on 15 March 2021. Each treatment had four replicates randomly. The incidence of foliar disease was investigated in May 2021, leaf samples were collected in April and May 2021, and the yields and tuberous roots were detected in August 2021.
The effect of strain B-BH16-1 on the quality of P. heterophylla
Chemical analyses were used to determine the crude polysaccharide and total saponin contents in harvested tuberous roots. The crude polysaccharide and total saponin contents were measured by the phenol‒sulfuric acid method [14] and the vanillin‒acetic acid-perchloric acid method [14], respectively. The glucose concentration was calculated based on the glucose standard curve. The following formula was used to calculate the polysaccharide content:
The letters a, b, f, and W indicate the glucose concentration in the test solution, the dilution factor, the conversion factor (2.97), and the weight of the sample, respectively.
The saponin concentration was calculated based on the ginsenoside Rb1 standard curve. The following formula was used to calculate the saponin content:
The letters a, b, f, and W indicate the concentration of ginsenoside Rb1 in the test solution, the dilution factor, the conversion factor (2.97), and the weight of the sample, respectively.
The effect of strain B-BH16-1 on the phyllosphere microbiome
Genomic DNA was extracted from the microbiota using the E.Z.N.A.® Soil DNA Kit (Omega Biotech, Norcross, GA, U.S.) following the manufacturer’s protocol. DNA quality and quantity were detected by electrophoresis on a 1% agarose gel and with a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). For taxonomic profiling, the V3V4 fragment of the 16 S rRNA gene was amplified with the primers 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The ITS2 fragment of the fungal rRNA gene was amplified with primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and ITS2R (5′-GCTGCGTTCTTCATCGATGC-3′) by Thermos Cycler PCR system (GeneAmp 9700, ABI, USA). PCR products were separated by electrophoresis on a 2% agarose gel and purified by an AxyPrep DNA gel recovery kit (Axygen, Shanghai, China). Furthermore, the sequencing of the purified amplicons was conducted on an Illumina MiSeq platform (Illumina, San Diego, USA) according to standard protocols by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The 250-bp paired-end reads were filtered to obtain high-quality clean reads using Trimmomatic (V0.33, http://www.usadellab.org/cms/?page=trimmomatic), and sequences were assigned to each sample based on its unique barcode. Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE software (version 7.1, http://drive5.com/uparse/). The taxonomy of the V3V4 16 S and ITS2 rRNA gene sequences was analyzed by the Ribosomal Database Project Classifier algorithm (RDP, V.2.2, http://rdp.cme.msu.edu/) against the Silva (SSU123) 16 S rRNA database and the UNITE database (Version 6, Vanemuise, Tartu, Estonia) with a 70% confidence threshold.
Alpha-diversity indices were used to evaluate the richness and species diversity of each sample. The observed species, Chao, ACE, Shannon, and Simpson indices, were calculated using QIIME software (V1.806). The beta diversity metrics were evaluated by hierarchical clustering analysis and principal component ordinated analysis (PCoA) based on the Bray–Curtis distance matrix using the “phyloseq” and “vegan” packages of R software (v3.6.27). The difference in the microbial community composition at the genus and species levels was assessed by the “statnet” package of R software and was visualized by Origin software (version 2018, Origin Lab Inc., Northampton, MA, USA).
Statistical analysis
The significant differences between the mean values were determined using Student’s t tests (p < 0.05) following one-way analysis of variance (ANOVA). Statistical analysis was performed using Origin software (Version 2018, OriginLab Inc., Northampton, MA, USA).
Results
The assemblage of beneficial bacteria was driven by disease
A neutral model from the field experiment was used to assess the microbial community assembly processes. The β-nearest taxon index (βNTI) between sample pairs showed that stochastic process selection occured in the diseased rhizosphere bacterial communities (|βNTI| < 2), while deterministic process selection occured within healthy samples (2 pairs of |βNTI| < 2) (Fig. 1A and B). Stochastic process (|βNTI| < 2) dominated phylogenetic turnover in the diseased rhizosphere microbiome, and homogenizing dispersal (RCbray < -0.95) was found to be dominant among the stochastic processes (Fig. 1B). Furthermore, a random forest (RF) model was used to analyze and compare the relative importance of different bacterial genera associated with different disease severities in P. heterophylla, and the results revealed that 32 genera (Pseudomonas, Chryseobacterium, Flavobacterium, etc.) were the essential bacteria that contributed to the differences in disease severity in P. heterophylla (Fig. 1C). Moreover, the high-throughput sequencing results revealed that the diseased rhizosphere soil had a greater relative abundance of both Pseudomonas (Fig. 1D) and Fusarium (Fig. 1E) than the healthy rhizosphere soil.

Pathogen drove the assemblage of beneficial bacteria in the P. heterophylla rhizosphere microbiome. The β-nearest taxon index (βNTI) (A) and the Raup-Crick metric (RCbray) (B) in the neutral model were used to assess the microbial community assembly processes. (C) A random forest (RF) model was used to screen the keystone taxa induced by disease and the relative abundances of Pseudomonas (D) and Fusarium (E) in the P. heterophylla rhizosphere soil
The Pseudomonas palleroniana strain B-BH16-1 exhibited broad-spectrum antifungal activity through the emission of VOCs to inhibit multiple pathogens
The culturable bacteria of diseased rhizosphere soil of P. heterophylla were isolated to identify the keystone bacteria involved in disease resistance. A total of 196 isolates belonging to fourteen genera were obtained (Fig. 2A). Among them, Pseudomonas, Chryseobacterium, and Flavobacterium, which were predicted to be the dominant bacteria affected by disease, were isolated and accounted for 12.24%, 4.59%, and 1.53%, respectively (Fig. 2B). The antifungal activity of the three genera was tested via an FTF antagonism assay against F. oxysporum; the antifungal activity of Pseudomonas was significantly greater than that of Chryseobacterium and Flavobacterium (Fig. 2C). Hence, we further detected the broad-spectrum antifungal activity of Pseudomonas palleroniana strain B-BH16-1 from the culturable bacteria on the seven primary pathogens of P. heterophylla and the results revealed that it has vigorous inhibitory activity on the seven pathogens (Fig. 2D).

Screening of pathogen-induced beneficial bacteria in the P. heterophylla rhizosphere microbiome. The phylogenetic tree of isolated bacteria (A) and their relative abundance (B); (C) the inhibitory ability of Pseudomonas, Chryseobacterium, and Flavobacterium; (D) the broad-spectrum antipathogens of Pseudomonas strain B-BH16-1 on seven pathogens of P. heterophylla foliar disease
In addition, volatile organic compounds (VOCs) were identified by GC-MS/MS, and the results showed that dimethyl disulfide, 1-undecene, and squalene were the primary metabolites emitted from B-BH16-1 strain (Fig. 3A). Further antifungal assays of VOCs revealed that these three compounds were the main inhibitory substances against the seven pathogens (Fig. 3B). Microscopy observation showed that B-BH16-1 strain significantly inhibited spore formation and the mycelium growth (Fig. 3C). qRT‒PCR showed that it substantially downregulated Smsp, iic, Fab, and PIG-L expression related to mycelia growth and spore formation (Fig. 3D). In addition, the in vitro biocontrol effect of strain B-BH16-1 against F. oxysporum revealed that it significantly inhibited the biosynthesis of fusaric acid (Fig. 3E and F, and 3G), a key pathogenesis factor of F. oxysporum, and downregulated the expression of Fub8, which is involved in the biosynthesis of fusaric acid (Fig. 3H).

Identification of volatile organic compounds emitted from strain B-BH16-1 and their antifungal effects on spore formation and virulence factor synthesis in F. oxysporum. (A) GC‒MS/MS analysis of volatile organic compounds emitted from strain B-BH16-1; (B) The inhibitory effect of VOCs on seven pathogens of P. heterophylla folia disease; VOCs inhibit the spore formation of F. oxysporum (C) and the expression of genes related to spore development (D); VOCs disrupt fusaric acid synthesis, a virulence factor of F. oxysporum (E, F, and G), and downregulate the expression of its vital synthesis gene (H) The broad-spectrum antipathogen effect of Pseudomonas strain B-BH16-1 on seven pathogens of P. heterophylla folia disease
P. palleroniana strain B-BH16-1 reshaped the phyllosphere microbial community on P. heterophylla
The effect of strain B-BH16-1 on the phyllosphere microbial community of P. heterophylla was evaluated by sequencing. Alpha diversity analysis revealed that strain B-BH16-1 could affect soil microbial richness and diversity (Figure S1). The application of strain B-BH16-1 resulted in a substantial increase in the Shannon index of both the bacterial and fungal communities, although the difference was not significant (Figures S1A and S1E). This result showed that strain B-BH16-1 might increase the diversity of both fungal and bacterial communities. Moreover, strain B-BH16-1 also increased the Ace, Chao, and Sobs indices of the bacterial communities, although not significantly (Figure S1B, S1C, and S1D). In addition, it significantly increased the Sobs indices of the fungal communities (Figure S1H). This result indicated that strain B-BH16-1 could increase the richness of microbial communities.
Hierarchical clustering analysis at the species level revealed that the bacterial communities in both CK and PP were entirely separated by strain B-BH16-1 (Fig. 4A). Similar results were also found for the fungal communities (Fig. 5A). Moreover, the principal component ordinated analysis (PCoA) using the Bray–Curtis metric based on species level revealed that strain B-BH16-1 can drive changes in the bacterial (p = 0.0340, Fig. 4B) and fungal (p = 0.0340, Fig. 5B) communities.

Strain B-BH16-1 reshaped the bacterial communities in the P. heterophylla phyllosphere. Strain B-BH16-1 separates bacterial communities between CK and PP based on hierarchical clustering analysis (A) and principal component ordinated analysis (B) at the species level; the B-BH16-1 strain induces changes in the bacterial community composition at the genus (C) and species (D) levels

Strain B-BH16-1 reshaped the fungal communities in the P. heterophylla phyllosphere. Strain B-BH16-1 separates fungal communities between CK and PP based on hierarchical clustering analysis (A) and principal component ordinated analysis (B) at the species level; the B-BH16-1 strain induces changes of fungal in community composition at the genus (C) and species (D) levels
LDA at the genus level and heatmap analysis at the OTU level showed that B-BH16-1 strain strongly promoted bacterial and fungal consortia assemblages on P. heterophylla folia. In the bacterial communities, the abundance of 42 genera significantly changed between the PP treatment and CK group, 40 of whichwere enriched in PP treatment group, and only two of which were increased in CK group (Fig. 4C). In addition, 69 significantly changed OTUs associated with B-BH16-1 strain belonged to 57 genera, of which 66 OTUs were enriched in the PP treatment group and only three OTUs were enriched in the CK group (Fig. 4D). In the fungal communities, the abundance of 31 genera significantly changed between the PP treatment and CK group, of which 28 were enriched in PP treatment group, and only three of which were increased in the CK group (Fig. 5C). Furthermore, B-BH16-1 strain induced 34 significantly changed OTUs belonging to 31 genera, in which 30 of which were enriched in the PP group, while only four OTUs were enriched in the CK group (Fig. 5D).
The P. palleroniana strain B-BH16-1 enhanced the disease resistance of P. heterophylla by obliterating pathogens and assembling beneficial microbiota
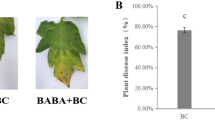
Pot experiments revealed that strain B-BH16-1 significantly decreased the disease incidence by 67.39% (Fig. 6A). Moreover, the field experiment also considerably reduced the disease incidence by 42.12% (Fig. 6B). Interestingly, the B-BH16-1 strain significantly improved the length of 50 tuberous roots by 3.84%. In addition, we detected the quality indicators of P. heterophylla, and the results showed that the application of strain B-BH16-1 greatly enhanced the polysaccharide and saponin contents by 13.97% and 6.91%, respectively (Fig. 6D and E). These results demonstrated that the B-BH16-1 strain enhances the disease resistance of P. heterophylla.

The B-BH16-1 strain enhances the disease resistance of P. heterophylla in pot (A) and continuous monocropping field (B) assays. The B-BH16-1 strain decreased the disease incidence in pot (A) and field (B) experiments and increased the diameter of 50 tuberous roots in field (C), and improved the polysaccharide (D) and saponin (E) contents; in addition, it increased the relative abundance of Pseudomonas (D) and decreased the relative abundance of Fusarium (E) in the P. heterophylla phyllosphere
The absolute abundance of Fusarium measured by qRT‒PCR was significantly lower in the B-BH16-1 treatment group than in the control group in April and May (Fig. 6F). In contrast, the absolute abundance of Pseudomonas determined by qRT‒PCR increased considerably in the PP treatment group in April and May (Fig. 6G). Microbial community sequencing analysis also revealed that the genus Fusarium was eliminated by the B-BH16-1 strain, whereas the genus Pseudomonas was significantly enriched in the PP treatment group (Figs. 4C and D and 5C, and 5D). In addition, the primary foliar fungal pathogen Botrytis [4] and the bacterial pathogen Robbsia [15] were also strongly repelled by strain B-BH16-1, except for Fusarium (Figs. 4C and 5C), and the OTU1843 belonging to Botrytis and the OTU2671 belonging to Robbsia were also eliminated (Figs. 4D and 5D).
In addition, beneficial bacteria, such as Bacillus (OTU784), Flexivirga (OTU830) [16], Pseudonocardia (OTU569) [17], Sphingobacterium (OTU592 and OTU2066) [18], Pseudarthrobacter (OTU641 and OTU2613) [19], Leucobacter (OTU1980) [20], Devosia [21], Exiguobacterium [22], Sinomonas (OTU2714) [23], Acinetobacter (OTU2675) [24], Catenulispora (OTU535) [25], and Ochrobactrum (OTU2729) [26], were significantly enriched in the strain B-BH16-1 treatment group (Fig. 4D). Moreover, the beneficial fungi at genus level, such as Sporobolomyces (OTU1815 and OTU258) [27], Paraphaeosphaeria (OTU313) [28], Lachnum (OTU447) [29], Coprinopsis [30], and Phlebia [31], were significantly enriched in the strain B-BH16-1 treatment group (Fig. 5D).
Discussion
Biologically important probiotics play essential roles in plant adaptation to diverse environments, especially environmental pressures such as drought, low temperatures, and pathogens, by promoting plant growth and enhancing plant disease resistance. Plant-associated microbial communities play an essential synergistic role in the effects of probiotics on disease resistance. Probiotics can reshape microbial communities to maintain their structural and functional stability, which is significant for plant health. In this study, pathogen-induced probiotics in P. heterophylla were screened and identified under continuous monocropping conditions. The broad-spectrum volatile antipathogen metabolites emitted by pathogen-induced Pseudomonas were explored. In addition, we revealed its role in reshaping the phyllosphere microbial communities of P. heterophylla, alleviating plant disease incidence, and improving P. heterophylla production.
Increasing evidence has shown that plant-associated microbial communities can be impacted by host factors, such as plant exudates and species; environmental factors, such as soil type and climate; agricultural management measures, such as cropping patterns; and stress, such as pathogen infection, as well as plant interaction to shape plant structure and function. Our previous study showed that Fusarium could extensively reshape the P. heterophylla rhizosphere microbiome under continuous monocropping conditions [1], which is in agreement with the finding that Fusarium-induced changes in pepper microbiome assembly and functional adaptation [32]. In the present study, 32 bacterial genera, as keystone taxa enriched in P. heterophylla rhizosphere soil, were induced by pathogens under continuous monocropping conditions (Fig. 1C). Some beneficial microbes, such as Pseudomonas, Chryseobacterium, and Flavobacterium, were significantly attracted by pathogen infection. This study differs considerably from previous findings that the population of beneficial bacteria decreased in the P. heterophylla rhizosphere with increasing years of monoculture [6]. In addition, pathogen-induced beneficial bacteria such as Pseudomonas, Chryseobacterium, and Flavobacterium were screened from P. heterophylla rhizosphere soil (Fig. 2A and B). Pathogen-reduced probiotics with strong specificity and persistence may be a disease management strategy with sound application prospects because they can effectively inhibit target pathogens and exist in soil or plants for a long time, continuously inhibiting pathogens growth and reproduction. This finding provides important insight for developing a self-adjustment biocontrol strategy using pathogen-mediated probiotics.
Increasing evidence has shown that probiotics can directly inhibit or indirectly compete with pathogens and hosts to occupy the ecological niche by producing a variety of metabolites. The present study confirmed that the P. palleroniana strain B-BH16-1 can directly inhibit the growth of seven primary pathogens of P. heterophylla foliar disease (i.e., Botrytis cinerea, Fusarium oxysporum, Alternaria altemata, Arcopilus aureus, Cladosporium cladosporioides, Nemania diffusa, and Whalleya mnicroplaca) [4] by emitting a variety of volatile organic compounds (VOCs), such as dimethyl disulfide, 1-undecene, and squalene (Fig. 3A and B, and 3C). This result is consistent with previous findings that beneficial endophytic Pseudomonas strains from olive roots strongly inhibit the mycelial growth of Verticillium dahlia by emitting VOCs [33]. Both qPCR analysis and amplicon sequencing analysis demonstrated that the population of Fusarium decreased in respose to strain B-BH16-1 (Fig. 5B and D, and 6F). Moreover, the abundance of Botrytis also decreased (Fig. 5B and D). These results demonstrated that the pathogen-mediated beneficial Pseudomonas strain B-BH16-1 can directly inhibit broad-spectrum pathogen growth and development to reshape plant-associated microbial communities involving the plant disease suppressors.
Previous evidence has shown that the occupying ecological niches of the pathogen infect the host mainly through the formation of spores for transmission [13] and the production of virulence factors such as phytotoxins that poison the host and inhibit the growth of other microorganisms [34,35,36]. In the present study, microscopy and qRT‒PCR analysis showed that the Fusarium spore formation was significantly inhibited by VOCs emitted from the B-BH16-1 strain (Fig. 3C and D), which is in agreement with the finding that Pseudomonas fluorescens inhibited the formation of F. oxysporum spores to prevent tomato disease [37]. A previous study revealed that disrupting the genes related to spore formation in Fusarium results in the dysfunction of its pathogenesis [38, 39]. This result indicated that the B-BH16-1 strain may restrict the occupation of ecological niches by inhibiting pathogen diffusion to control the spore formation, indirectly through the control of plant disease development.
In addition, fusaric acid, a virulence factor produced by Fusarium, breaks through the plant microbial barrier and becomes a dominant microbial community, assisting pathogens in infecting the host. In the present study, strain B-BH16-1 significantly inhibited fusaric acid production (Fig. 3F and G) and its vital synthesis gene expression (Fig. 3H) by emitting VOCs. Previous evidence has shown that fusaric acid can disrupt beneficial microbial communities such as Bacillus, Penicillium, Ruminococcus, and Methanobacter by inhibiting their growth and metabolite production [35, 36, 40,41,42,43,44]. Moreover, it can compete with beneficial microbiota ecological niches by blocking the quorum sensing system (i.e., biofilm formation and secretion system), inhibiting their reproduction and colonization, and reducing their protection of the host [45, 46]. In this study, the beneficial microbial communities were enriched by theB-BH16-1 strain (Figs. 4B and D and 5B and D). These results indicate that the B-BH16-1 strains may disrupt virulence factor biosynthesis to enhance ecological niches of beneficial microorganisms, indirectly involving the prevention of plant disease. These results show that inhibiting pathogen virulence biosynthesis to reshape the plant microbial community using disease-inducing probiotics will be an innovative strategy for managing plant disease, especially under continuous monoculture conditions.
The results of the present study suggest that pathogen-induced Pseudomonas reshaped phyllosphere microbial community in P. heterophylla via direct antagonism of the pathogen and indirect disruption of pathogen virulence factor biosynthesis to enhance the ecological niches of beneficial microorganisms, which is helpful for plant fitness, endowing plants with suppression abilities and improved the yields. Thirty-two keystone bacterial genera enriched in P. heterophylla rhizosphere soil were induced by pathogens under continuous monocropping conditions. Some beneficial microbes, such as Pseudomonas, Chryseobacterium, and Flavobacterium, were significantly attracted by pathogen infection. The P. palleroniana strain B-BH16-1 can directly inhibit the growth and spore formation of seven primary pathogens of P. heterophylla folia disease (i.e., Botrytis cinerea, Fusarium oxysporum, Alternaria altemata, Arcopilus aureus, Cladosporium cladosporioides, Nemania diffusa, and Whalleya mnicroplaca) by emitting a variety of volatile organic compounds (VOCs), such as dimethyl disulfide, 1-undecene, and squalene. Moreover, it can significantly inhibit fusaric acid production and vital synthesis gene expression by emitting VOCs. In addition, strain B-BH16-1 enhances the disease resistance of P. heterophylla by obliterating the pathogen and assembling beneficial microbiota. These results indicate that the B-BH16-1 strain may disrupt virulence factor biosynthesis to enhance the ecological niches of beneficial microorganisms, indirectly involving the prevention of plant disease. These results show that inhibiting pathogen virulence biosynthesis to reshape the plant microbial community using disease-inducing probiotics with strong specificity and persistence capabilities will be an innovative strategy for managing plant disease, especially under continuous monoculture conditions.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA803322).
References
Yuan Q-S, Wang L, Wang H, Wang X, Jiang W, Ou X, Xiao C, Gao Y, Xu J, Yang Y, et al. Pathogen-Mediated Assembly of Plant-Beneficial Bacteria to alleviate fusarium wilt in pseudostellaria heterophylla. Front microbiol. 2022;13:842372–86.
Wu L, Chen J, Wu H, Wang J, Wu Y, Lin S, Khan MU, Zhang Z, Lin W. Effects of consecutive monoculture of Pseudostellaria heterophylla on soil fungal community as determined by pyrosequencing. Sci Rep. 2016;6:26601–10.
Wu L, Chen J, Wu H, Qin X, Wang J, Wu Y, Khan MU, Lin S, Xiao Z, Luo X, et al. Insights into the regulation of rhizosphere bacterial communities by application of bio-organic fertilizer in Pseudostellaria heterophylla monoculture regime. Front Microbiol. 2016;7:1788–801.
Wang X-A, Gao Y, Jiang W, Wang L, Wang H, Ou X, Yang Y, Wu H, Guo L, Zhou T, et al. Comparative analysis of the expression of resistance-related genes respond to the Diversity Foliar Pathogens of Pseudostellaria heterophylla. Curr Microbiol. 2023;80(9):298–312.
Ma CY, Zhang W, Luo DL, Jiang HJ, Wu XH, Sun K, Dai CC. Fungal endophyte promotes plant growth and disease resistance of Arachis hypogaea L. by reshaping the core root microbiome under monocropping conditions. Microbiol Res. 2023;277:127491.
Zhao Y-P, Lin S, Chu L, Gao J, Azeem S, Lin W. Insight into structure dynamics of soil microbiota mediated by the richness of replanted Pseudostellaria heterophylla. Sci Rep. 2016;6:26175–83.
Wu H, Qin X, Wang J, Wu L, Chen J, Fan J, Zheng L, Tangtai H, Arafat Y, Lin W. Rhizosphere responses to environmental conditions in Radix pseudostellariae under continuous monoculture regimes. Agric Ecosyst Environ. 2019;270:19–31.
Prabhukarthikeyan SR, Keerthana U, Raguchander T. Antibiotic-producing Pseudomonas fluorescens mediates rhizome rot disease resistance and promotes plant growth in turmeric plants. Microbiol Res. 2018;210:65–73.
Tagele SB, Lee HG, Kim SW, Lee YS. Phenazine and 1-Undecene Producing Pseudomonas chlororaphis subsp. aurantiaca strain KNU17Pc1 for Growth Promotion and Disease suppression in Korean maize cultivars. J Microbiol Biotechnol. 2019;29(1):66–78.
Zhuang L, Li Y, Wang Z, Yu Y, Zhang N, Yang C, Zeng Q, Wang Q. Synthetic community with six Pseudomonas strains screened from garlic rhizosphere microbiome promotes plant growth. Microb Biotechnol. 2021;14(2):488–502.
Xun W, Li W, Xiong W, Ren Y, Liu Y, Miao Y, Xu Z, Zhang N, Shen Q, Zhang R. Diversity-triggered deterministic bacterial assembly constrains community functions. Nat Commun. 2019;10(1):3833.
Wen T, Xie P, Penton CR, Hale L, Thomashow LS, Yang S, Ding Z, Su Y, Yuan J, Shen Q. Specific metabolites drive the deterministic assembly of diseased rhizosphere microbiome through weakening microbial degradation of autotoxin. Microbiome. 2022;10(1):177–91.
Zhou S, Yuan Q-S, Wang X, Jiang W, Ou X, Yang C, Gao Y, Wang Y, Guo L, Huang L, et al. Volatiles from Pseudomonas palleroniana strain B-BH16-1 suppress aflatoxin production and growth of aspergillus flavus on Coix lacryma-jobi during storage. Toxins. 2023;15(1):77–92.
Ng CWW, Wang YC, Ni JJ, So PS. Effects of phosphorus-modified biochar as a soil amendment on the growth and quality of Pseudostellaria heterophylla. Sci Rep. 2022;12(1):7268–81.
Cui X, Cai Y, Chen R, Liu Q. First report of bacterial leaf spot disease on Pueraria montana var. Thomsonii caused by Robbsia Andropogonis in China. Plant Dis 2022.
Kan Y, Zhang L, Wang Y, Ma Q, Zhou Y, Jiang X, Zhang W, Ruan Z. Endophytic bacterium Flexivirga meconopsidis sp. nov. with plant growth-promoting function, isolated from the seeds of Meconopsis Integrifolia. Microorganisms 2023, 11(12).
Goldstein SL, Klassen JL. Pseudonocardia symbionts of Fungus-growing ants and the evolution of defensive secondary metabolism. Front Microbiol. 2020;11:621041.
Ahmed I, Ehsan M, Sin Y, Paek J, Khalid N, Hayat R, Chang YH. Sphingobacterium pakistanensis sp. nov., a novel plant growth promoting rhizobacteria isolated from rhizosphere of Vigna mungo. Antonie Van Leeuwenhoek. 2014;105(2):325–33.
Ham SH, Yoon AR, Oh HE, Park YG. Plant Growth-promoting microorganism pseudarthrobacter sp. NIBRBAC000502770 enhances the growth and flavonoid content of Geum aleppicum. Microorganisms 2022, 10(6).
Zhu J, Che J, Jiang X, Ma M, Guan D, Li L, Cao F, Zhao B, Kang Y, Zhao J et al. Leucobacter chinensis sp. nov., with plant growth-promoting potential isolated from field soil after seven-years continuous maize cropping. Int J Syst Evol MicroBiol 2022, 72(8).
Shah A, Subramanian S, Smith DL. Flavonoids and Devosia sp SL43 cell-free supernatant increase early plant growth under salt stress and optimal growth conditions. Front Plant Sci. 2022;13:1030985.
Kasana RC, Pandey CB. Exiguobacterium: an overview of a versatile genus with potential in industry and agriculture. Crit Rev Biotechnol. 2018;38(1):141–56.
Fu Y, Yan R, Liu D, Zhao J, Song J, Wang X, Cui L, Zhang J, Xiang W. Characterization of Sinomonas gamaensis sp. nov., a Novel Soil Bacterium with antifungal activity against Exserohilum turcicum. Microorganisms 2019, 7(6).
Silambarasan S, Vangnai AS. Biodegradation of 4-nitroaniline by plant-growth promoting Acinetobacter sp. AVLB2 and toxicological analysis of its biodegradation metabolites. J Hazard Mater. 2016;302:426–36.
Starr EP, Shi S, Blazewicz SJ, Koch BJ, Probst AJ, Hungate BA, Pett-Ridge J, Firestone MK, Banfield JF. Stable-Isotope-Informed, genome-resolved Metagenomics uncovers potential cross-kingdom interactions in Rhizosphere Soil. mSphere. 2021;6(5):e0008521.
Sipahutar MK, Vangnai AS. Role of plant growth-promoting Ochrobactrum sp. MC22 on triclocarban degradation and toxicity mitigation to legume plants. J Hazard Mater. 2017;329:38–48.
Jędrzejczyk RJ, Gustab M, Ważny R, Domka A, Jodłowski PJ, Sitarz M, Bezkosty P, Kowalski M, Pawcenis D, Jarosz K, et al. Iron inactivation by Sporobolomyces ruberrimus and its potential role in plant metal stress protection. An in vitro study. Sci Total Environ. 2023;870:161887.
Chen Q, Yu JJ, He J, Feng T, Liu JK. Isobenzofuranones and isocoumarins from Kiwi endophytic fungus paraphaeosphaeria sporulosa and their antibacterial activity against Pseudomonas syringae Pv. Actinidiae. Phytochemistry. 2022;195:113050.
Lou H, Guo C, Fan B, Fu R, Su H, Zhang J, Sun L. Lingonberry (Vaccinium vitis-idaea L.) Interact with Lachnum pygmaeum to Mitigate Drought and promote growth. Front Plant Sci. 2022;13:920338.
Bleuler-Martinez S, Varrot A, Olieric V, Schubert M, Vogt E, Fetz C, Wohlschlager T, Plaza DF, Wälti M, Duport Y, et al. Structure-function relationship of a novel fucoside-binding fruiting body lectin from Coprinopsis cinerea exhibiting nematotoxic activity. Glycobiology. 2022;32(7):600–15.
Ishihara A, Ashida C, Ube N, Abe M, Hiyoshi H, Umezu K, Endo N, Sotome K, Maekawa N, Nakagiri A, et al. Isolation of isolactarane sesquiterpenes from a Phlebia tremellosa culture filtrate and their growth promotion effects on lettuce roots. J Pesticide Sci. 2019;44(1):9–14.
Gao M, Xiong C, Gao C, Tsui CKM, Wang MM, Zhou X, Zhang AM, Cai L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome. 2021;9(1):187.
Montes-Osuna N, Cernava T, Gómez-Lama Cabanás C, Berg G, Mercado-Blanco J. Identification of volatile Organic compounds emitted by two beneficial endophytic Pseudomonas strains from Olive roots. Plants (Basel Switzerland) 2022, 11(3).
Liu S, Li J, Zhang Y, Liu N, Viljoen A, Mostert D, Zuo C, Hu C, Bi F, Gao H, et al. Fusaric acid instigates the invasion of banana by Fusarium oxysporum f. sp. cubense TR4. New Phytol. 2020;225(2):913–29.
Van Rij ET, Girard G, Lugtenberg BJJ, Bloemberg GV. Influence of fusaric acid on phenazine-1-carboxamide synthesis and gene expression of Pseudomonas chlororaphis strain PCL1391. Microbiology 2005, 151(8):2805–2814.
Quecine MC, Kidarsa TA, Goebel NC, Shaffer BT, Henkels MD, Zabriskie TM, Loper JE. An Interspecies Signaling System mediated by Fusaric Acid has parallel effects on Antifungal Metabolite production by Pseudomonas protegens strain Pf-5 and Antibiosis of Fusarium spp. Appl Environ Microbiol. 2015;82(5):1372–82.
Kamilova F, Lamers G, Lugtenberg B. Biocontrol strain Pseudomonas fluorescens WCS365 inhibits germination of Fusarium oxysporum spores in tomato root exudate as well as subsequent formation of new spores. Environ Microbiol. 2008;10(9):2455–61.
Wang G, Wang C, Hou R, Zhou X, Li G, Zhang S, Xu J-R. The AMT1 arginine methyltransferase gene is important for plant infection and normal hyphal growth in Fusarium Graminearum. PLoS ONE. 2012;7(5):e38324.
Lysøe E, Pasquali M, Breakspear A, Kistler HC. The transcription factor FgStuAp influences Spore Development, pathogenicity, and secondary metabolism in Fusarium Graminearum. Mol Plant Microbe Interact. 2010;24(1):54–67.
Landa BB, Cachinero-Díaz JM, Lemanceau P, Jiménez-Díaz RM, Alabouvette C. Effect of fusaric acid and phytoanticipins on growth of rhizobacteria and fusarium oxysporum. Can J Microbiol. 2002;48(11):971–85.
May HD, Wu Q, Blake CK. Effects of the Fusarium spp. mycotoxins fusaric acid and deoxynivalenol on the growth of Ruminococcus albus and Methanobrevibacter ruminantium. Can J Microbiol. 2000;46(8):692–9.
Notz R, Maurhofer M, Dubach H, Haas D, Défago G. Fusaric acid-producing strains of Fusarium oxysporum alter 2,4-diacetylphloroglucinol biosynthetic gene expression in Pseudomonas fluorescens CHA0 in vitro and in the rhizosphere of wheat. Appl Environ Microbiol. 2002;68(5):2229–35.
Van Rij ET, Wesselink M, C-A-WTF C, Bloemberg GV, Lugtenberg BJJ. Influence of environmental conditions on the production of phenazine-1-carboxamide by Pseudomonas chlororaphis PCL1391. Mol Plant Microbe Interact. 2004;17(5):557–66.
Crutcher FK, Puckhaber LS, Stipanovic RD, Bell AA, Nichols RL, Lawrence KS, Liu J. Microbial Resistance mechanisms to the antibiotic and Phytotoxin Fusaric Acid. J Chem Ecol. 2017;43(10):996–1006.
Li J, Sun W, Guo Z, Lu C, Shen Y. Fusaric acid modulates Type three Secretion System of Salmonella enterica Serovar Typhimurium. Biochem Biophys Res Commun. 2014;449(4):455–9.
Tung TT, Jakobsen TH, Dao TT, Fuglsang AT, Givskov M, Christensen SB, Nielsen J. Fusaric acid and analogues as Gram-negative bacterial quorum sensing inhibitors. Eur J Med Chem. 2017;126:1011–20.
Acknowledgements
Not applicable.
Funding
This work was supported by National Key R&D Program of China [2023YFC3503803], Guizhou Provincial Major Scientific and Technological Program [Qian Ke He Zhi Cheng (2022) Yi Ban 136], and Guizhou Provincial Major Scientific and Technological Program [Qian Ke He Zhi Cheng (2022) Yi Ban 136]
Author information
Authors and Affiliations
Contributions
Q.S.Y.: investigation, methodology, writing original draft, writing review and editing, and validation. Y.P.G.: investigation and validation. L.L.W.: visualization. L.W.: investigation, and methodology. X.A.W.: investigation, and methodology. L.L.W.: data curation. J.Y.R.: data curation. X.H.O. and C.H.X.: supervision. Y.H.W.: methodology. Y.W.: methodology. W.K.J.: conceptualization and supervision. L.P.G.: conceptualization and supervision. T.Z.: resources and project administration. L.Q.H.: conceptualization, and supervision. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yuan, QS., Gao, Y., Wang, L. et al. Pathogen-driven Pseudomonas reshaped the phyllosphere microbiome in combination with Pseudostellaria heterophylla foliar disease resistance via the release of volatile organic compounds. Environmental Microbiome 19, 61 (2024). https://doi.org/10.1186/s40793-024-00603-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-024-00603-3